
PPARγ in Atherosclerotic Endothelial Dysfunction: Regulatory Compounds and PTMs



Abstract
:1. Introduction
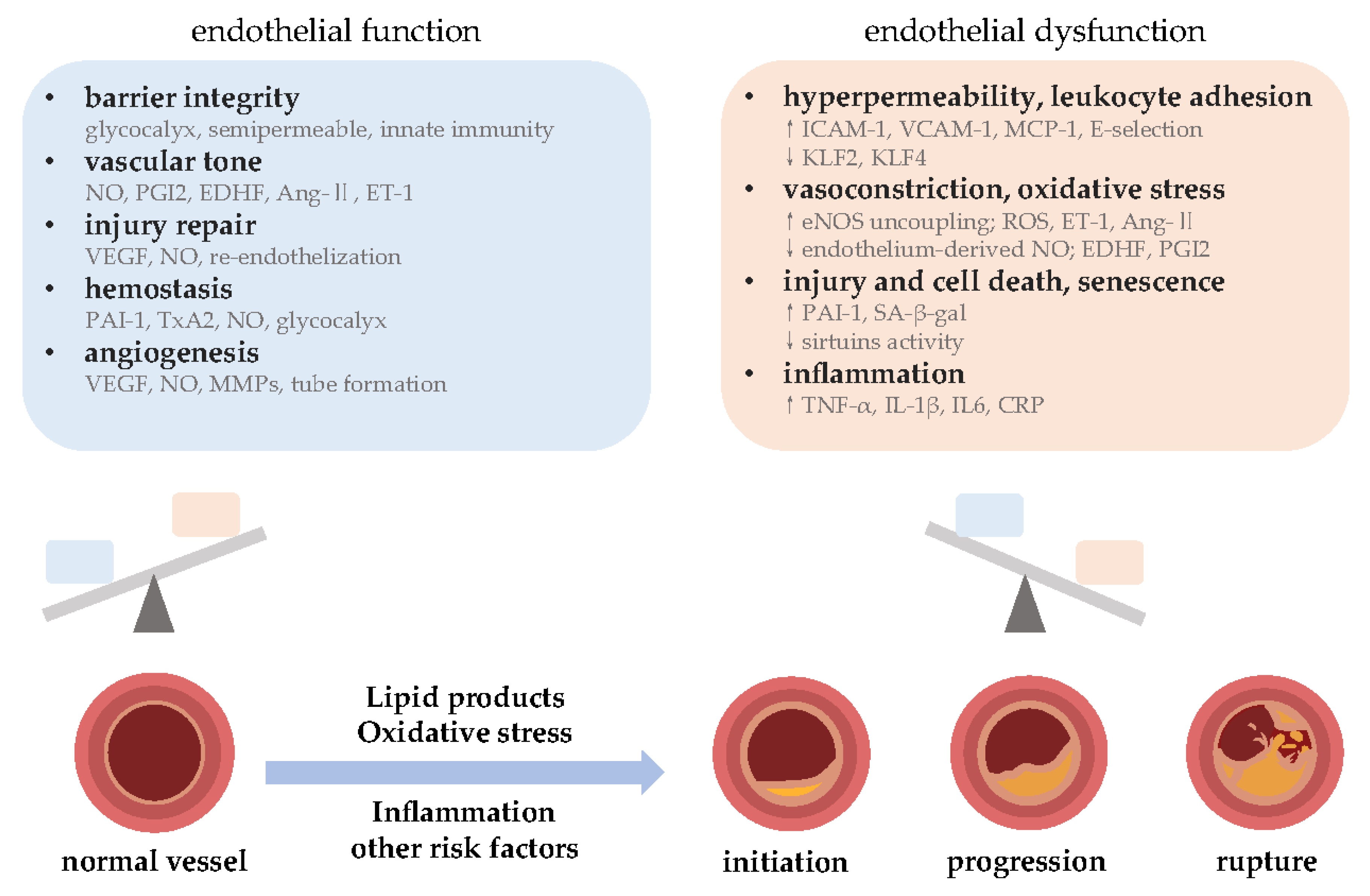
2. Endothelial Function and Atherosclerosis
2.1. Endothelial Function and Associated Molecular Pathways
2.2. Endothelial Dysfunction and Its Role in Atherosclerosis Development
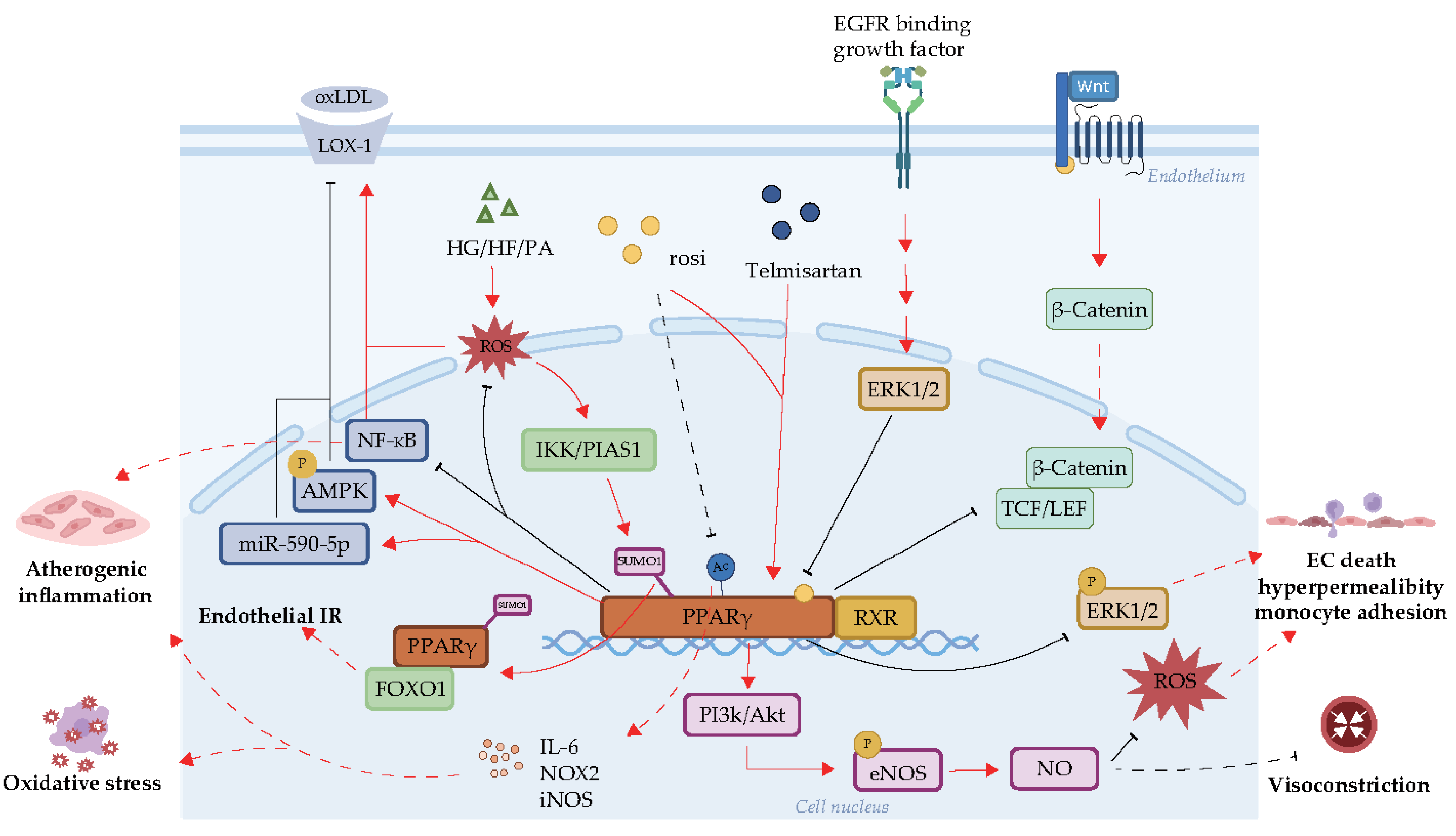
3. Effects of PPARγ on Endothelial Function
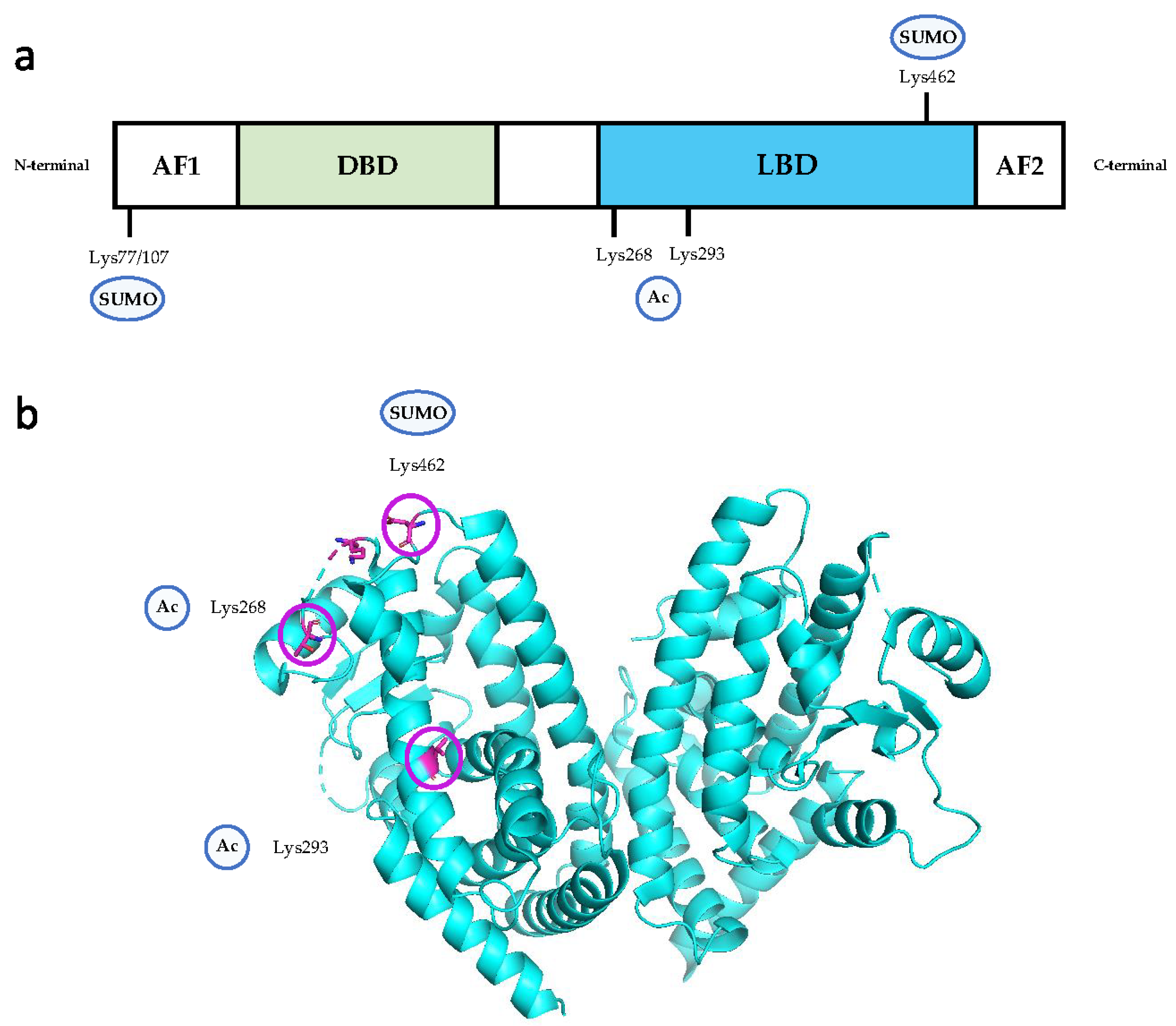
3.1. PTMs of PPARγ
3.2. Molecular Pathways of Endothelial Protection by PPARγ
3.3. Compounds Regulating PPARγ and Their Role in Endothelial Function
3.3.1. TZDs
3.3.2. Alkaloids
3.3.3. Saponins and Triterpenes
3.3.4. Isoflavones
3.3.5. Biological Metabolites
3.3.6. Dual PPARα/γ Agonists
3.3.7. Others

{kind=link}
{kind=link}
{kind=link}
| Class | Compound | Endothelial Change | Mechanism | References |
|---|---|---|---|---|
| TZDs | rosiglitazone | ↓oxidative and nitrative stresses, angiogenesis, leukocyte accumulation, inflammation ↑endothelium-dependent vasodilation, apoptosis | ↑PPARγ expression, eNOS activity, NO production ↓gp91phox and iNOS expression, superoxide and total NOx, nitrotyrosine, ERK activation ↑PPARγ-mediated maxi-K channel opening ↓diacylglycerol-protein kinase C signaling pathway ↓AT1-ROS-MAPK signal pathway | [90,95,96,97] |
| troglitazone | ↑Autophagy | independent of EGFR transactivation and PPARγ activation ↓ROS/NF-κB signaling pathway | [81,98] | |
| pioglitazone | ↓endothelial regrowth ↑apoptosis, endothelium-dependent dilation | ↑eNOS activity, VEGF protein levels, p38 MAPK activation ↓ERK activation ↓VEGF/FGF stimulation of the ERK 1/2 pathways | [99,100,101] | |
| ciglitazone | ↓inflammation ↑vasodilation | ↓NF-κB pathway through a PPARγ-dependent mechanism ↑NO independent of eNOS expression | [71,82] | |
| lobeglitazone | ↓leukocyte recruitment, inflammation, intima-media ratio | ↓adhesion molecules, NF-κB p65 nuclear translocation | [111] | |
| Alkaloids | hypaphorine | ↓inflammation | ↓TNF-α, IL-1β, MCP-1, VCAM-1 and TLR4 ↑PPARγ protein levels, phosphorylation of AMPK and ACC, Akt and mTOR ↓TLR4 and ↑PPARγ, dependent on PI3K/Akt/mTOR signal pathway | [102,103] |
| berberine | ↓oxidative stress ↑vasodilation, cell viabilities | ↑PPARγ ↓ROS | [104] | |
| Saponins and triterpenoids | notoginsenoside Fc | ↓inflammation, apoptosis ↑proliferation | ↑PPARγ ↓pro-inflammatory cytokines | [105] |
| ginsenoside-Rb1 | ↓angiogenesis | ↑PPARγ, PEDF protein ↓miR-33a expression | [106] | |
| 7,8-didehydrocimigenol (7,8-DHC) | ↓leukocyte recruitment, inflammation | ↑PPARγ ↓VCAM-1 (but not ICAM-1), NF-kB activity | [83] | |
| Isoflavones | genistein | ↓leukocyte recruitment, inflammation | ↑PPARγ Dependent on flow rather than regulation of the adhesion molecules | [74] |
| formononetin | ↓inflammation, oxidative stress, apoptosis | Stimulates PPARγ signaling | [54] | |
| Biological metabolites | 2-methoxyestradiol | ↑vasodilation | ↑p-Akt, p-eNOS, NO Via the PPARγ/PI3K/Akt pathway | [72] |
| Urolithin A(UA) | ↓monocyte adhesion, inflammation | ↓microRNA-27 expression ↑ERK/PPARγ pathway | [107] | |
| Dual PPARα/γ agonists | TAK-559 | ↓leukocyte recruitment, intimal thickness | ↓MCP-1 mRNA expression and secretion | [108] |
| conjugated linoleic acid | Normalizes EC responses to hemodynamic change | ↑PPAR and eNOS expression ↓Pro-atherogenic ET-1 response | [109] | |
| Others | 15d-PGJ2 | ↓endothelial cell activation, inflammation ↑vasodilation | ↑NO independent of eNOS expression ↑diacylglycerol kinase ↓diacylglycerol-protein kinase C signaling pathway ↓proinflammatory adhesion molecule expression ↓NF-κB pathway through a PPARγ-independent mechanism ↓ROS/NF-κB signaling pathway | [71,76,81,118] |
| tert-Butylhydroquinone | ↑vasodilation | ↑HO-1 to ↓ROS ↑DDAH to ↓asymmetric dimethylarginine ↑PPARγ/eNOS | [73] | |
| Magnolol | ↑insulin-induced vasodilation | ↑PPARγ, insulin-induced phosphorylated Akt and eNOS levels, NO production ↓TRB3 | [114] | |
| Telmisartan | ↑vasodilation ↓medial thickness, oxidative stress | ↑PPARγ, eNOS, p-eNOS, p-Akt, NO production ↓nitrotyrosine Via a PPARγ-dependent PI3K/Akt/eNOS pathway Related to the PPARγ/eNOS pathway and Rho-kinase pathway | [52,69,70] | |
| Cyclic phosphatidic acid (cPA) | ↓neointima formation | ↓VEGF-mediated growth and migration ↓PPARγ, cPA-PPARγ axis | [115] | |
| 1,8-Cineole | ↓inflammation | Via the PPARγ/NF-κB pathway ↑PPARγ expression ↓VCAM-1, phosphorylation of NF-κB p65, E-selectin, IL-6, and IL-8 | [79] | |
| carbon monoxide | ↓leukocyte recruitment | ↓endothelial ICAM-1 ↑AMPK/PPARγ pathway | [116] | |
| simvastatin | ↓leukocyte recruitment | ↓Neutrophil adhesion, ROS associated with ↑PPAR-γ and ↓RAGEs expression | [117] | |
| KR-62980 | ↓angiogenesis, EC proliferation and chemotactic migration ↑apoptosis | ↓VEGFR-2 ↑chromosome 10 (PTEN) | [113] |
3.4. PTMs of PPARγ Responsible for Endothelial Dysfunction
3.4.1. Acetylation
3.4.2. SUMOylation
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsumoto, T.; Kobayashi, T.; Kamata, K. Relationships among ET-1, PPARgamma, oxidative stress and endothelial dysfunction in diabetic animals. J. Smooth Muscle Res. 2008, 44, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.Z.; Usher, M.G.; Mortensen, R.M. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ. Res. 2008, 102, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. 2016 Russell Ross Memorial Lecture in Vascular Biology: Molecular-Cellular Mechanisms in the Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47 (Suppl. S8), C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109 (Suppl. S1), III27–III32. [Google Scholar] [CrossRef]
- Wang, X.; Wang, R.; Jiang, L.; Xu, Q.; Guo, X. Endothelial repair by stem and progenitor cells. J. Mol. Cell. Cardiol. 2022, 163, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Bhat, O.M.; Uday Kumar, P.; Harishankar, N.; Ravichandaran, L.; Bhatia, A.; Dhawan, V. Interleukin-18-induced cell adhesion molecule expression is associated with feedback regulation by PPAR-gamma and NF-kappaB in ApoE−/− mice. Mol. Cell. Biochem. 2017, 428, 119–128. [Google Scholar] [CrossRef]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M.; Guarino, P.D.; Lovejoy, A.M.; Peduzzi, P.N.; Conwit, R.; et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K.; Doi, T. The Role of PPARs in Cancer. PPAR Res. 2008, 2008, 102737. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.Z.; Althagafi, I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Panunti, B.; Fonseca, V. Effects of PPAR gamma agonists on cardiovascular function in obese, non-diabetic patients. Vascul. Pharmacol. 2006, 45, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Bruder-Nascimento, T.; Faulkner, J.L.; Haigh, S.; Kennard, S.; Antonova, G.; Patel, V.S.; Fulton, D.J.R.; Chen, W.; Belin de Chantemele, E.J. Leptin Restores Endothelial Function via Endothelial PPARgamma-Nox1-Mediated Mechanisms in a Mouse Model of Congenital Generalized Lipodystrophy. Hypertension 2019, 74, 1399–1408. [Google Scholar] [CrossRef]
- Martens, F.M.; Visseren, F.L.; Lemay, J.; de Koning, E.J.; Rabelink, T.J. Metabolic and additional vascular effects of thiazolidinediones. Drugs 2002, 62, 1463–1480. [Google Scholar] [CrossRef]
- Heikkinen, S.; Auwerx, J.; Argmann, C.A. PPARgamma in human and mouse physiology. Biochim. Biophys. Acta 2007, 1771, 999–1013. [Google Scholar] [CrossRef]
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARgamma signaling and emerging opportunities for improved therapeutics. Pharmacol. Res. 2016, 111, 76–85. [Google Scholar] [CrossRef]
- Sugawara, A.; Uruno, A.; Kudo, M.; Matsuda, K.; Yang, C.W.; Ito, S. PPARgamma agonist beyond glucose lowering effect. Korean J. Intern. Med. 2011, 26, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, A.; Uruno, A.; Matsuda, K.; Saito-Ito, T.; Funato, T.; Saito-Hakoda, A.; Kudo, M.; Ito, S. Effects of PPARgamma agonists against vascular and renal dysfunction. Curr. Mol. Pharmacol. 2012, 5, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef]
- Ji, X.; Zhang, W.; Yin, L.; Shi, Z.; Luan, J.; Chen, L.; Liu, L. The Potential Roles of Post-Translational Modifications of PPARgamma in Treating Diabetes. Biomolecules 2022, 12, 1832. [Google Scholar] [CrossRef]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, S.; Costa, V.; Ciccodicola, A.; Aprile, M. PPARgamma and Diabetes: Beyond the Genome and Towards Personalized Medicine. Curr. Diab. Rep. 2021, 21, 18. [Google Scholar] [CrossRef]
- Yin, L.; Wang, L.; Shi, Z.; Ji, X.; Liu, L. The Role of Peroxisome Proliferator-Activated Receptor Gamma and Atherosclerosis: Post-translational Modification and Selective Modulators. Front. Physiol. 2022, 13, 826811. [Google Scholar] [CrossRef]
- Reiterer, M.; Branco, C.M. Endothelial cells and organ function: Applications and implications of understanding unique and reciprocal remodelling. FEBS J. 2020, 287, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef]
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Ricard, N.; Bailly, S.; Guignabert, C.; Simons, M. The quiescent endothelium: Signalling pathways regulating organ-specific endothelial normalcy. Nat. Rev. Cardiol. 2021, 18, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef]
- Malik, A.B.; Lynch, J.J.; Cooper, J.A. Endothelial barrier function. J. Investig. Dermatol. 1989, 93 (Suppl. S2), 62S–67S. [Google Scholar] [CrossRef] [PubMed]
- Alphonsus, C.S.; Rodseth, R.N. The endothelial glycocalyx: A review of the vascular barrier. Anaesthesia 2014, 69, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C.t.; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell. Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef]
- Dennis, J.; Johnson, C.Y.; Adediran, A.S.; de Andrade, M.; Heit, J.A.; Morange, P.E.; Tregouet, D.A.; Gagnon, F. The endothelial protein C receptor (PROCR) Ser219Gly variant and risk of common thrombotic disorders: A HuGE review and meta-analysis of evidence from observational studies. Blood 2012, 119, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Boehme, M.W.; Gao, I.K.; Norden, C.; Lemmel, E.M. Decrease in circulating endothelial cell adhesion molecule and thrombomodulin levels during oral iloprost treatment in rheumatoid arthritis patients: Preliminary results. Rheumatol. Int. 2006, 26, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Andrulis, M.; Dietrich, S.; Longerich, T.; Koschny, R.; Burian, M.; Schmitt-Graf, A.; Schirmacher, P.; Ho, A.D.; Dreger, P.; Luft, T. Loss of endothelial thrombomodulin predicts response to steroid therapy and survival in acute intestinal graft-versus-host disease. Haematologica 2012, 97, 1674–1677. [Google Scholar] [CrossRef]
- Boehme, M.W.; Deng, Y.; Raeth, U.; Bierhaus, A.; Ziegler, R.; Stremmel, W.; Nawroth, P.P. Release of thrombomodulin from endothelial cells by concerted action of TNF-alpha and neutrophils: In vivo and in vitro studies. Immunology 1996, 87, 134–140. [Google Scholar] [PubMed]
- Pistrosch, F.; Matschke, J.B.; Schipp, D.; Schipp, B.; Henkel, E.; Weigmann, I.; Sradnick, J.; Bornstein, S.R.; Birkenfeld, A.L.; Hanefeld, M. Rivaroxaban compared with low-dose aspirin in individuals with type 2 diabetes and high cardiovascular risk: A randomised trial to assess effects on endothelial function, platelet activation and vascular biomarkers. Diabetologia 2021, 64, 2701–2712. [Google Scholar] [CrossRef]
- Ott, C.; Schneider, M.P.; Delles, C.; Schlaich, M.P.; Schmieder, R.E. Reduction in basal nitric oxide activity causes albuminuria. Diabetes 2011, 60, 572–576. [Google Scholar] [CrossRef]
- Adams, M.R.; McCredie, R.; Jessup, W.; Robinson, J.; Sullivan, D.; Celermajer, D.S. Oral L-arginine improves endothelium-dependent dilatation and reduces monocyte adhesion to endothelial cells in young men with coronary artery disease. Atherosclerosis 1997, 129, 261–269. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.; van Nieuw Amerongen, G.P.; Koolwijk, P.; van Hinsbergh, V.W.; Groeneveld, A.B. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax 2008, 63, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Han, B.; Zhang, R.; Liu, Q.; Wang, X.; Huang, X.; Liu, D.; Qiao, W.; Yang, M.; Luo, X.; et al. C1q/TNF-Related Protein 9 Attenuates Atherosclerosis by Inhibiting Hyperglycemia-Induced Endothelial Cell Senescence Through the AMPKalpha/KLF4 Signaling Pathway. Front. Pharmacol. 2021, 12, 758792. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Du, H.; Chai, Q.; Jia, Q.; Liu, L.; Zhao, M.; Li, J.; Tang, H.; Chen, W.; Zhao, L.; et al. Blocking mitochondrial cyclophilin D ameliorates TSH-impaired defensive barrier of artery. Redox Biol. 2018, 15, 418–434. [Google Scholar] [CrossRef]
- Turner, A.L.; Michaelson, L.V.; Shewry, P.R.; Lovegrove, A.; Spencer, J.P.E. Increased bioavailability of phenolic acids and enhanced vascular function following intake of feruloyl esterase-processed high fibre bread: A randomized, controlled, single blind, crossover human intervention trial. Clin. Nutr. 2021, 40, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Yoshida, M.; Nakano, Y.; Suganami, T.; Satoh, N.; Mita, T.; Azuma, K.; Itoh, M.; Yamamoto, Y.; Kamei, Y.; et al. In vivo and in vitro inhibition of monocyte adhesion to endothelial cells and endothelial adhesion molecules by eicosapentaenoic acid. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2173–2179. [Google Scholar] [CrossRef]
- O’Byrne, D.; Devaraj, S.; Islam, K.N.; Collazo, R.; McDonald, L.; Grundy, S.; Jialal, I. Low-density lipoprotein (LDL)-induced monocyte-endothelial cell adhesion, soluble cell adhesion molecules, and autoantibodies to oxidized-LDL in chronic renal failure patients on dialysis therapy. Metabolism 2001, 50, 207–215. [Google Scholar] [CrossRef]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef]
- Ikejima, H.; Imanishi, T.; Tsujioka, H.; Kuroi, A.; Kobayashi, K.; Shiomi, M.; Muragaki, Y.; Mochizuki, S.; Goto, M.; Yoshida, K.; et al. Effects of telmisartan, a unique angiotensin receptor blocker with selective peroxisome proliferator-activated receptor-gamma-modulating activity, on nitric oxide bioavailability and atherosclerotic change. J. Hypertens. 2008, 26, 964–972. [Google Scholar] [CrossRef]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar] [CrossRef]
- Zhang, B.; Hao, Z.; Zhou, W.; Zhang, S.; Sun, M.; Li, H.; Hou, N.; Jing, C.; Zhao, M. Formononetin protects against ox-LDL-induced endothelial dysfunction by activating PPAR-gamma signaling based on network pharmacology and experimental validation. Bioengineered 2021, 12, 4887–4898. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Mintz, G.S. What have we learned about plaque rupture in acute coronary syndromes? Curr. Cardiol. Rep. 2010, 12, 338–343. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Quillard, T.; Araujo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef]
- Chapman, M.J.; Redfern, J.S.; McGovern, M.E.; Giral, P. Niacin and fibrates in atherogenic dyslipidemia: Pharmacotherapy to reduce cardiovascular risk. Pharmacol. Ther. 2010, 126, 314–345. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Sacks, F.; Hermans, M.P.; Assmann, G.; Brown, W.V.; Ceska, R.; Chapman, M.J.; Dodson, P.M.; Fioretto, P.; Ginsberg, H.N.; et al. The Residual Risk Reduction Initiative: A call to action to reduce residual vascular risk in patients with dyslipidemia. Am. J. Cardiol. 2008, 102 (Suppl. S10), 1K–34K. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7A–11A. [Google Scholar] [CrossRef] [PubMed]
- Adamiec, R.; Gacka, M.; Dobosz, T.; Szymaniec, S.; Bednarska-Chabowska, D.; Sadakierska-Chudy, A. Stimulation of the peroxisome proliferator-activated receptor gamma (PPAR gamma) and the expression of selected blood monocyte cytokine genes in diabetic macroangiopathy. Atherosclerosis 2007, 194, e108–e115. [Google Scholar] [CrossRef]
- Moreno, P.R.; Fuster, V. New aspects in the pathogenesis of diabetic atherothrombosis. J. Am. Coll. Cardiol. 2004, 44, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Ugusman, A.; Kumar, J.; Aminuddin, A. Endothelial function and dysfunction: Impact of sodium-glucose cotransporter 2 inhibitors. Pharmacol. Ther. 2021, 224, 107832. [Google Scholar] [CrossRef]
- Luconi, M.; Cantini, G.; Serio, M. Peroxisome proliferator-activated receptor gamma (PPARgamma): Is the genomic activity the only answer? Steroids 2010, 75, 585–594. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, J.; Li, Y. Chronic central miR-29b antagonism alleviates angiotensin II-induced hypertension and vascular endothelial dysfunction. Life Sci. 2019, 235, 116862. [Google Scholar] [CrossRef]
- Li, H.; Lu, W.; Cai, W.W.; Wang, P.J.; Zhang, N.; Yu, C.P.; Wang, D.L.; Liu, B.C.; Sun, W. Telmisartan attenuates monocrotaline-induced pulmonary artery endothelial dysfunction through a PPAR gamma-dependent PI3K/Akt/eNOS pathway. Pulm. Pharmacol. Ther. 2014, 28, 17–24. [Google Scholar] [CrossRef]
- Kobayashi, N.; Ohno, T.; Yoshida, K.; Fukushima, H.; Mamada, Y.; Nomura, M.; Hirata, H.; Machida, Y.; Shinoda, M.; Suzuki, N.; et al. Cardioprotective mechanism of telmisartan via PPAR-gamma-eNOS pathway in dahl salt-sensitive hypertensive rats. Am. J. Hypertens. 2008, 21, 576–581. [Google Scholar] [CrossRef]
- Calnek, D.S.; Mazzella, L.; Roser, S.; Roman, J.; Hart, C.M. Peroxisome proliferator-activated receptor gamma ligands increase release of nitric oxide from endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cui, Y.; Zheng, S.; Huang, J.; Li, P.; Simoncini, T.; Zhang, Y.; Fu, X. 2-methoxyestradiol induces vasodilation by stimulating NO release via PPARgamma/PI3K/Akt pathway. PLoS ONE 2015, 10, e0118902. [Google Scholar] [CrossRef]
- Luo, Z.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Activation of nuclear factor erythroid 2-related factor 2 coordinates dimethylarginine dimethylaminohydrolase/PPAR-gamma/endothelial nitric oxide synthase pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension 2015, 65, 896–902. [Google Scholar] [CrossRef]
- Chacko, B.K.; Chandler, R.T.; Mundhekar, A.; Khoo, N.; Pruitt, H.M.; Kucik, D.F.; Parks, D.A.; Kevil, C.G.; Barnes, S.; Patel, R.P. Revealing anti-inflammatory mechanisms of soy isoflavones by flow: Modulation of leukocyte-endothelial cell interactions. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H908–H915. [Google Scholar] [CrossRef] [PubMed]
- Franceschelli, S.; De Cecco, F.; Pesce, M.; Ripari, P.; Guagnano, M.T.; Nuevo, A.B.; Grilli, A.; Sancilio, S.; Speranza, L. Hydroxytyrosol Reduces Foam Cell Formation and Endothelial Inflammation Regulating the PPARgamma/LXRalpha/ABCA1 Pathway. Int. J. Mol. Sci. 2023, 24, 2057. [Google Scholar] [CrossRef]
- Verrier, E.; Wang, L.; Wadham, C.; Albanese, N.; Hahn, C.; Gamble, J.R.; Chatterjee, V.K.; Vadas, M.A.; Xia, P. PPARgamma agonists ameliorate endothelial cell activation via inhibition of diacylglycerol-protein kinase C signaling pathway: Role of diacylglycerol kinase. Circ. Res. 2004, 94, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Scott, D.W.; Chandler, R.T.; Patel, R.P. Endothelial surface N-glycans mediate monocyte adhesion and are targets for anti-inflammatory effects of peroxisome proliferator-activated receptor gamma ligands. J. Biol. Chem. 2011, 286, 38738–38747. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhan, R.X.; Chen, J.Q.; Gao, Y.; Chen, L.; Kong, Y.; Zhong, X.J.; Liu, M.Q.; Chu, J.J.; Yan, G.Q.; et al. Pharmacological activation of PPAR gamma ameliorates vascular endothelial insulin resistance via a non-canonical PPAR gamma-dependent nuclear factor-kappa B trans-repression pathway. Eur. J. Pharmacol. 2015, 754, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Linghu, K.G.; Wu, G.P.; Fu, L.Y.; Yang, H.; Li, H.Z.; Chen, Y.; Yu, H.; Tao, L.; Shen, X.C. 1,8-Cineole Ameliorates LPS-Induced Vascular Endothelium Dysfunction in Mice via PPAR-gamma Dependent Regulation of NF-kappaB. Front. Pharmacol. 2019, 10, 178. [Google Scholar] [CrossRef]
- Marcone, S.; Haughton, K.; Simpson, P.J.; Belton, O.; Fitzgerald, D.J. Milk-derived bioactive peptides inhibit human endothelial-monocyte interactions via PPAR-gamma dependent regulation of NF-kappaB. J. Inflamm. 2015, 12, 1–13. [Google Scholar] [CrossRef]
- Tomita, H.; Osanai, T.; Toki, T.; Sasaki, S.; Maeda, N.; Murakami, R.; Magota, K.; Yasujima, M.; Okumura, K. Troglitazone and 15-deoxy-delta(12,14)-prostaglandin J2 inhibit shear-induced coupling factor 6 release in endothelial cells. Cardiovasc. Res. 2005, 67, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Cook, J.A.; O’Connor, M.; Zingarelli, B. Peroxisome proliferator-activated receptor gamma is required for the inhibitory effect of ciglitazone but not 15-deoxy-Delta 12,14-prostaglandin J2 on the NFkappaB pathway in human endothelial cells. Shock 2007, 28, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Mun, L.; Jun, M.S.; Kim, Y.M.; Lee, Y.S.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Son, K.H.; Lee, D.H.; Kim, Y.S.; et al. 7,8-didehydrocimigenol from Cimicifugae rhizoma inhibits TNF-alpha-induced VCAM-1 but not ICAM-1expression through upregulation of PPAR-gamma in human endothelial cells. Food Chem. Toxicol. 2011, 49, 166–172. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, K.; Peng, J.; Wang, C.; Kang, L.; Chang, N.; Sun, H. Rhizoma Dioscoreae Nipponicae polysaccharides protect HUVECs from H2O2-induced injury by regulating PPARgamma factor and the NADPH oxidase/ROS-NF-kappaB signal pathway. Toxicol. Lett. 2015, 232, 149–158. [Google Scholar] [CrossRef]
- Hofmann, A.; Brunssen, C.; Poitz, D.M.; Langbein, H.; Strasser, R.H.; Henle, T.; Ravens, U.; Morawietz, H. Lectin-like oxidized low-density lipoprotein receptor-1 promotes endothelial dysfunction in LDL receptor knockout background. Atheroscler. Suppl. 2017, 30 (Suppl. S30), 294–302. [Google Scholar] [CrossRef]
- Inoue, K.; Arai, Y.; Kurihara, H.; Kita, T.; Sawamura, T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ. Res. 2005, 97, 176–184. [Google Scholar] [CrossRef]
- Chiba, Y.; Ogita, T.; Ando, K.; Fujita, T. PPARgamma ligands inhibit TNF-alpha-induced LOX-1 expression in cultured endothelial cells. Biochem. Biophys. Res. Commun. 2001, 286, 541–546. [Google Scholar] [CrossRef]
- Mehta, J.L.; Hu, B.; Chen, J.; Li, D. Pioglitazone inhibits LOX-1 expression in human coronary artery endothelial cells by reducing intracellular superoxide radical generation. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2203–2208. [Google Scholar] [CrossRef]
- Ho, T.C.; Chen, S.L.; Yang, Y.C.; Liao, C.L.; Cheng, H.C.; Tsao, Y.P. PEDF induces p53-mediated apoptosis through PPAR gamma signaling in human umbilical vein endothelial cells. Cardiovasc. Res. 2007, 76, 213–223. [Google Scholar] [CrossRef]
- Kim, K.Y.; Cheon, H.G. Antiangiogenic effect of rosiglitazone is mediated via peroxisome proliferator-activated receptor gamma-activated maxi-K channel opening in human umbilical vein endothelial cells. J. Biol. Chem. 2006, 281, 13503–13512. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhao, G.; Zhu, H.; Wang, S.; Sun, A.; Zou, Y.; Ge, J. Peroxisome Proliferator-Activated Receptor-gamma Antagonizes LOX-1-Mediated Endothelial Injury by Transcriptional Activation of miR-590-5p. PPAR Res. 2019, 2019, 2715176. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Shah, Y.M.; Manna, S.K.; Gonzalez, F.J. Disruption of endothelial peroxisome proliferator-activated receptor gamma accelerates diet-induced atherogenesis in LDL receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.M.; de Lange, W.J.; Halabi, C.M.; Modrick, M.L.; Keen, H.L.; Faraci, F.M.; Sigmund, C.D. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ. Res. 2008, 103, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Mukohda, M.; Stump, M.; Ketsawatsomkron, P.; Hu, C.; Quelle, F.W.; Sigmund, C.D. Endothelial PPAR-gamma provides vascular protection from IL-1beta-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H39–H48. [Google Scholar] [CrossRef]
- Tao, L.; Liu, H.R.; Gao, E.; Teng, Z.P.; Lopez, B.L.; Christopher, T.A.; Ma, X.L.; Batinic-Haberle, I.; Willette, R.N.; Ohlstein, E.H.; et al. Antioxidative, antinitrative, and vasculoprotective effects of a peroxisome proliferator-activated receptor-gamma agonist in hypercholesterolemia. Circulation 2003, 108, 2805–2811. [Google Scholar] [CrossRef]
- Lombardi, A.; Cantini, G.; Piscitelli, E.; Gelmini, S.; Francalanci, M.; Mello, T.; Ceni, E.; Varano, G.; Forti, G.; Rotondi, M.; et al. A new mechanism involving ERK contributes to rosiglitazone inhibition of tumor necrosis factor-alpha and interferon-gamma inflammatory effects in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Han, C.J.; Liu, J.T.; Li, M.; Cui, M.; Pang, X.M.; Mao, J.J.; Liu, X.F. Rosiglitazone inhibits angiotensin II-induced C-reactive protein production in human aortic endothelial cells through regulating AT(1)-ROS-MAPK signal pathway. Inflamm. Res. 2012, 61, 1031–1037. [Google Scholar] [CrossRef]
- Yan, J.; Yang, H.; Wang, G.; Sun, L.; Zhou, Y.; Guo, Y.; Xi, Z.; Jiang, X. Autophagy augmented by troglitazone is independent of EGFR transactivation and correlated with AMP-activated protein kinase signaling. Autophagy 2010, 6, 67–73. [Google Scholar] [CrossRef]
- Desouza, C.V.; Gerety, M.; Hamel, F.G. Effects of a PPAR-γ agonist, on growth factor and insulin stimulated endothelial cells. Vascul. Pharmacol. 2009, 51, 162–168. [Google Scholar] [CrossRef]
- Campia, U.; Matuskey, L.A.; Panza, J.A. Peroxisome proliferator-activated receptor-γ activation with pioglitazone improves endothelium-dependent dilation in nondiabetic patients with major cardiovascular risk factors. Circulation 2006, 113, 867–875. [Google Scholar] [CrossRef]
- Huang, P.H.; Sata, M.; Nishimatsu, H.; Sumi, M.; Hirata, Y.; Nagai, R. Pioglitazone ameliorates endothelial dysfunction and restores ischemia-induced angiogenesis in diabetic mice. Biomed. Pharmacother. 2008, 62, 46–52. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Lin, W.; Zhou, Y.; Cai, W.; Qiu, L. Interactions of TLR4 and PPARγ, Dependent on AMPK Signalling Pathway Contribute to Anti-Inflammatory Effects of Vaccariae Hypaphorine in Endothelial Cells. Cell. Physiol. Biochem. 2017, 42, 1227–1239. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Cai, W.; Qiu, L. Hypaphorine Attenuates Lipopolysaccharide-Induced Endothelial Inflammation via Regulation of TLR4 and PPAR-γ Dependent on PI3K/Akt/mTOR Signal Pathway. Int. J. Mol. Sci. 2017, 18, 844. [Google Scholar] [CrossRef]
- Li, H.; He, C.; Wang, J.; Li, X.; Yang, Z.; Sun, X.; Fang, L.; Liu, N. Berberine activates peroxisome proliferator-activated receptor gamma to increase atherosclerotic plaque stability in Apoe−/− mice with hyperhomocysteinemia. J. Diabetes Investig. 2016, 7, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, C.; Ma, X.; Wang, J. Notoginsenoside Fc attenuates high glucose-induced vascular endothelial cell injury via upregulation of PPAR-γ in diabetic Sprague-Dawley rats. Vascul. Pharmacol. 2018, 109, 27–35. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, X.; Kwok, H.H.; Dong, M.; Liu, Z.; Poon, P.Y.; Luan, X.; Ngok-Shun Wong, R. Ginsenoside-Rb1-Mediated Anti-angiogenesis via Regulating PEDF and miR-33a through the Activation of PPAR-γ Pathway. Front. Pharmacol. 2017, 8, 783. [Google Scholar] [CrossRef]
- Han, Q.A.; Yan, C.; Wang, L.; Li, G.; Xu, Y.; Xia, X. Urolithin A attenuates ox-LDL-induced endothelial dysfunction partly by modulating microRNA-27 and ERK/PPAR-γ pathway. Mol. Nutr. Food. Res. 2016, 60, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Seki, N.; Bujo, H.; Jiang, M.; Shibasaki, M.; Takahashi, K.; Hashimoto, N.; Saito, Y. A potent activator of PPARalpha and gamma reduces the vascular cell recruitment and inhibits the intimal thickning in hypercholesterolemic rabbits. Atherosclerosis 2005, 178, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dancu, M.B.; Berardi, D.E.; Vanden Heuvel, J.P.; Tarbell, J.M. Atherogenic Endothelial Cell eNOS and ET-1 Responses to Asynchronous Hemodynamics are Mitigated by Conjugated Linoleic Acid. Ann. Biomed. Eng. 2007, 35, 1111–1119. [Google Scholar] [CrossRef]
- Nanjan, M.J.; Mohammed, M.; Prashantha Kumar, B.R.; Chandrasekar, M.J.N. Thiazolidinediones as antidiabetic agents: A critical review. Bioorg. Chem. 2018, 77, 548–567. [Google Scholar] [CrossRef]
- Lim, S.; Lee, K.S.; Lee, J.E.; Park, H.S.; Kim, K.M.; Moon, J.H.; Choi, S.H.; Park, K.S.; Kim, Y.B.; Jang, H.C. Effect of a new PPAR-gamma agonist, lobeglitazone, on neointimal formation after balloon injury in rats and the development of atherosclerosis. Atherosclerosis 2015, 243, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef]
- Kim, K.Y.; Ahn, J.H.; Cheon, H.G. Anti-angiogenic action of PPARgamma ligand in human umbilical vein endothelial cells is mediated by PTEN upregulation and VEGFR-2 downregulation. Mol. Cell. Biochem. 2011, 358, 375–385. [Google Scholar] [CrossRef]
- Liang, X.; Xing, W.; He, J.; Fu, F.; Zhang, W.; Su, F.; Liu, F.; Ji, L.; Gao, F.; Su, H.; et al. Magnolol administration in normotensive young spontaneously hypertensive rats postpones the development of hypertension: Role of increased PPAR gamma, reduced TRB3 and resultant alleviative vascular insulin resistance. PLoS ONE 2015, 10, e0120366. [Google Scholar] [CrossRef]
- Tsukahara, T.; Tsukahara, R.; Haniu, H.; Matsuda, Y.; Murakami-Murofushi, K. Cyclic phosphatidic acid inhibits the secretion of vascular endothelial growth factor from diabetic human coronary artery endothelial cells through peroxisome proliferator-activated receptor gamma. Mol. Cell. Endocrinol. 2015, 412, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Nizamutdinova, I.T.; Kim, Y.M.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. Carbon monoxide (from CORM-2) inhibits high glucose-induced ICAM-1 expression via AMP-activated protein kinase and PPAR-gamma activations in endothelial cells. Atherosclerosis 2009, 207, 405–411. [Google Scholar] [CrossRef]
- Spadaccio, C.; De Marco, F.; Di Domenico, F.; Coccia, R.; Lusini, M.; Barbato, R.; Covino, E.; Chello, M. Simvastatin attenuates the endothelial pro-thrombotic shift in saphenous vein grafts induced by Advanced glycation endproducts. Thromb. Res. 2014, 133, 418–425. [Google Scholar] [CrossRef]
- Imaizumi, T.; Kumagai, M.; Nishi, N.; Hirashima, M.; Hatakeyama, M.; Tamo, W.; Yoshida, H.; Nakamura, T.; Okumura, K.; Satoh, K. 15-deoxy-Δ12,14-prostaglandin J2 inhibits IFN-gamma-induced galectin-9 expression in cultured human umbilical vein endothelial cells. Int. Arch. Allergy Immunol. 2003, 131, 57–61. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; B’Nai Taub, A.; Yu, L.; Yao, Y.; Zhang, R.; Zahr, T.; Aaron, N.; LeSauter, J.; Fan, L.; Liu, L.; et al. PPARgamma Acetylation Orchestrates Adipose Plasticity and Metabolic Rhythms. Adv. Sci. 2023, 10, e2204190. [Google Scholar] [CrossRef]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef]
- Kumar, J.; Kumar, S. Sirtuin1 in vascular endothelial function, an overview. Epigenetics 2022, 17, 953–969. [Google Scholar] [CrossRef]
- Takahashi, N.; Kawada, T.; Yamamoto, T.; Goto, T.; Taimatsu, A.; Aoki, N.; Kawasaki, H.; Taira, K.; Yokoyama, K.K.; Kamei, Y.; et al. Overexpression and ribozyme-mediated targeting of transcriptional coactivators CREB-binding protein and p300 revealed their indispensable roles in adipocyte differentiation through the regulation of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2002, 277, 16906–16912. [Google Scholar] [CrossRef] [PubMed]
- Gelman, L.; Zhou, G.; Fajas, L.; Raspe, E.; Fruchart, J.C.; Auwerx, J. p300 interacts with the N- and C-terminal part of PPARγ2 in a ligand-independent and -dependent manner, respectively. J. Biol. Chem. 1999, 274, 7681–7688. [Google Scholar] [CrossRef] [PubMed]
- Zahr, T.; Liu, L.; Chan, M.; Zhou, Q.; Cai, B.; He, Y.; Aaron, N.; Accili, D.; Sun, L.; Qiang, L. PPARgamma (Peroxisome Proliferator-Activated Receptor gamma) Deacetylation Suppresses Aging-Associated Atherosclerosis and Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fan, L.; Chan, M.; Kraakman, M.J.; Yang, J.; Fan, Y.; Aaron, N.; Wan, Q.; Carrillo-Sepulveda, M.A.; Tall, A.R.; et al. PPARgamma Deacetylation Confers the Antiatherogenic Effect and Improves Endothelial Function in Diabetes Treatment. Diabetes 2020, 69, 1793–1803. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, Z.; Zhang, F.; Huang, L.; Xing, C.; Liu, N.; Xu, Y.; Wang, X. HDAC3 inhibition prevents oxygen glucose deprivation/reoxygenation-induced transendothelial permeability by elevating PPARgamma activity in vitro. J. Neurochem. 2019, 149, 298–310. [Google Scholar] [CrossRef]
- Katafuchi, T.; Holland, W.L.; Kollipara, R.K.; Kittler, R.; Mangelsdorf, D.J.; Kliewer, S.A. PPARγ-K107 SUMOylation regulates insulin sensitivity but not adiposity in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 12102–12111. [Google Scholar] [CrossRef]
- Lim, S.; Ahn, B.Y.; Chung, S.S.; Park, H.S.; Cho, B.J.; Kim, M.; Choi, S.H.; Lee, I.K.; Lee, S.W.; Choi, S.J.; et al. Effect of a peroxisome proliferator-activated receptor gamma sumoylation mutant on neointimal formation after balloon injury in rats. Atherosclerosis 2009, 206, 411–417. [Google Scholar] [CrossRef]
- Lan, D.; Shen, X.; Yuan, W.; Zhou, Y.; Huang, Q. Sumoylation of PPARγ contributes to vascular endothelium insulin resistance through stabilizing the PPARγ-NcoR complex. J. Cell. Physiol. 2019, 234, 19663–19674. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Niu, A.; Yuan, W.; Zhou, Y.; Xia, M.; Xiong, X.; Lu, Y.; Yin, T.; Zhang, Y.; Chen, S.; et al. Interaction of FOXO1 and SUMOylated PPARγ1 induced by hyperlipidemia and hyperglycemia favors vascular endothelial insulin resistance and dysfunction. Vascul. Pharmacol. 2022, 147, 107125. [Google Scholar] [CrossRef]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.; Zushin, P.J.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.P.; Spiegelman, B.M. An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Stechschulte, L.A.; Czernik, P.J.; Rotter, Z.C.; Tausif, F.N.; Corzo, C.A.; Marciano, D.P.; Asteian, A.; Zheng, J.; Bruning, J.B.; Kamenecka, T.M.; et al. PPARG Post-translational Modifications Regulate Bone Formation and Bone Resorption. EBioMedicine 2016, 10, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Dong, Y.G.; Li, H.L. PPARgamma phosphorylation mediated by JNK MAPK: A potential role in macrophage-derived foam cell formation. Acta Pharmacol. Sin. 2006, 27, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luan, J.; Ji, X.; Liu, L. PPARγ in Atherosclerotic Endothelial Dysfunction: Regulatory Compounds and PTMs. Int. J. Mol. Sci. 2023, 24, 14494. https://doi.org/10.3390/ijms241914494
Luan J, Ji X, Liu L. PPARγ in Atherosclerotic Endothelial Dysfunction: Regulatory Compounds and PTMs. International Journal of Molecular Sciences. 2023; 24(19):14494. https://doi.org/10.3390/ijms241914494
Chicago/Turabian StyleLuan, Jinwen, Xiaohui Ji, and Longhua Liu. 2023. "PPARγ in Atherosclerotic Endothelial Dysfunction: Regulatory Compounds and PTMs" International Journal of Molecular Sciences 24, no. 19: 14494. https://doi.org/10.3390/ijms241914494
APA StyleLuan, J., Ji, X., & Liu, L. (2023). PPARγ in Atherosclerotic Endothelial Dysfunction: Regulatory Compounds and PTMs. International Journal of Molecular Sciences, 24(19), 14494. https://doi.org/10.3390/ijms241914494