
DNA Damage Induced by T-2 Mycotoxin in Human Skin Fibroblast Cell Line—Hs68

 , ,
, ,
Abstract
1. Introduction
2. Results
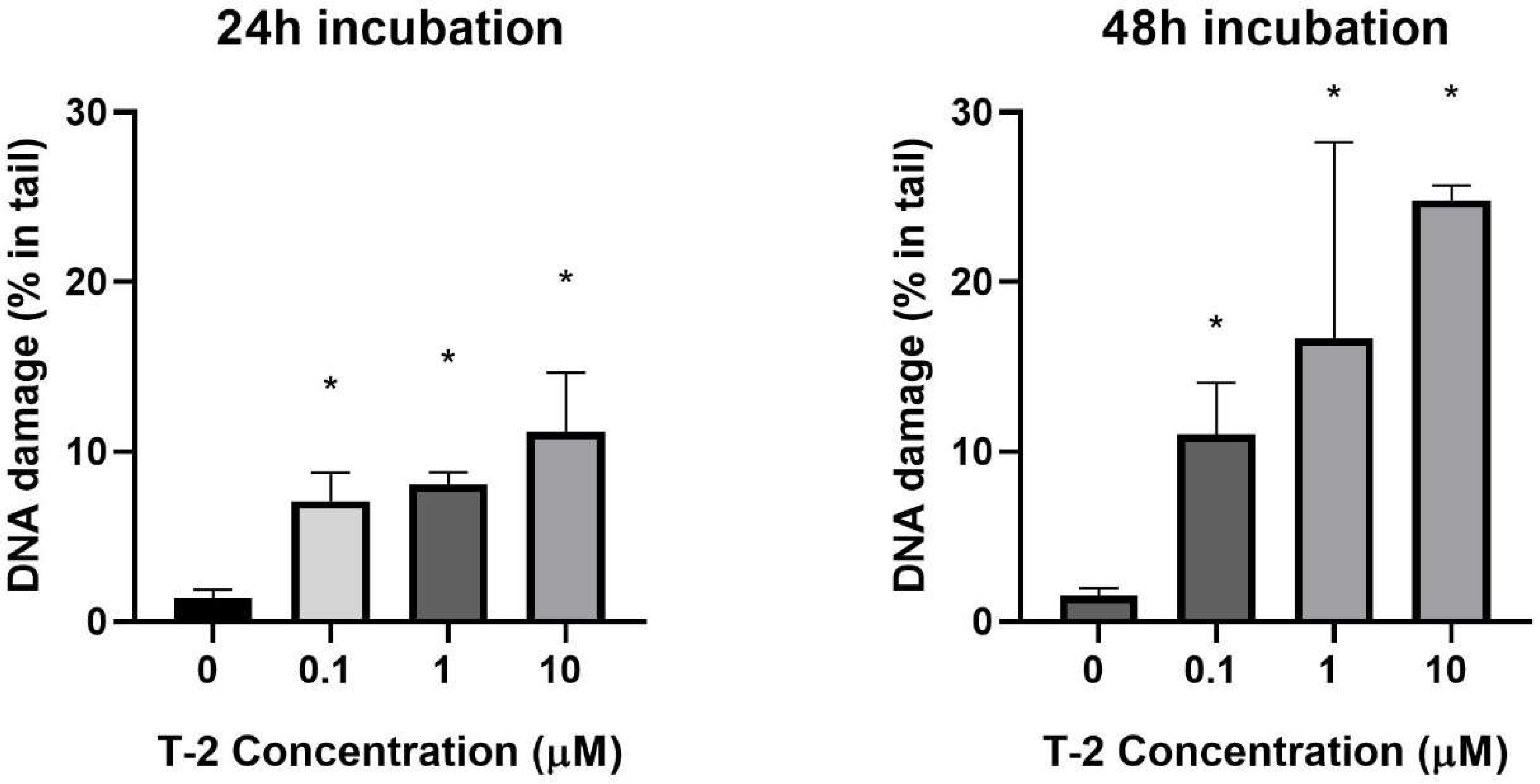
2.1. DNA Damage—Comet Assay
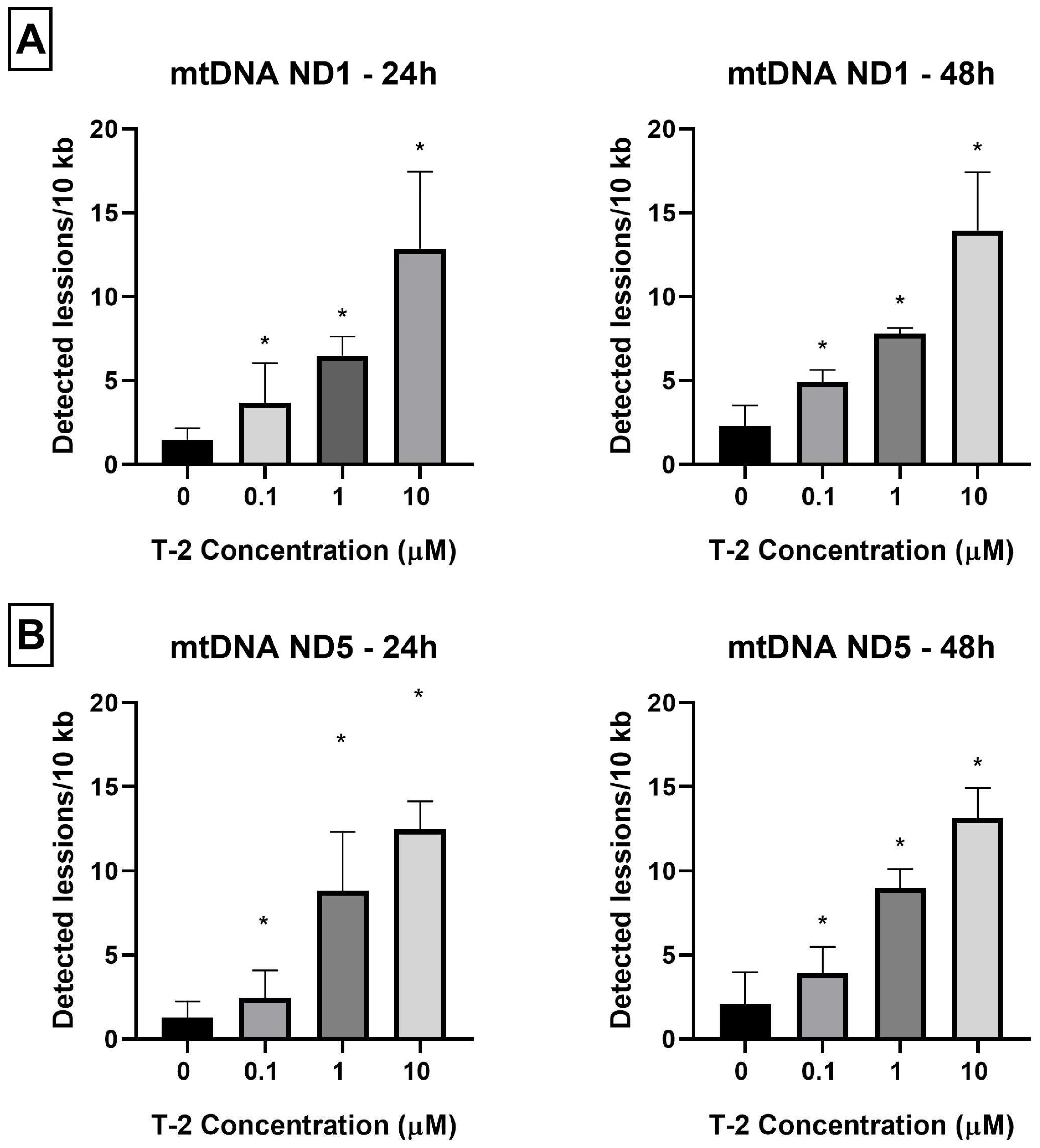
2.2. Determination of Nuclear DNA Damage—Semi-Long Run qRT-PCR (SLR-qRT-PCR)
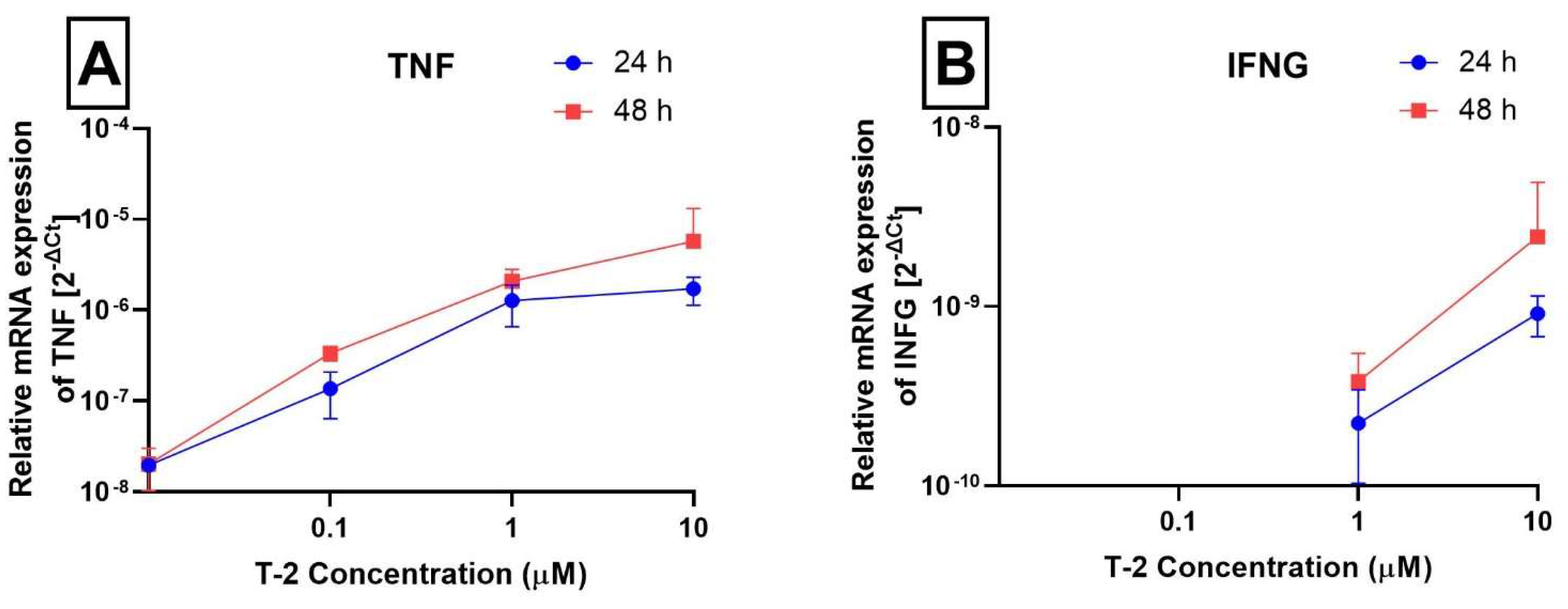
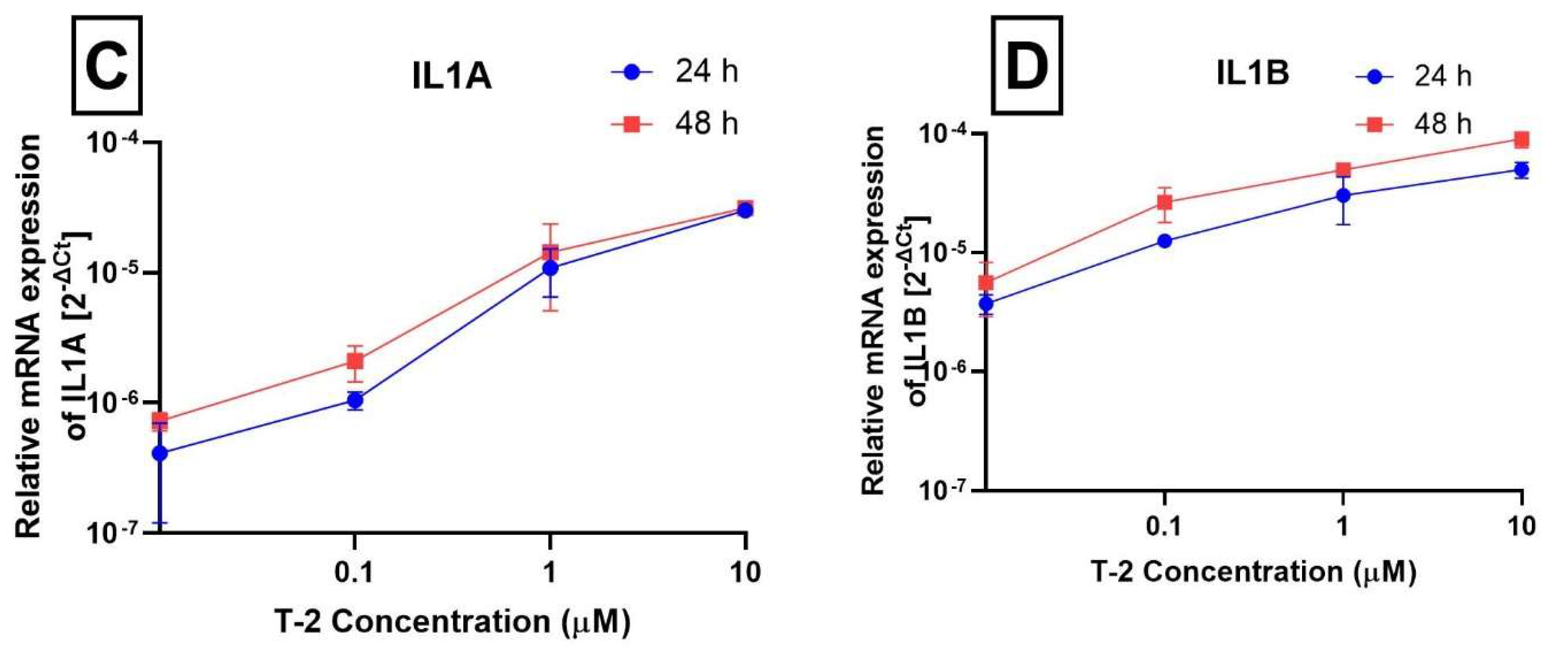
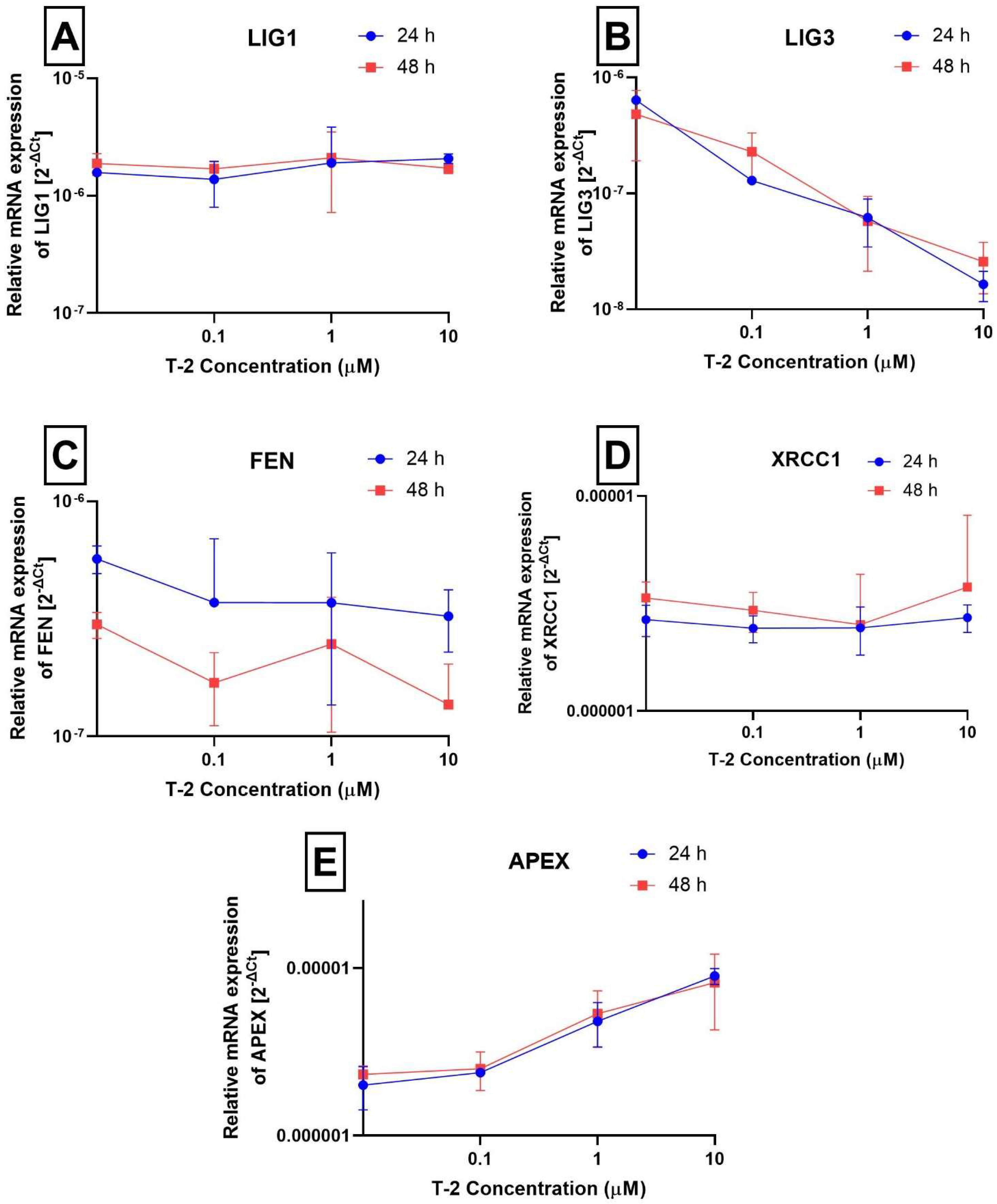
2.3. Analysis of Gene Expression
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. DNA Damage—Comet Assay
4.4. Isolation of Total Genomic DNA from Cell Lines
4.5. Determination of Nuclear DNA Damage—Semi-Long Run qRT-PCR (SLR-qRT-PCR)
4.6. Gene Expression Analysis Using Real-Time PCR
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grovey, J.F. The trichothecenes and their biosynthesis. In Progress in the Chemistry of Organic Natural Products; Springer: Vienna, Austia, 2007; pp. 63–130. [Google Scholar]
- Chaudhary, M.; Bhaskar, A.S.B.; Rao, P.V.L. Differential effects of route of T-2 toxin exposure on hepatic oxidative damage in mice. Environ. Toxicol. 2015, 30, 64–73. [Google Scholar] [CrossRef]
- Nayakwadi, S.; Ramu, R.; Kumar Sharma, A.; Kumar Gupta, V.; Rajukumar, K.; Kumar, V.; Shirahatti, P.S.; Basalingappa, K.M. Toxicopathological studies on the effects of T-2 mycotoxin and their interaction in juvenile goats. PLoS ONE 2020, 15, e0229463. [Google Scholar]
- Rahman, S.; Sharma, A.K.; Singh, N.; Prawez, S. T-2 toxin induced nephrotoxicity in Wistar rats. Indian J. Vet. Pathol. 2016, 40, 320. [Google Scholar] [CrossRef]
- Lin, R.; Sun, Y.; Ye, W.; Zheng, T.; Wen, J.; Deng, Y. T-2 toxin inhibits the production of mucin via activating the IRE1/XBP1 pathway. Toxicology 2019, 424, 152230. [Google Scholar] [CrossRef]
- Weidner, M.; Lenczyk, M.; Schwerdt, G.; Gekle, M.; Humpf, H.-U. Neurotoxic Potential and Cellular Uptake of T-2 Toxin in Human Astrocytes in Primary Culture. Chem. Res. Toxicol. 2013, 26, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bhavanishankar, T.N.; Ramesh, H.P.; Shantha, T. Dermal toxicity of Fusarium toxins in combinations. Arch. Toxicol. 1988, 61, 241–244. [Google Scholar] [CrossRef]
- Janik-Karpinska, E.; Ceremuga, M.; Wieckowska, M.; Szyposzynska, M.; Niemcewicz, M.; Synowiec, E.; Sliwinski, T.; Bijak, M. Direct T-2 Toxicity on Human Skin-Fibroblast Hs68 Cell Line-In Vitro Study. Int. J. Mol. Sci. 2022, 23, 4929. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yang, D.; Huang, Y.; Wang, S. Study on the apoptosis mechanism induced by T-2 toxin. PLoS ONE 2013, 8, e83105. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.F.; Atroshi, F.; Ahotupa, M.; Sankari, S.; Elovaara, E. Protective Effect of Antioxidants against Free Radical-Mediated Lipid Peroxidation Induced by DON or T-2 Toxin. J. Vet. Med. Ser. A 1994, 41, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Guo, X.; Pang, Q.; Wu, S.; Wu, C.; Xu, P.; Bai, Y. The role of mitochondria in T-2 toxin-induced human chondrocytes apoptosis. PLoS ONE 2014, 9, e108394. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, J.; Zhu, C.-C.; Tang, F.; Cui, X.-S.; Kim, N.-H.; Sun, S.-C. Exposure to HT-2 toxin causes oxidative stress induced apoptosis/autophagy in porcine oocytes. Sci. Rep. 2016, 6, 33904. [Google Scholar] [CrossRef]
- Chaudhari, M.; Jayaraj, R.; Bhaskar, A.S.B.; Lakshmana Rao, P.V. Oxidative stress induction by T-2 toxin causes DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology 2009, 262, 153–161. [Google Scholar] [CrossRef]
- Yuan, Z.; Matias, F.B.; Yi, J.-e.; Wu, J. T-2 toxin-induced cytotoxicity and damage on TM3 Leydig cells. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2016, 181–182, 47–54. [Google Scholar] [CrossRef]
- Frankič, T.; Salobir, J.; Rezar, V. The effect of vitamin E supplementation on reduction of lymphocyte DNA damage induced by T-2 toxin and deoxynivalenol in weaned pigs. Anim. Feed. Sci. Technol. 2008, 141, 274–286. [Google Scholar] [CrossRef]
- Frankič, T.; Pajk, T.; Rezar, V.; Levart, A.; Salobir, J. The role of dietary nucleotides in reduction of DNA damage induced by T-2 toxin and deoxynivalenol in chicken leukocytes. Food Chem. Toxicol. 2006, 44, 1838–1844. [Google Scholar] [CrossRef]
- Rezar, V.; Frankič, T.; Narat, M.; Levart, A.; Salobir, J. Dose-Dependent Effects of T-2 Toxin on Performance, Lipid Peroxidation, and Genotoxicity in Broiler Chickens. Poult. Sci. 2007, 86, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Szabó, R.T.; Kovács-Weber, M.; Erdélyi, M.; Balogh, K.; Fazekas, N.; Horváth, Á.; Mézes, M.; Kovács, B. Comet assay study of the genotoxic effect of T-2 and HT-2 toxins in chicken hepatocytes. Biol. Futur. 2019, 70, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Gholampour Azizi, I.; Azarmi, M.; Danesh Pouya, N.; Rouhi, S. T-2 toxin Analysis in Poultry and Cattle Feedstuff. Jundishapur J. Nat. Pharm. Prod. 2014, 9, e13734. [Google Scholar] [CrossRef]
- Yin, H.; Han, S.; Chen, Y.; Wang, Y.; Li, D.; Zhu, Q. T-2 Toxin Induces Oxidative Stress, Apoptosis and Cytoprotective Autophagy in Chicken Hepatocytes. Toxins 2020, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, G.; Wang, X.; Wang, Y.; Lin, X.; Yu, F.; Mary, B.G.; Guo, X.; Mikko, J.L. Cellular responses to T-2 toxin and/or deoxynivalenol that induce cartilage damage are not specific to chondrocytes. Sci. Rep. 2017, 7, 2231. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 2005, 17, 568–575. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Saini, N.; Giacobone, C.K.; Klimczak, L.J.; Papas, B.N.; Burkholder, A.B.; Li, J.-L.; Fargo, D.C.; Bai, R.; Gerrish, K.; Innes, C.L.; et al. UV-exposure, endogenous DNA damage, and DNA replication errors shape the spectra of genome changes in human skin. PLOS Genet. 2021, 17, e1009302. [Google Scholar] [CrossRef]
- Friedberg, E.C.; McDaniel, L.D.; Schultz, R.A. The role of endogenous and exogenous DNA damage and mutagenesis. Curr. Opin. Genet. Dev. 2004, 14, 5–10. [Google Scholar] [CrossRef]
- Borrego-Soto, G.; Ortiz-López, R.; Rojas-Martínez, A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet. Mol. Biol. 2015, 38, 420–432. [Google Scholar] [CrossRef]
- Greinert, R.; de Vries, E.; Erdmann, F.; Espina, C.; Auvinen, A.; Kesminiene, A.; Schüz, J. European Code against Cancer 4th Edition: Ultraviolet radiation and cancer. Cancer Epidemiol. 2015, 39, S75–S83. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-L.; Lee, S.-K. Ultraviolet radiation: DNA damage, repair, and human disorders. Mol. Cell. Toxicol. 2017, 13, 21–28. [Google Scholar] [CrossRef]
- Puyo, S.; Montaudon, D.; Pourquier, P. From old alkylating agents to new minor groove binders. Crit. Rev. Oncol./Hematol. 2014, 89, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Kehe, K.; Schrettl, V.; Thiermann, H.; Steinritz, D. Modified immunoslotblot assay to detect hemi and sulfur mustard DNA adducts. Chem.-Biol. Interact. 2013, 206, 523–528. [Google Scholar] [CrossRef]
- Janik-Karpinska, E.; Ceremuga, M.; Niemcewicz, M.; Synowiec, E.; Sliwiński, T.; Bijak, M. Mitochondrial Damage Induced by T-2 Mycotoxin on Human Skin—Fibroblast Hs68 Cell Line. Molecules 2023, 28, 2408. [Google Scholar] [CrossRef] [PubMed]
- Cordelli, E.; Bignami, M.; Pacchierotti, F. Comet assay: A versatile but complex tool in genotoxicity testing. Toxicol. Res. 2021, 10, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Copp, M.E.; Chubinskaya, S.; Bracey, D.N.; Shine, J.; Sessions, G.; Loeser, R.F.; Diekman, B.O. Comet assay for quantification of the increased DNA damage burden in primary human chondrocytes with aging and osteoarthritis. Aging Cell 2022, 21, e13698. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.M.; Pinto, M.; Vicente, A.M.; Gonçalves, C.; Rodrigo, A.P.; Louro, H.; Costa, M.H.; Caeiro, S.; Silva, M.J. An integrative assessment to determine the genotoxic hazard of estuarine sediments: Combining cell and whole-organism responses. Front. Genet. 2014, 5, 437. [Google Scholar] [CrossRef]
- Islam, J.; Kabir, Y. Measurement of DNA damage in bladder cancer patients by alkaline comet assay. Ann. Urol. Oncol. 2019, 2, 64–70. [Google Scholar] [CrossRef]
- Horvatovich, K.; Hafner, D.; Bodnár, Z.; Berta, G.; Hancz, C.; Dutton, M.; Kovács, M. Dose-related genotoxic effect of T-2 toxin measured by comet assay using peripheral blood mononuclear cells of healthy pigs. Acta Vet. Hung. 2013, 61, 175–186. [Google Scholar] [CrossRef][Green Version]
- Kang, T.H.; Park, Y.; Bader, J.S.; Friedmann, T. The housekeeping gene hypoxanthine guanine phosphoribosyltransferase (HPRT) regulates multiple developmental and metabolic pathways of murine embryonic stem cell neuronal differentiation. PLoS ONE 2013, 8, e74967. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef]
- Miller, T.; Tokarz-Deptuła, B.; Deptuła, W. Cytokiny z rodziny interleukiny 1. Postep. Mikrobiol. 2011, 50, 217–221. [Google Scholar]
- Kim, J.-M.; Yeo, M.-K.; Lim, J.S.; Song, I.-S.; Chun, K.; Kim, K.-H. APEX1 Expression as a Potential Diagnostic Biomarker of Clear Cell Renal Cell Carcinoma and Hepatobiliary Carcinomas. J. Clin. Med. 2019, 8, 1151. [Google Scholar] [CrossRef]
- Tomkinson, A.E.; Sallmyr, A. Structure and function of the DNA ligases encoded by the mammalian LIG3 gene. Gene 2013, 531, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Wang, Z.; Klaunig, J.E. Alkaline Comet Assay for Assessing DNA Damage in Individual Cells. Curr. Protoc. Toxicol. 2015, 65, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M.; Saluk, J.; Antosik, A.; Ponczek, M.B.; Żbikowska, H.M.; Borowiecka, M.; Nowak, P. Aronia melanocarpa as a protector against nitration of fibrinogen. Int. J. Biol. Macromol. 2013, 55, 264–268. [Google Scholar] [CrossRef]
- Zbikowska, H.M.; Antosik, A.; Szejk, M.; Bijak, M.; Olejnik, A.K.; Saluk, J.; Nowak, P. Does quercetin protect human red blood cell membranes against γ-irradiation? Redox Rep. 2014, 19, 65–71. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene Symbol | Forward Primer Sequences (5′→3′) | Reverse Primer Sequence (5′→3′) | Amplicon Length (bp) |
|---|---|---|---|
| TP53 (tumor protein p53) | Long fragment: GGGTGTAGATGATGGGGATG | Long fragment: AACTGCGGAATGAAACAACC | 1172 |
| Small fragment: AAGCTGCTAAGGTCCCACAA | Small fragment: GGAAAGATCGCTCCAGGAA | 56 | |
| HPRT1 (hypoxanthine phosphoribosyltransferase 1) | Long fragment: AGGGCAAAGGATGTGTTACG | Long fragment: AGTGGTTTCTGGTGCGACTT | 1018 |
| Small fragment: TGCTGACCTGCTGGATTACA | Small fragment: TCTACAGTCATAGGAATGGATCTATCA | 69 |
| Gene Name | Gene Symbol | Entrez Gene ID | RefSeq | TaqMan® Gene Expression Assay IDs * |
|---|---|---|---|---|
| Eukaryotic 18S rRNA | 18S rRNA | HSRRN18S | X03205.1 (GenBank mRNA) | Hs99999901_s1 |
| Tumor necrosis factor | TNF | 7124 | NM_000594.3 | Hs00174128_m1 |
| Interferon gamma | INFG | 3458 | NM_000619.2 | Hs00989291_m1 |
| Interleukin 1 alpha | IL1A | 3552 | NM_000575.4 | Hs00174092_m1 |
| Interleukin 1 beta | IL1B | 3553 | NM_000576.2 | Hs01555410_m1 |
| DNA ligase 1 | LIG1 | 3978 | NM_000234.2 | Hs01553527_m1 |
| DNA ligase 3 | LIG3 | 3980 | NM_002311.4 | Hs00242692_m1 |
| Flap structure-specific endonuclease 1 | FEN1 | 2237 | NM_004111.5 | Hs00748727_s1 |
| X-ray repair cross complementing 1 | XRCC1 | 7515 | NM_006297.2 | Hs00959834_m1 |
| Apurinic/apyrimidinic endodeoxyribonuclease 1 | APEX1 | 328 | NM_001244249.1 | Hs00959050_g1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janik-Karpinska, E.; Ceremuga, M.; Niemcewicz, M.; Synowiec, E.; Sliwinski, T.; Stela, M.; Bijak, M. DNA Damage Induced by T-2 Mycotoxin in Human Skin Fibroblast Cell Line—Hs68. Int. J. Mol. Sci. 2023, 24, 14458. https://doi.org/10.3390/ijms241914458
Janik-Karpinska E, Ceremuga M, Niemcewicz M, Synowiec E, Sliwinski T, Stela M, Bijak M. DNA Damage Induced by T-2 Mycotoxin in Human Skin Fibroblast Cell Line—Hs68. International Journal of Molecular Sciences. 2023; 24(19):14458. https://doi.org/10.3390/ijms241914458
Chicago/Turabian StyleJanik-Karpinska, Edyta, Michal Ceremuga, Marcin Niemcewicz, Ewelina Synowiec, Tomasz Sliwinski, Maksymilian Stela, and Michal Bijak. 2023. "DNA Damage Induced by T-2 Mycotoxin in Human Skin Fibroblast Cell Line—Hs68" International Journal of Molecular Sciences 24, no. 19: 14458. https://doi.org/10.3390/ijms241914458
APA StyleJanik-Karpinska, E., Ceremuga, M., Niemcewicz, M., Synowiec, E., Sliwinski, T., Stela, M., & Bijak, M. (2023). DNA Damage Induced by T-2 Mycotoxin in Human Skin Fibroblast Cell Line—Hs68. International Journal of Molecular Sciences, 24(19), 14458. https://doi.org/10.3390/ijms241914458