

Review of Applications of Density Functional Theory (DFT) Quantum Mechanical Calculations to Study the High-Pressure Polymorphs of Organic Crystalline Materials



Abstract
:1. Introduction
2. Brief Theoretical Background
3. The Classes of Modeled Systems
3.1. Organic Materials with Metal Additives
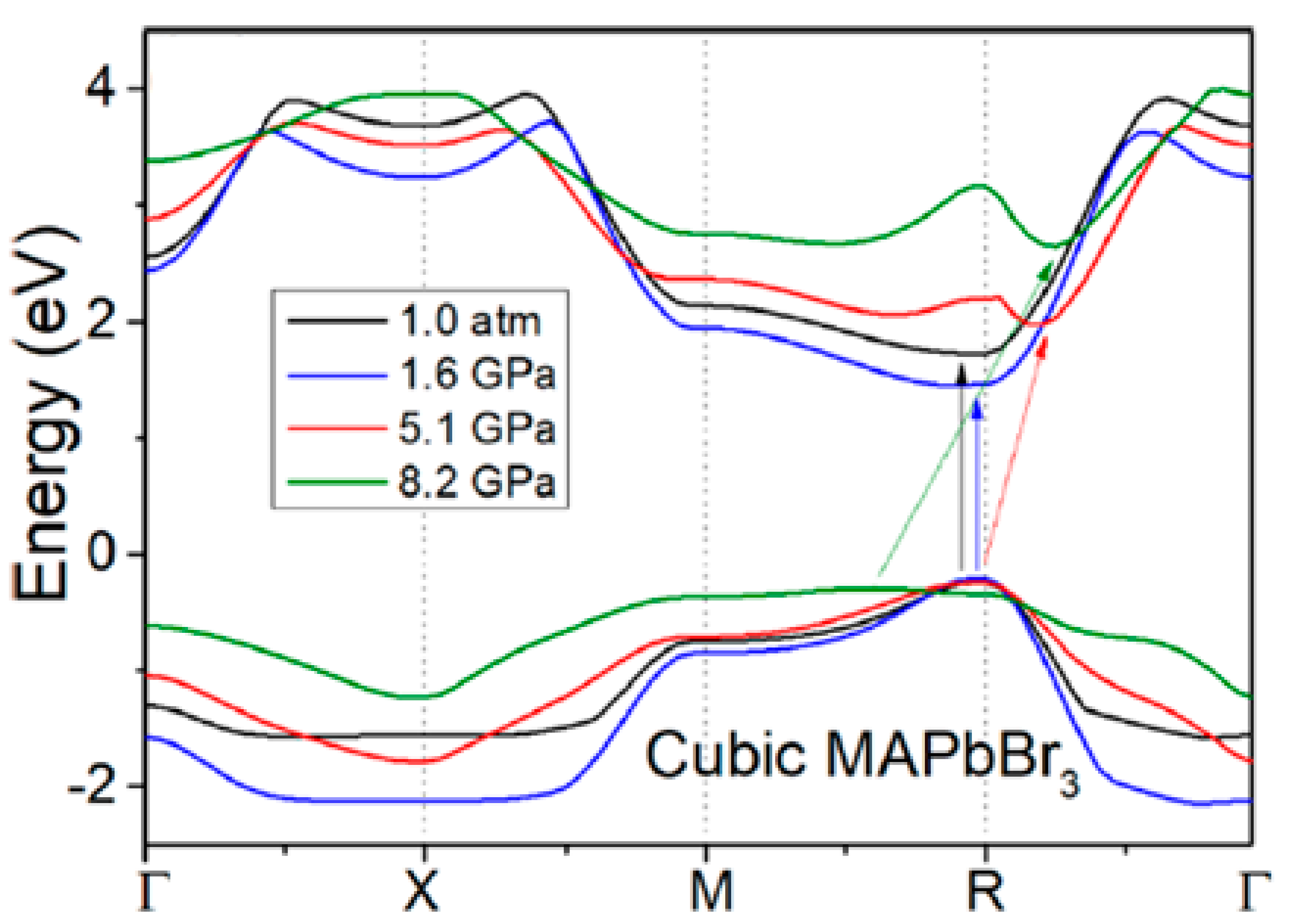
3.1.1. Methylammonium Lead Bromide (MAPbBr3)
3.1.2. Methylammonium Lead Iodide (MAPbI3)
3.1.3. Chloroindium(III) Hybrid Perovskite (IPy)4[In2Cl]10
3.1.4. Zn(μ-Cl)2(3,5-Dichloropyridine)2]n
3.1.5. Pt(bpy)Cl2
3.2. High-Energetic Organic Materials
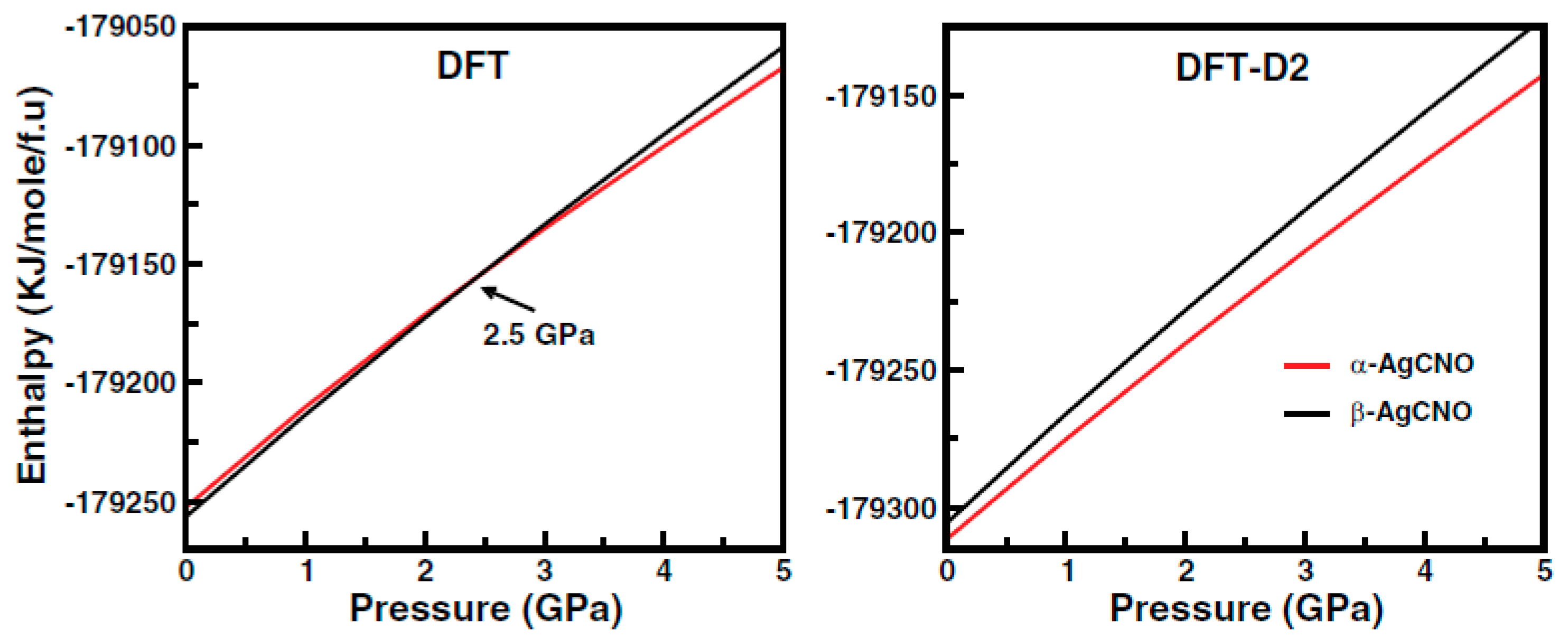
3.2.1. Silver Fulminate (AgCNO)
3.2.2. 3,5-Trinitrohexahydro-S-Triazine (RDX)
3.2.3. 2,6-Diamino-3,5-Dinitropyrazine-1-Oxide (LLM-105)
3.2.4. Cyclic Aliphatic Nitramine Octahydro-1,3,5,7-Tetranitro-1,3,5,7-Tetrazocin (HMX)
3.2.5. 1,1-Diamino-2,2-Dinitroethene (FOX-7)
3.2.6. 2,4,6-Trinitrotoluene (TNT)
3.2.7. Pentazolates
3.2.8. 2,4,6-Trinitro-3-Bromoanisole (TNBA)
3.2.9. Hexanitrohexaazaisowurtzitane (HNIW or CL-20)
3.2.10. Triaminotrinitrobenzene (TATB)
3.2.11. RDX, HMX, CL-20, NM, TATB, and PETN
3.3. Pharmaceuticals
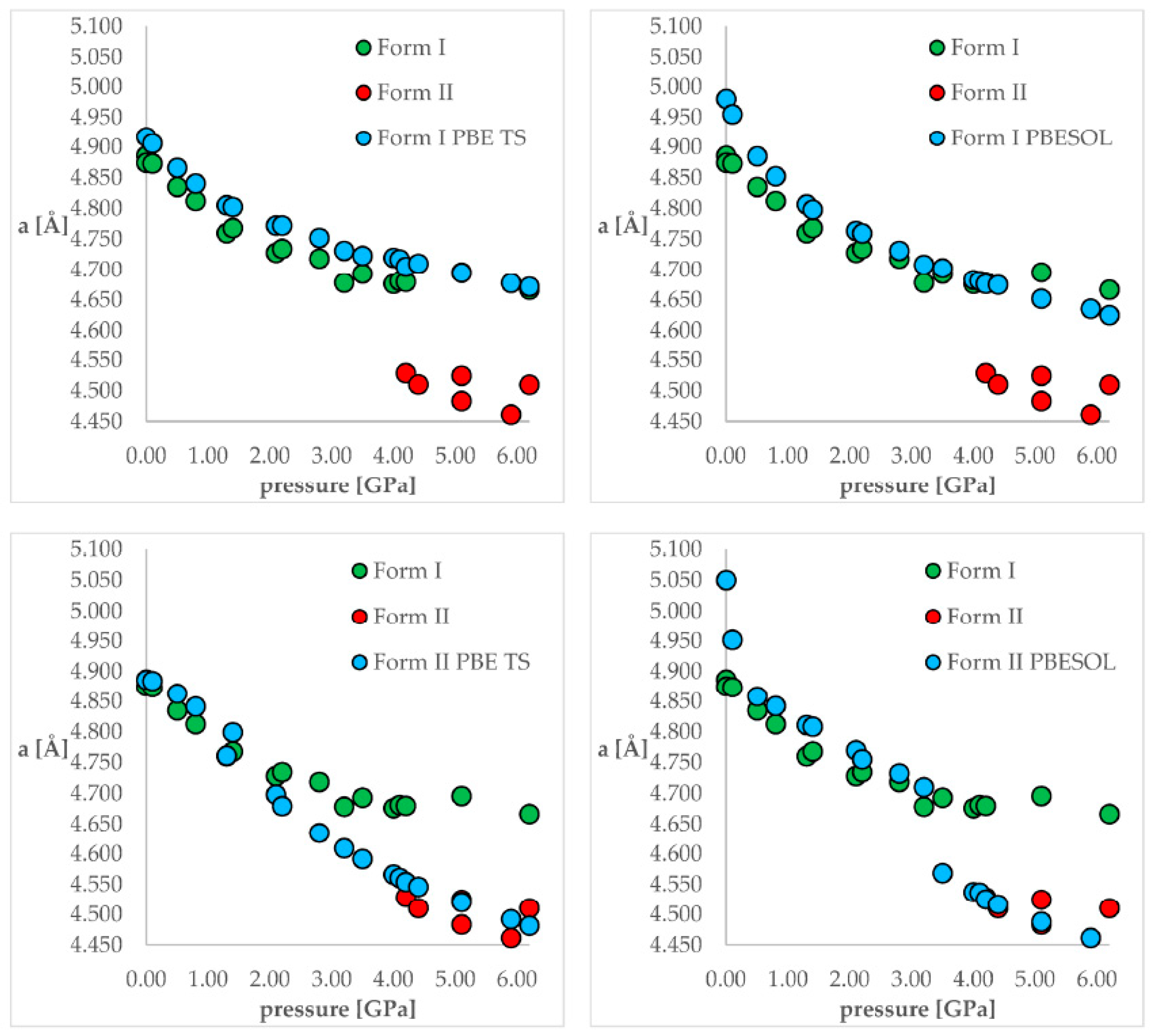
3.3.1. Chlorothiazide
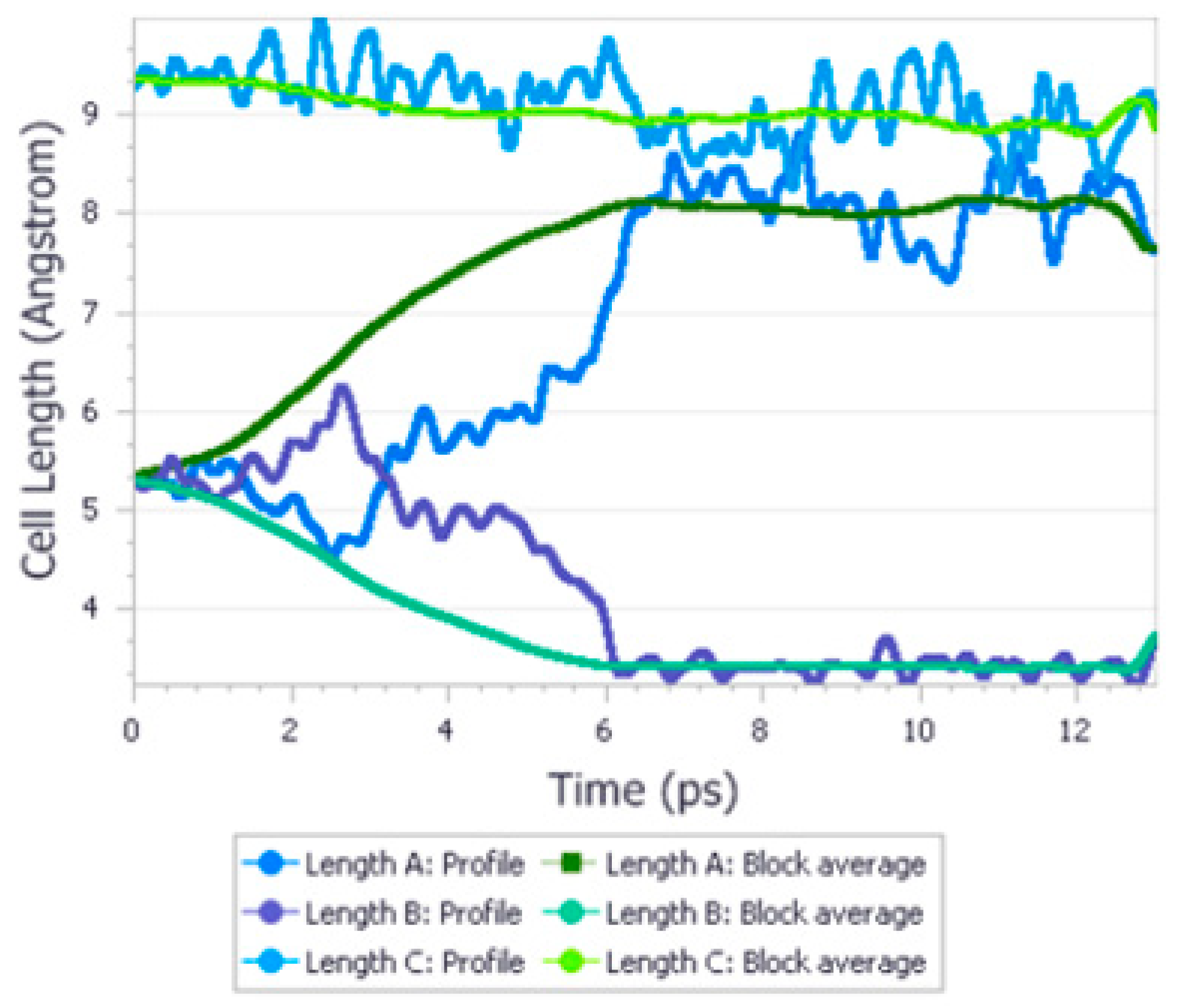
3.3.2. Urea
3.3.3. Tolazamide
3.3.4. Aspirin
3.3.5. Triclabendazole
3.3.6. Resorcinol
3.3.7. Glycine
4. Fundamental Aspects of DFT Calculations at High Pressure
4.1. Geomtry Optimization at Various Pressure Conditions and Crystal Structure Prediction
4.2. Vibrational Spectra
4.3. Enthalpy (ΔH) Calculations
4.4. Gibbs Free Energy (ΔG) Calculations
4.5. Phonon Calculations
5. Other Aspects Associated with DFT Calculations at High Pressure
5.1. Determination of Pressure-Induced Phase Transition Conditions
5.1.1. Common Tangent to the Two E(V) Curves, p = −dE/dV
5.1.2. Changes in Properties Observed upon Compression
5.2. Lack of Pressure-Induced Phase Transition
5.3. Anisotropic Compression
5.4. Polymorphic Transition Energy Barrier Calculations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Description |
| 2D PES | Two-Dimensional Potential Energy Surface |
| AIM | Atoms in Molecules |
| AS | Absorption Spectra Calculations |
| aiMD | Ab Initio Molecular Dynamics |
| BG | Bandgap |
| CL-20 | Hexanitrohexaazaisowurtzitane |
| CM | Center-of-Mass Fractional Position Calculations |
| COT | 1,3,5,7-Cyclooctatetraene |
| CSP | Crystal Structure Prediction |
| DFT | Density Functional Theory |
| DOS | Density of States |
| EDAB | Ethylenediamine Bisborane |
| EOS | Equation of State |
| ES | Excited State Calculation |
| FOX-7 | 1,1-Diamino-2,2-Dinitroethene |
| FPMD | First-Principles Molecular Dynamics |
| GD | Grimme Dispersion |
| GGA | Generalized Gradient Approximations |
| GO | Geometry Optimization |
| HF | Hartree–Fock |
| HMX | Cyclic Aliphatic Nitramine Octahydro-1,3,5,7-Tetranitro-1,3,5,7-Tetrazocin |
| HS | Hirshfeld Surface |
| IGM | Intramolecular Gradients Method |
| INS | Inelastic Neutron Scattering |
| IR | Infrared |
| KS | Kohn–Sham |
| LLM-105 | 2,6-Diamino-3,5-Dinitropyrazine-1-Oxide |
| MA | Methylammonium |
| MBD | Many-Body Dispersion |
| MD | Molecular dynamics |
| MO | Molecular Orbitals |
| MPD | Mutual Penetration Distance |
| MSST | Multi-Scale Shock Technique |
| NA | Not Applicable |
| NBO | Natural Bond Orbitals |
| NMR | Nuclear Magnetic Resonance |
| NP | Not Provided |
| NPT | Isothermal–Isobaric Ensemble |
| NVT | Canonical Ensemble |
| OP | Optical Properties |
| PC | Phonon DOS Calculation |
| PD | Phase Diagram |
| PF | Phonon Frequency |
| PL | Photoluminescence |
| pV | Pressure–Volume Terms |
| PXRD | Powder X-Ray Diffraction |
| QHA | Quasi Harmonic Approximation |
| QMD | Quantum Molecular Dynamics |
| RDX | 3,5-Trinitrohexahydro-S-Triazine |
| RMSD | Root Mean Square Deviation |
| SCC—DFTB | Self-Consistent Charge Density Functional Tight Binding |
| SCXRD | Single-Crystal X-Ray Diffraction |
| SOC | Spin–Orbit Coupling |
| SP | Single-Point Calculations |
| TATB | Triaminotrinitrobenzene |
| TB | Transition Barrier Calculation |
| TD | Thermodynamics |
| TD-DFT | Time-Dependent Density Functional Theory Calculations |
| TNBA | 2,4,6-Trinitro-3-Bromoanisole |
| TNT | 2,4,6-Trinitrotoluene |
| TS | Tkatchenko–Scheffler |
| USPEX | Universal Structure Predictor: Evolutionary Xtallography |
| VTST | Variational Transition-State Theory |
| XRD | X-Ray Diffraction |
| ZPVE | Zero-Point Vibrational Energy |
References
- Moggach, S.; Parsons, S. Molecular Solids at Extreme Pressure. CrystEngComm 2010, 12, 2515. [Google Scholar] [CrossRef]
- Boldyreva, E. High-Pressure Polymorphs of Molecular Solids: When Are They Formed, and When Are They Not? Some Examples of the Role of Kinetic Control. Cryst. Growth Des. 2007, 7, 1662–1668. [Google Scholar] [CrossRef]
- Hoja, J.; Reilly, A.M.; Tkatchenko, A. First-Principles Modeling of Molecular Crystals: Structures and Stabilities, Temperature and Pressure. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1294. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. Periodic DFT Calculations—Review of Applications in the Pharmaceutical Sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef]
- Wu, J.; Yu, D.; Liu, S.; Rong, C.; Zhong, A.; Chattaraj, P.K.; Liu, S. Is It Possible To Determine Oxidation States for Atoms in Molecules Using Density-Based Quantities? An Information-Theoretic Approach and Conceptual Density Functional Theory Study. J. Phys. Chem. A 2019, 123, 6751–6760. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; He, X.; Rong, C.; Zhong, A.; Liu, S.; Zhao, D. On the Origin and Nature of Internal Methyl Rotation Barriers: An Information-Theoretic Approach Study. Theor. Chem. Acc. 2022, 141, 68. [Google Scholar] [CrossRef]
- Luo, C.; He, X.; Zhong, A.; Liu, S.; Zhao, D. What Dictates Alkane Isomerization? A Combined Density Functional Theory and Information-Theoretic Approach Study. Theor. Chem. Acc. 2023, 142, 78. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- van Mourik, T.; Bühl, M.; Gaigeot, M.-P. Density Functional Theory across Chemistry, Physics and Biology. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2014, 372, 20120488. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, A.D.; Zhao, J.; Slebodnick, C.; Ross, N.L.; Cahill, C.L. High-Pressure Structural and Optical Property Evolution of a Hybrid Indium Halide Perovskite. J. Solid. State Chem. 2021, 300, 122262. [Google Scholar] [CrossRef]
- Liu, X.; Michalchuk, A.A.L.; Bhattacharya, B.; Yasuda, N.; Emmerling, F.; Pulham, C.R. High-Pressure Reversibility in a Plastically Flexible Coordination Polymer Crystal. Nat. Commun. 2021, 12, 3871. [Google Scholar] [CrossRef]
- Yesudhas, S.; Burns, R.; Lavina, B.; Tkachev, S.N.; Sun, J.; Ullrich, C.A.; Guha, S. Coupling of Organic Cation and Inorganic Lattice in Methylammonium Lead Halide Perovskites: Insights into a Pressure-Induced Isostructural Phase Transition. Phys. Rev. Mater. 2020, 4, 105403. [Google Scholar] [CrossRef]
- Jiang, H.; Xue, H.; Wang, L.; Tang, F.; Si, F. Effect of Pressure-Induced Structural Phase Transition on Electronic and Optical Properties of Perovskite CH3NH3PbI3. Mater. Sci. Semicond. Process 2019, 96, 59–65. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, L.; Ma, Z.; Wang, F. Pressure-Induced Band Structure Evolution of Halide Perovskites: A First-Principles Atomic and Electronic Structure Study. J. Phys. Chem. C 2019, 123, 739–745. [Google Scholar] [CrossRef]
- Cai, W.; Bi, T.; Zarifi, N.; Terpstra, T.; Zhang, C.; Verdeny, Z.V.; Zurek, E.; Deemyad, S. Effects of Nonhydrostatic Stress on Structural and Optoelectronic Properties of Methylammonium Lead Bromide Perovskite. J. Phys. Chem. Lett. 2017, 8, 3457–3465. [Google Scholar] [CrossRef]
- Jiang, S.; Fang, Y.; Li, R.; Xiao, H.; Crowley, J.; Wang, C.; White, T.J.; Goddard, W.A.; Wang, Z.; Baikie, T.; et al. Pressure-Dependent Polymorphism and Band-Gap Tuning of Methylammonium Lead Iodide Perovskite. Angew. Chem.-Int. Ed. 2016, 55, 6540–6544. [Google Scholar] [CrossRef]
- Valiente, R.; García-Lastra, J.M.; García-Fernández, P.; García-Revilla, S.; Wenger, O.S. Red-to-Yellow Pressure-Induced Phase Transition in Pt(Bpy)Cl2: Spectroscopic Study Supported by DFT Calculations. Eur. J. Inorg. Chem. 2007, 2007, 5735–5742. [Google Scholar] [CrossRef]
- Childs, C.M.; Steele, B.A.; Grivickas, P.; Zhang, D.; Crowhurst, J.; Kuo, I.W.; Bastea, S.; Clarke, S.M. High-Pressure Investigation of 2,4,6-Trinitro-3-Bromoanisole (TNBA): Structural Determination and Piezochromism. J. Phys. Chem. C 2022, 126, 1176–1187. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Dong, W.; Zhang, J.-G. Pressure-Induced Phase Transition of a Series of Energetic Pentazolate Anion Salts: A DFT Study. New J. Chem. 2022, 46, 5653–5662. [Google Scholar] [CrossRef]
- Gao, C.; Wang, J.; Zhang, Y.; Su, H.; Xu, Z.; Dai, R.; Wang, Z.; Zhang, Z. Pressure-Induced Phase Transition of β-RDX Single Crystals. J. Phys. Chem. C 2021, 125, 6418–6426. [Google Scholar] [CrossRef]
- Guan, R.; Wang, P.; Song, Y.; Staroverov, V.N. Pressure-Induced Polymorphic Transformations of Ethylenediamine Bisborane. J. Phys. Chem. C 2021, 125, 18614–18622. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Q.; Li, X.; Wang, J.; Wang, X.; Gao, C.; Dai, R.; Wang, Z.; Huang, S.; Liu, Y.; et al. Electronic Structure of LLM-105 Crystal under High Pressure and Low Temperature. J. Phys. Chem. C 2020, 124, 2399–2405. [Google Scholar] [CrossRef]
- Su, Y.; Fan, J.; Zheng, Z.; Zhao, J. Uniaxial Compression Behavior and Spectroscopic Properties of Energetic 1,1-Diamino-2,2-Dinitroethylene (FOX-7) Crystals from Density Functional Theory Calculations. Prog. Prog. Nat. Sci. Mater. Int. 2019, 29, 329–334. [Google Scholar] [CrossRef]
- Konar, S.; Michalchuk, A.A.L.; Sen, N.; Bull, C.L.; Morrison, C.A.; Pulham, C.R. High-Pressure Study of Two Polymorphs of 2,4,6-Trinitrotoluene Using Neutron Powder Diffraction and Density Functional Theory Methods. J. Phys. Chem. C 2019, 123, 26095–26105. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, W. Possible Pre-Phase Transition of the α-HMX Crystal Observed by the Variation of Hydrogen-Bonding Network under High Pressures. CrystEngComm 2019, 22, 330–348. [Google Scholar] [CrossRef]
- Landerville, A.C.; Crowhurst, J.C.; Grant, C.D.; Zaug, J.M.; Oleynik, I. Experimental and Theoretical Investigation of Pressuredependent Raman Spectra of Triaminotrinitrobenzene (TATB) at High Pressures. In Proceedings of the Conference of the American Physical Society Topical Group on Shock Compression of Condensed Matter, Tampa Bay, FL, USA, 14–19 June 2015; Volume 1793. [Google Scholar]
- Dreger, Z.A.; Stash, A.I.; Yu, Z.-G.Z.G.Z.-G.; Chen, Y.-S.Y.S.Y.-S.; Tao, Y.; Gupta, Y.M. High-Pressure Structural Response of an Insensitive Energetic Crystal: 1, 1-Diamino-2, 2-Dinitroethene (FOX-7). J. Phys. Chem. C 2016, 120, 27600–27607. [Google Scholar] [CrossRef]
- Cawkwell, M.J.; Luscher, D.J.; Addessio, F.L.; Ramos, K.J. Equations of State for the α and γ Polymorphs of Cyclotrimethylene Trinitramine. J. Appl. Phys. 2016, 119, 185106. [Google Scholar] [CrossRef]
- Stavrou, E.; Riad Manaa, M.; Zaug, J.M.; Kuo, I.F.W.; Pagoria, P.F.; Kalkan, B.; Crowhurst, J.C.; Armstrong, M.R. The High Pressure Structure and Equation of State of 2,6-Diamino-3,5-Dinitropyrazine-1-Oxide (LLM-105) up to 20 GPa: X-Ray Diffraction Measurements and First Principles Molecular Dynamics Simulations. J. Chem. Phys. 2015, 143, 144506. [Google Scholar] [CrossRef]
- Manaa, M.R.; Kuo, I.-F.W.; Fried, L.E. First-Principles High-Pressure Unreacted Equation of State and Heat of Formation of Crystal 2,6-Diamino-3, 5-Dinitropyrazine-1-Oxide (LLM-105). J. Chem. Phys. 2014, 141, 064702. [Google Scholar] [CrossRef] [PubMed]
- Ge, N.-N.; Wei, Y.-K.; Zhao, F.; Chen, X.-R.; Ji, G.-F. Pressure-Induced Metallization of Condensed Phase β-HMX under Shock Loadings via Molecular Dynamics Simulations in Conjunction with Multi-Scale Shock Technique. J. Mol. Model. 2014, 20, 2350. [Google Scholar] [CrossRef] [PubMed]
- Yedukondalu, N.; Vaitheeswaran, G. Polymorphism and Thermodynamic Ground State of Silver Fulminate Studied from van Der Waals Density Functional Calculations. J. Chem. Phys. 2014, 140, 224705. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.C.; Rice, B.M. Theoretical Predictions of Energetic Molecular Crystals at Ambient and Hydrostatic Compression Conditions Using Dispersion Corrections to Conventional Density Functionals (DFT-D). J. Phys. Chem. C 2010, 114, 6734–6748. [Google Scholar] [CrossRef]
- Tkachev, S.N.; Pravica, M.; Kim, E.; Romano, E.; Week, P.F. High-Pressure Studies of 1,3,5,7-Cyclooctatetraene: Experiment and Theory. J. Phys. Chem. A 2008, 112, 11501–11507. [Google Scholar] [CrossRef]
- Xu, X.-J.; Zhu, W.-H.; Xiao, H.-M. DFT Studies on the Four Polymorphs of Crystalline CL-20 and the Influences of Hydrostatic Pressure on ε-CL-20 Crystal. J. Phys. Chem. B 2007, 111, 2090–2097. [Google Scholar] [CrossRef]
- Ali, I.; Tang, J.; Han, Y.; Wei, Z.; Zhang, Y.; Li, J. A Solid-Solid Phase Transformation of Triclabendazole at High Pressures. Crystals 2022, 12, 300. [Google Scholar] [CrossRef]
- Szeleszczuk, Ł.; Mazurek, A.H.; Milcarz, K.; Napiórkowska, E.; Pisklak, D.M. Can We Predict the Isosymmetric Phase Transition ? Application of DFT Calculations to Study the Pressure Induced Transformation of Chlorothiazide. Int. J. Mol. Sci. 2021, 22, 10100. [Google Scholar] [CrossRef]
- Silva, R.; de Castro, A.J.; da Silva Filho, J.G.; de Sousa, F.F.; Paraguassu, W.; Freire, P.T.C.; Façanha Filho, P.F. Pressure-Induced Phase Transition in Glycinium Maleate Crystal. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 262, 120076. [Google Scholar] [CrossRef]
- Cook, C.; McKinley, J.L.; Beran, G.J.O.O. Modeling the α-and β-Resorcinol Phase Boundary via Combination of Density Functional Theory and Density Functional Tight-Binding. J. Chem. Phys. 2021, 154, 134109. [Google Scholar] [CrossRef]
- Xavier, N.F.; Da Silva, A.M.; Bauerfeldt, G.F. What Rules the Relative Stability of α-, β-, and γ-Glycine Polymorphs? Cryst. Growth Des. 2020, 20, 4695–4706. [Google Scholar] [CrossRef]
- Novelli, G.; Maynard-Casely, H.E.; McIntyre, G.J.; Warren, M.R.; Parsons, S. Effect of High Pressure on the Crystal Structures of Polymorphs of L-Histidine. Cryst. Growth Des. 2020, 20, 7788–7804. [Google Scholar] [CrossRef]
- Novelli, G.; Maynard-Casely, H.E.; McIntyre, G.J.; Warren, M.R.; Parsons, S. Correction to “The Effect of High Pressure on the Crystal Structures of Polymorphs of L-Histidine”. Cryst. Growth Des. 2022, 22, 937–938. [Google Scholar] [CrossRef]
- Mazurek, A.; Szeleszczuk, Ł.; Pisklak, D.M. Can We Predict the Pressure Induced Phase Transition of Urea? Application of Quantum Molecular Dynamics. Molecules 2020, 25, 2021. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.M.; Steele, B.A.; Kroonblawd, M.P.; Zhang, D.; Kuo, I.-F.W.; Stavrou, E. An Isosymmetric High-Pressure Phase Transition in α-Glycylglycine: A Combined Experimental and Theoretical Study. J. Phys. Chem. B 2020, 124, 1–10. [Google Scholar] [CrossRef]
- Giordano, N.; Beavers, C.M.; Kamenev, K.V.; Marshall, W.G.; Moggach, S.A.; Patterson, S.D.; Teat, S.J.; Warren, J.E.; Wood, P.A.; Parsons, S. High-Pressure Polymorphism in l-Threonine between Ambient Pressure and 22 GPa. CrystEngComm 2019, 21, 4444–4456. [Google Scholar] [CrossRef]
- Prazyan, T.L.; Zhuravlev, Y.N.; Golovko, O.V.; Obolonskaya, O.S. DFT-Study of Pressure-Induced Phase Transition in L-Threonine. J. Mol. Struct. 2019, 1196, 271–279. [Google Scholar] [CrossRef]
- Szeleszczuk, Ł.; Pisklak, D.M.; Zielińska-Pisklak, M. Can We Predict the Structure and Stability of Molecular Crystals under Increased Pressure? First-Principles Study of Glycine Phase Transitions. J. Comput. Chem. 2018, 39, 1300–1306. [Google Scholar] [CrossRef]
- Rychkov, D.A.; Stare, J.; Boldyreva, E. V Pressure-Driven Phase Transition Mechanisms Revealed by Quantum Chemistry: L-Serine Polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 6671–6676. [Google Scholar] [CrossRef]
- Fedorov, A.Y.; Rychkov, D.A.; Losev, E.A.; Zakharov, B.A.; Stare, J.; Boldyreva, E.V. Effect of Pressure on Two Polymorphs of Tolazamide: Why No Interconversion? CrystEngComm 2017, 19, 2243–2252. [Google Scholar] [CrossRef]
- Leblanc, L.M.; Otero-de-La-Roza, A.; Johnson, E.R. Evaluation of Shear-Slip Transitions in Crystalline Aspirin by Density-Functional Theory. Cryst. Growth Des. 2016, 16, 6867–6873. [Google Scholar] [CrossRef]
- Adhikari, K.; Flurchick, K.M.; Valenzano, L. A Hybrid Density Functional Study on the Effects of Pressure on Paracetamol and Aspirin Polymorphs. Comput. Theor. Chem. 2015, 1062, 90–98. [Google Scholar] [CrossRef]
- Druzbicki, K.; Mikuli, E.; Pałka, N.; Zalewski, S.; Ossowska-Chruściel, M.D. Polymorphism of Resorcinol Explored by Complementary Vibrational Spectroscopy (FT-RS, THz-TDS, INS) and First-Principles Solid-State Computations (Plane-Wave DFT). J. Phys. Chem. B 2015, 119, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, R.; Küçükbenli, E.; Kolb, B.; Thonhauser, T.; De Gironcoli, S. Structural Evolution of Amino Acid Crystals under Stress from a Non-Empirical Density Functional. J. Phys. Condens. Matter 2012, 24, 424209. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.A.; Francis, D.; Marshall, W.G.; Moggach, S.A.; Parsons, S.; Pidcock, E.; Rohl, A.L. A Study of the High-Pressure Polymorphs of L-Serine Using Ab Initio Structures and PIXEL Calculations. CrystEngComm 2008, 10, 1154–1166. [Google Scholar] [CrossRef]
- Howard, C.M.; Wood, I.G.; Knight, K.S.; Fortes, A.D. Ab Initio Simulations of α- and β-Ammonium Carbamate (NH4·NH2CO2), and the Thermal Expansivity of Deuterated α-Ammonium Carbamate from 4.2 to 180 K by Neutron Powder Diffraction. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2022, 78, 459–475. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Chumak, T.; Rogoża, J. Phase Stability of Chloroform and Dichloromethane at High Pressure. Crystals 2020, 10, 920. [Google Scholar] [CrossRef]
- Batyrev, I.G. The Modeling the Structure of Croconic and Squaric Acids under Pressure. In Proceedings of the Conference of the American Physical Society Topical Group on Shock Compression of Condensed Matter, Portland, OR, USA, 16–21 June 2019; Lane, J.M.D., Germann, T.C., A.M.R.W.R.D.D.Z.J., Eds.; American Institute of Physics Inc.: College Park, MD, USA, 2020; Volume 2272. [Google Scholar]
- Sahoo, S.; Ravindran, T.R.; Rajaraman, R.; Srihari, V.; Pandey, K.K.; Chandra, S. Pressure-Induced Amorphization of Diisopropylammonium Perchlorate Studied by Raman Spectroscopy and X-Ray Diffraction. J. Phys. Chem. A 2020, 124, 1993–2000. [Google Scholar] [CrossRef]
- Zhou, J.; Lian, J.; Ding, Y.; Gao, Y.; Zhu, W. Pressure-Induced Variation of Structural, Elastic, Vibrational, Thermodynamic Properties and Hardness of C11N4 Polymorphs. Results Phys. 2019, 14, 102453. [Google Scholar] [CrossRef]
- Colmenero, F. Mechanical Properties of Anhydrous Oxalic Acid and Oxalic Acid Dihydrate. Phys. Chem. Chem. Phys. 2019, 21, 2673–2690. [Google Scholar] [CrossRef]
- Saraiva, G.D.; Maia, J.R.; Lima, J.A., Jr.; Nogueira, C.E.S.; Freire, P.T.C.; de Sousa, F.F.; Teixeira, A.M.R.; Mendes Filho, J. Pressure Induced Transformations in Sorbic Acid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2017, 184, 327–334. [Google Scholar] [CrossRef]
- Wong, J.W.L.; Mailman, A.; Lekin, K.; Winter, S.M.; Yong, W.; Zhao, J.; Garimella, S.V.; Tse, J.S.; Secco, R.A.; Desgreniers, S.; et al. Pressure Induced Phase Transitions and Metallization of a Neutral Radical Conductor. J. Am. Chem. Soc. 2014, 136, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Lekin, K.; Leitch, A.A.; Tse, J.S.; Bao, X.; Secco, R.A.; Desgreniers, S.; Ohishi, Y.; Oakley, R.T. A Pressure Induced Structural Dichotomy in Isostructural Bis-1,2,3-Thiaselenazolyl Radical Dimers. Cryst. Growth Des. 2012, 12, 4676–4684. [Google Scholar] [CrossRef]
- Schatschneider, B.; Liang, J.-J.J.J. Accurate Modeling of Molecular Crystal through Dispersion-Corrected Density Functional Theory (DFT-D) Method. In Materials Research Society Symposium Proceedings; MRS Online Proceedings Library (OPL): Warrendale, PA, USA, 2011; Volume 1301, pp. 125–130. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- CASTEP. Available online: http://www.castep.org/ (accessed on 10 August 2023).
- Hunter, S.; Sutinen, T.; Parker, S.F.; Morrison, C.A.; Williamson, D.M.; Thompson, S.; Gould, P.J.; Pulham, C.R. Experimental and DFT-D Studies of the Molecular Organic Energetic Material RDX. J. Phys. Chem. C 2013, 117, 8062–8071. [Google Scholar] [CrossRef]
- VASP-Vienna Ab Initio Simulation Package. Available online: https://www.vasp.at/ (accessed on 10 August 2023).
- Sun, G.; Kürti, J.; Rajczy, P.; Kertesz, M.; Hafner, J.; Kresse, G. Performance of the Vienna Ab Initio Simulation Package (VASP) in Chemical Applications. J. Mol. Struct. Theochem. 2003, 624, 37–45. [Google Scholar] [CrossRef]
- Quantum Espresso. Available online: https://www.quantum-espresso.org/ (accessed on 10 August 2023).
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced Capabilities for Materials Modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Gaussian.Com|Expanding the Limits of Computational Chemistry. Available online: https://gaussian.com/ (accessed on 10 August 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01 2016. Available online: https://gaussian.com/citation/ (accessed on 12 September 2023).
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An Electronic Structure and Molecular Dynamics Software Package -Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]
- CP2K. Available online: https://www.cp2k.org/ (accessed on 10 August 2023).
- CRYSTAL. Available online: https://www.crystal.unito.it/index.html (accessed on 10 August 2023).
- Erba, A.; Desmarais, J.K.; Casassa, S.; Civalleri, B.; Donà, L.; Bush, I.J.; Searle, B.; Maschio, L.; Edith-Daga, L.; Cossard, A.; et al. CRYSTAL23: A Program for Computational Solid State Physics and Chemistry. J. Chem. Theory Comput. 2022. [Google Scholar] [CrossRef]
- DMol3-BIOVIA. Available online: https://www.3ds.com/products-services/biovia/ (accessed on 10 August 2023).
- Delley, B. DMol3 DFT Studies: From Molecules and Molecular Environments to Surfaces and Solids. Comput. Mater. Sci. 2000, 17, 122–126. [Google Scholar] [CrossRef]
- Andreoni, W.; Curioni, A. New Advances in Chemistry and Materials Science with CPMD and Parallel Computing. Parallel Comput. 2000, 26, 819–842. [Google Scholar] [CrossRef]
- CPMD. Available online: http://www.cpmd.org/ (accessed on 10 August 2023).
- DFTB+. Available online: https://dftbplus.org/ (accessed on 10 August 2023).
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitric, T.; Dominguez, A.; et al. DFTB+, a Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA Method for Ab Initio Order-N Materials Simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Siesta. Available online: https://siesta-project.org/siesta/ (accessed on 10 August 2023).
- WIEN2k. Available online: http://www.wien2k.at/index.html (accessed on 10 August 2023).
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APW+lo Program for Calculating the Properties of Solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef]
- Bučko, T.; Lebègue, S.; Hafner, J.; Ángyán, J.G. Tkatchenko-Scheffler van Der Waals Correction Method with and without Self-Consistent Screening Applied to Solids. Phys. Rev. B 2013, 87, 064110. [Google Scholar] [CrossRef]
- Grimme, S.; Steinmetz, M. Effects of London Dispersion Correction in Density Functional Theory on the Structures of Organic Molecules in the Gas Phase. Phys. Chem. Chem. Phys. 2013, 15, 16031. [Google Scholar] [CrossRef]
- Xu, P.; Alkan, M.; Gordon, M.S. Many-Body Dispersion. Chem. Rev. 2020, 120, 12343–12356. [Google Scholar] [CrossRef]
- Winkler, B.; Milman, V. Density Functional Theory Based Calculations for High Pressure Research. Z. Krist. Cryst. Mater. 2014, 229, 112–122. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and Related Crystal Properties from Density-Functional Perturbation Theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Gelpi, J.; Hospital, A.; Goñi, R.; Orozco, M. Molecular Dynamics Simulations: Advances and Applications. Adv. Appl. Bioinform. Chem. 2015, 37, 37–47. [Google Scholar] [CrossRef]
- Starodumov, I.; Ankudinov, V.; Nizovtseva, I. A Review of Continuous Modeling of Periodic Pattern Formation with Modified Phase-Field Crystal Models. Eur. Phys. J. Spec. Top. 2022, 231, 1135–1145. [Google Scholar] [CrossRef]
- Hasnip, P.J.; Refson, K.; Probert, M.I.J.; Yates, J.R.; Clark, S.J.; Pickard, C.J. Density Functional Theory in the Solid State. Philos. Trans. R. Soc. A: Math. Phys. Eng. Sci. 2014, 372, 20130270. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, P. NMR Crystallography of Molecular Organics. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118, 10–53. [Google Scholar] [CrossRef] [PubMed]
- Rychkov, D.A. A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals. Crystals 2020, 10, 81. [Google Scholar] [CrossRef]
- Fabbiani, F.P.A.; Pulham, C.R. High-Pressure Studies of Pharmaceutical Compounds and Energetic Materials. Chem. Soc. Rev. 2006, 35, 932. [Google Scholar] [CrossRef]
- Holy, J.A. Raman Spectroscopy of Aminated and Ultrafine 1,3,5-Triamino-2,4,6-Trinitrobenzene and PBX 9502 as a Function of Pressing Pressure. J. Phys. Chem. B 2008, 112, 7489–7498. [Google Scholar] [CrossRef]
- Oswald, I.D.H.; Lennie, A.R.; Pulham, C.R.; Shankland, K. High-Pressure Structural Studies of the Pharmaceutical, Chlorothiazide. CrystEngComm 2010, 12, 2533. [Google Scholar] [CrossRef]
- Reilly, A.M.; Tkatchenko, A. Role of Dispersion Interactions in the Polymorphism and Entropic Stabilization of the Aspirin Crystal. Phys. Rev. Lett. 2014, 113, 055701. [Google Scholar] [CrossRef]
- Crowell, E.L.; Dreger, Z.A.; Gupta, Y.M. High-Pressure Polymorphism of Acetylsalicylic Acid (Aspirin): Raman Spectroscopy. J. Mol. Struct. 2015, 1082, 29–37. [Google Scholar] [CrossRef]
- Marom, N.; DiStasio, R.A.; Atalla, V.; Levchenko, S.; Reilly, A.M.; Chelikowsky, J.R.; Leiserowitz, L.; Tkatchenko, A. Many-Body Dispersion Interactions in Molecular Crystal Polymorphism. Angew. Chem. Int. Ed. 2013, 52, 6629–6632. [Google Scholar] [CrossRef]
- Rodríguez, J.S.; Costa, G.; da Silva, M.B.; Silva, B.P.; Honório, L.J.; de Lima-Neto, P.; Santos, R.C.R.; Caetano, E.W.S.; Alves, H.W.L.; Freire, V.N. Structural and Optoelectronic Properties of the α-, β-, and γ-Glycine Polymorphs and the Glycine Dihydrate Crystal: A DFT Study. Cryst. Growth Des. 2019, 19, 5204–5217. [Google Scholar] [CrossRef]
- Dudek, M.K.; Drużbicki, K. Along the Road to Crystal Structure Prediction (CSP) of Pharmaceutical-like Molecules. CrystEngComm 2022, 24, 1665–1678. [Google Scholar] [CrossRef]
- Nyman, J.; Reutzel-Edens, S.M. Crystal Structure Prediction Is Changing from Basic Science to Applied Technology. Faraday Discuss. 2018, 211, 459–476. [Google Scholar] [CrossRef] [PubMed]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the Sixth Blind Test of Organic Crystal Structure Prediction Methods. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Conclusions of the 7th Crystal Structure Prediction Blind Test|CCDC. Available online: https://www.ccdc.cam.ac.uk/discover/news/conclusions-of-the-7th-crystal-structure-prediction-blind-test/ (accessed on 4 September 2023).
- Bhardwaj, R.M.; McMahon, J.A.; Nyman, J.; Price, L.S.; Konar, S.; Oswald, I.D.H.; Pulham, C.R.; Price, S.L.; Reutzel-Edens, S.M. A Prolific Solvate Former, Galunisertib, under the Pressure of Crystal Structure Prediction, Produces Ten Diverse Polymorphs. J. Am. Chem. Soc. 2019, 141, 13887–13897. [Google Scholar] [CrossRef]
- Neumann, M.A.; van de Streek, J.; Fabbiani, F.P.A.; Hidber, P.; Grassmann, O. Combined Crystal Structure Prediction and High-Pressure Crystallization in Rational Pharmaceutical Polymorph Screening. Nat. Commun. 2015, 6, 7793. [Google Scholar] [CrossRef]
- Jaffe, A.; Lin, Y.; Mao, W.L.; Karunadasa, H.I. Pressure-Induced Conductivity and Yellow-to-Black Piezochromism in a Layered Cu–Cl Hybrid Perovskite. J. Am. Chem. Soc. 2015, 137, 1673–1678. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Molecule | Polymorphs Studied | Pressure Range | Type of Calculation | Software Applied | Methods—DFT Functional and Dispersion Correction | Year | Ref. in Article |
|---|---|---|---|---|---|---|---|---|
| A. Organic materials with metal additives | ||||||||
| 1. | (IPy)4(In2Cl10) IPy = 4-iodopyridinium | NP | 0–1.51 GPa | GO, HS, NBO, DOS, OP, MO | Crystal Explorer; Gaussian | B3LYP | 2021 | [12] |
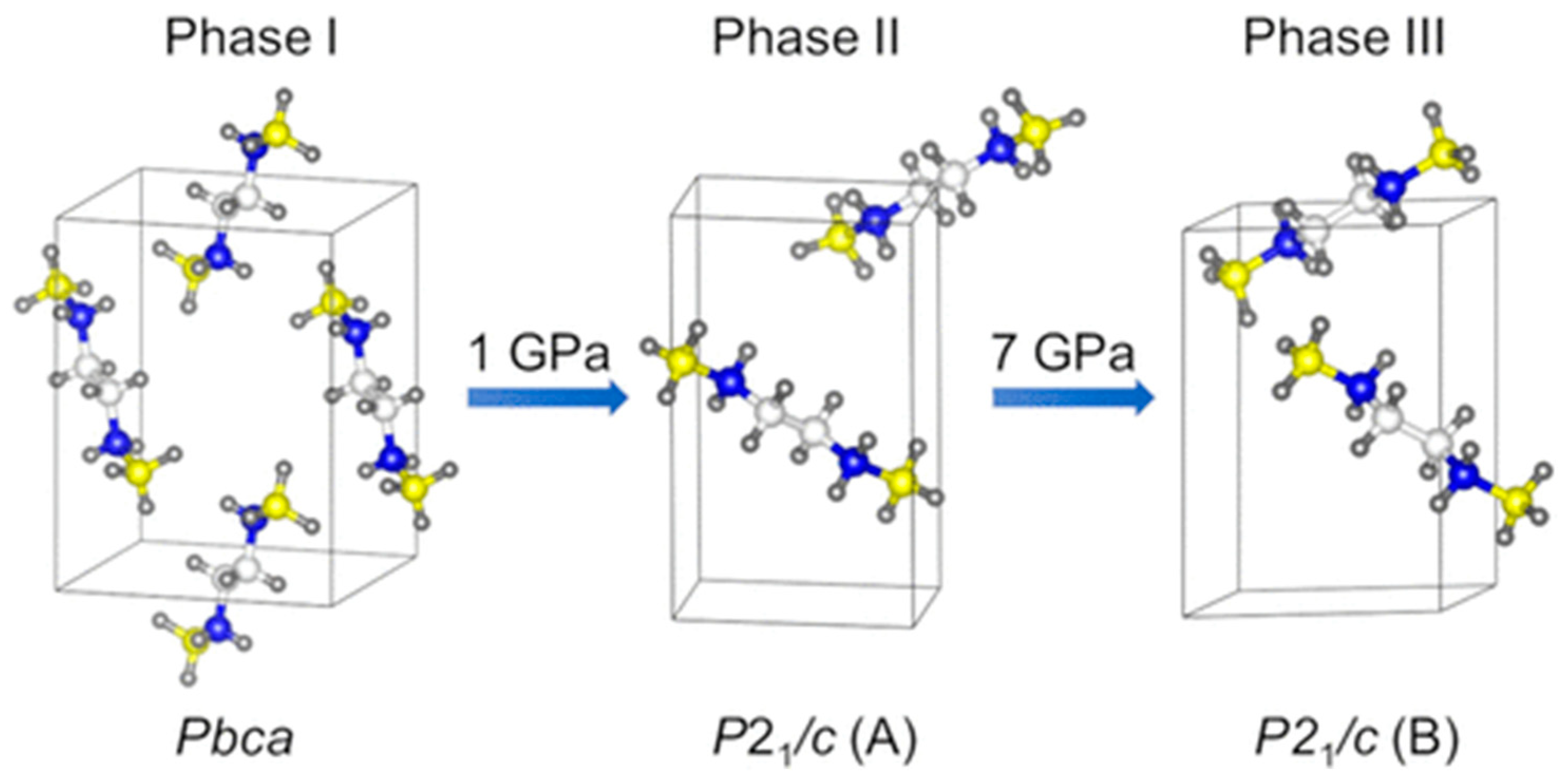
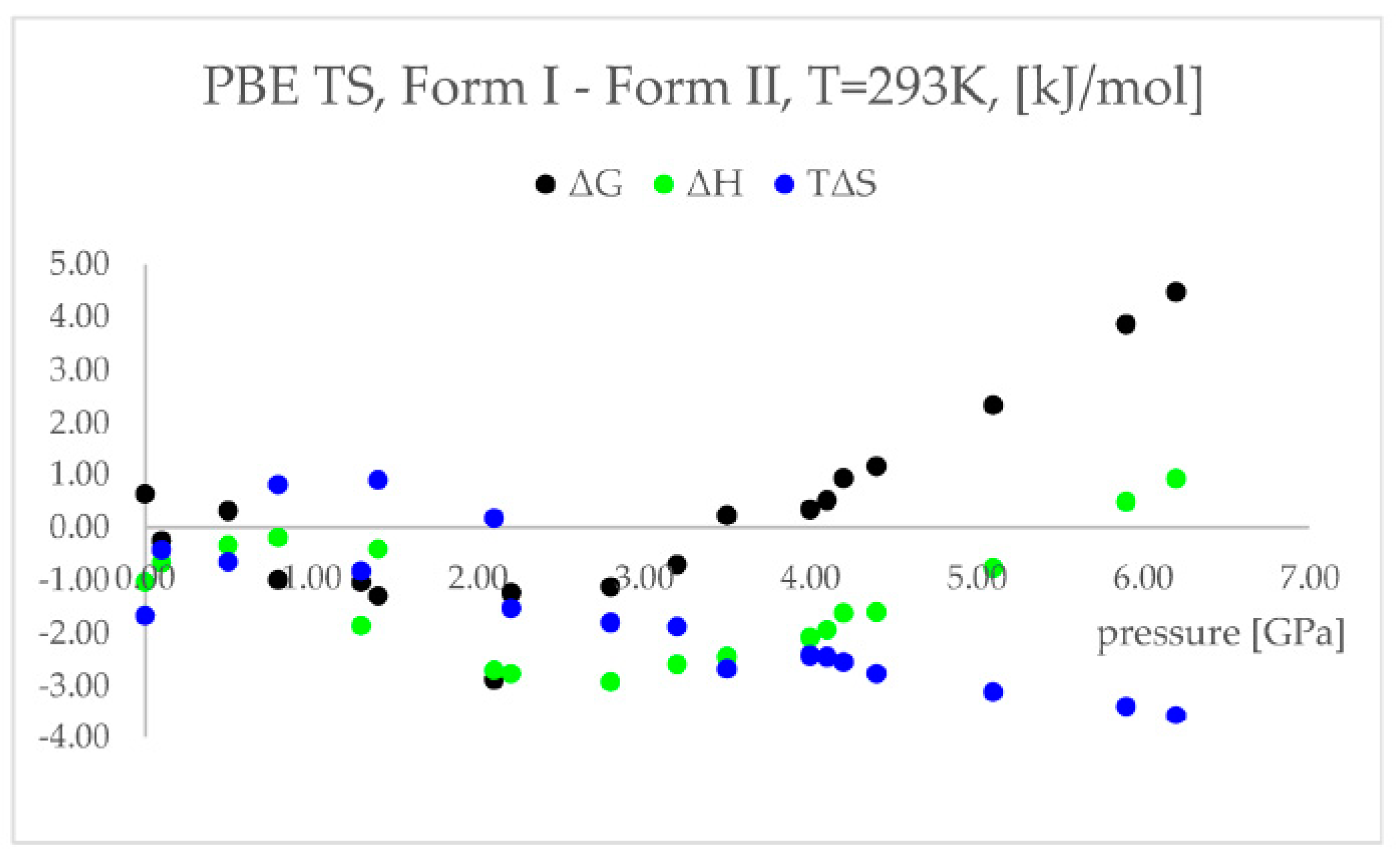
| 2. | [Zn(μ-Cl)2(3,5-dichloropyridine)2]n | , | 0–9.34 GPa | GO (compression and decompression), Raman | CASTEP | PBE TS | 2021 | [13] |
| 3. | Methylammonium lead bromide (MAPbBr3) | , Im | 0–2.5 GPa | GO, BG | Quantum ESPRESSO | PBE-D3 Grimme | 2020 | [14] |
| 4. | CH3NH3PbI3 (MAPbI3) | Tetragonal, orthorhombic, and cubic structures | 0–2 GPa | BG, DOS | VASP | PBE | 2019 | [15] |
| 5. | Methylammonium lead bromide (MAPbBr3) | , R3m, R3 | 0–130 GPa | GO, BG | VASP | PBE | 2019 | [16] |
| 6. | Methylammonium lead bromide (MAPbBr3) | I () II (Im), III (Im), IV (Pnma) | 0–3 GPa | GO, BG, aiMD | VASP | PBE | 2017 | [17] |
| 7. | Methylammonium lead iodide (MAPbI3) | I4/mcm, ImmmIm | 0–1.95 GPa | GO, BG | NP | PBE-D3, B3PW91 + SOCPBE | 2016 | [18] |
| 8. | Pt(bpy)Cl2, bpy = 2,2′-bipyridine | Yellow and red form | 0–3.8 GPa | GO, MO, TD-DFT | Gaussian | B3LYP, LDA, BLYP | 2007 | [19] |
| B. High-energetic organic materials | ||||||||
| 9. | 2,4,6-Trinitro-3-bromoanisole (TNBA) | P21/c, P212121 | 0–10 GPa | USPEX, GO, BG, DOS | VASP | PBE-D2 | 2022 | [20] |
| 10. | Pentazolate anion (cyclo-N5−) salt 3,9-diamino-6,7-dihydro-5H-bis([1,2,4]triazolo)[4,3-e:30,40-g][1,2,4,5]tetrazepine-2,10-diium ((N5−)2DABTT2+) | NP | 0–50 GPa | DOS, BG, PC, IR | CASTEP | PBE/G06 | 2022 | [21] |
| 11. | Pentazolate anion (cyclo-N5−) salt N-carbamoylguanidinium (N5−GU+) | NP | 0–50 GPa | DOS, BG, PC, IR | CASTEP | PBE/G06 | 2022 | [21] |
| 12. | 1,3,5-Trinitrohexahydro-s-triazine (RDX) | α, β, ε′ | 0–20.7 GPa | GO | VASP | PBE vdW correction | 2021 | [22] |
| 13. | Ethylenediamine bisborane (EDAB) | I, II, III | 0–17 GPa | USPEX, XRD, GO, PC, PF | DIAMOND; Quantum ESPRESSO | vdW-DF, PBE | 2021 | [23] |
| 14. | 2,6-Diamino-3,5-dinitropyrazine-1-oxide (LLM-105) | NP | 0–25.7 GPa | GO, DOS, BG AS, ES | NP; Gaussian | PBE; B3LYP | 2020 | [24] |
| 15. | 1,1-Diamino-2,2-dinitroethene (FOX-7) | α, α′, β, γ, δ, and ε | 0–30 GPa | GO, PC, Raman | CASTEP | PBE | 2019 | [25] |
| 16. | 2,4,6-Trinitrotoluene (TNT) | m-TNT and o-TNT | 0–5 GPa | GO | CASTEP | PBE-D2 PBE TS | 2019 | [26] |
| 17. | Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) | α, β | 0–50 GPa | GO, HS AIM, IGM. MPD, MP, PC, PF, IR, DOS | CASTEP | PBE | 2019 | [27] |
| 18. | Triaminotrinitrobenzene (TATB) | NP | 0–27 GPa | GO, ZPE, Raman | VASP | PBE-D2 Grimme | 2017 | [28] |
| 19. | 1,1-Diamino-2,2-dinitroethene (FOX-7) | α, α′, ε | 0–12.8 GPa | GO | CASTEP | PBE Grimme | 2016 | [29] |
| 20. | Cyclotrimethylenetrinitramine (RDX) | α, γ | 0–10 GPa | GO, TD, PC | CP2K | PBE-D3(BJ) | 2016 | [30] |
| 21. | 2,6-Diamino-3,5-dinitropyrazine-1-oxide (LLM-105) | NP | 0–20 GPa | GO, aiMD | CP2K | PBE-D2 | 2015 | [31] |
| 22. | 2,6-Diamino-3,5-dinitropyrazine-1-oxide (LLM-105) | NP | 0–45 GPa | GO, MO, aiMD (at 0 Gpa) | CASTEP; CP2K | PBE-D2 | 2014 | [32] |
| 23. | Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) | Β-HMX, insulator, metal | 0–130 GPa | BG, QMD+ MSST | CP2K | SCC-DFTB | 2014 | [33] |
| 24. | Silver fulminate (AgCNO) | α, β | 0–5 GPa | GO, PC, TD, BG, DOS | CASTEP; WIEN2k | PWSCF; PBE, PBE-D2; TB-mBJ | 2014 | [34] |
| 25. | 2,4,6-Trinitro-1,3,5-benzenetriamine (TATB) | NP | 0–7.02 GPa | GO, CM | Quantum ESPRESSO | PBE, PBE Grimme | 2010 | [35] |
| 26. | Cyclotrimethylenetrinitramine (RDX) | α, γ | 0–3.36 GPa and 3.9–7.99 GPa | GO, CM | Quantum ESPRESSO | PBE, PBE Grimme | 2010 | [35] |
| 27. | Hexanitrohexaazaisowurtzitane (CL20, HNIW) | β, γ, ε | 0–2.7 GPa | GO, CM | Quantum ESPRESSO | PBE, PBE Grimme | 2010 | [35] |
| 28. | Nitromethane (NM) | NP | 0–7.6 GPs | GO, CM | Quantum ESPRESSO | PBE, PBE Grimme | 2010 | [35] |
| 29. | Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) | α, β, δ | 0–7.47 GPa | GO, CM | Quantum ESPRESSO | PBE, PBE Grimme | 2010 | [35] |
| 30. | Pentaerythritol tetranitrate (PETN) | NP | 0–9.04 GPa | GO, CM | Quantum ESPRESSO | PBE, PBE Grimme | 2010 | [35] |
| 31. | 1,3,5,7-Cyclooctatetraene (COT) | NP | 0 and 3.8 GPa | GO, PC, Raman, XRD, aiMD | DMol3; Gaussian; CPMD | PW91 | 2008 | [36] |
| 32. | Hexanitrohexaazaisowurtzitane (CL-20, HNIW) | α· H2O, β, γ, and ε | 0–400 GPa | GO, SP, DOS, BG | CASTEP; DMol3 | PBE; rPBE | 2007 | [37] |
| 33. | Triclabendazole | I, II | 0–10 GPa | Supercell approach combined with the embedded fragment method, GO, PC, IR, Raman, TD | Gaussian | ωB97XD | 2022 | [38] |
| C. Pharmaceuticals | ||||||||
| 34. | Chlorothiazide | I, II | 0–6.2 GPa | GO, PC, TD, aiMD | CASTEP | PBE TS, PBESOL | 2021 | [39] |
| 35. | Glycinium maleate | NP | 0–5.6 Gpa | GO, PC, Raman | Quantum ESPRESSO | LDA | 2021 | [40] |
| 36. | Resorcinol | α, β | 0–4 GPa | GO, DFT, and DFTB3-D3(BJ) approach: vibrational frequencies | Quantum ESPRESSO; Phonopy; DFTB+ | B86bPBE-XDM | 2021 | [41] |
| 37. | Glycine | α, β, γ | 0–50 GPa | GO, TD, BG | Quantum ESPRESSO | PBE-D3 | 2020 | [42] |
| 38. | L-Histidine | I, I′, II, II′ | 0–7 GPa | GO | CASTEP | PBE TS | 2020 | [43,44] |
| 39. | Urea | Form I and IV | 0 and 3.1 GPa | GO, PC, TD, aiMD | CASTEP | PBE TS, PBESOL, WC | 2020 | [45] |
| 40 | A-Glycylglycine | α, α′, P212121 | 0–18GPa | USPEX, GO, ZPE, PXRD | VASP, VASP VTST tools for ZPE | PBE-D2 | 2020 | [46] |
| 41. | L-Threonine | I, I′, II, III | 0–22.31 GPa | GO | CASTEP | PBE | 2019 | [47] |
| 42. | L-Threonine | α, β | 0–5 GPa | GO | CRYSTAL | PBE-D3(BJ) | 2019 | [48] |
| 43. | Glycine | γ, δ | 0–7.8 GPa | GO, PC, TD | CASTEP | PBE, PBE Grimme, PBE TS, PBESOL, PW91, PW91 OBS, RPBE, WC, CA-PZ, CA-PZ OBS | 2018 | [49] |
| 44. | L-Serine | I, II, III | 0–8.2 GPa | GO (compression and decompression) | VASP; Gaussian | PBE-D; M06-2X | 2017 | [50] |
| 45. | Tolazamide | I, II | 0–20 GPa | GO, ZPE, TB | Gaussian; VASP | M062X; PBE-D3(BJ) | 2017 | [51] |
| 46. | Aspirin | I, II | 0–12 GPa | GO, 2D PES, PC, ZPE, TD | Quantum ESPRESSO, Phonopy | B86Bpbe, B86bPBE-XDM | 2016 | [52] |
| 47. | Aspirin | I, II | 0–5 GPa | GO, PC, IR | CRYSTAL | B3LYP-2D | 2015 | [53] |
| 48. | Paracetamol | I, II | 0–5 GPa | GO, PC, IR | CRYSTAL | B3LYP-2D | 2015 | [53] |
| 49. | Resorcinol | α, β | 0–4.5 GPa | GO, INS, PC, TD, Raman | CASTEP; CRYSTAL | WC, PBESOL, PW91, PBE, rPBE, PBE-D2, PBE TS, PBE/pob-TZVP | 2015 | [54] |
| 50. | Glycine | α, β, γ, δ, ε | 0–10 GPa | GO | Quantum ESPRESSO | PBE, revPBE, vdW-DF, vdW-DF-c09x | 2012 | [55] |
| 51. | L-Alanine | NP | 0–10 GPa | GO | Quantum ESPRESSO | PBE, revPBE, vdW-DF, vdW-DF-c09x | 2012 | [55] |
| 52. | L-Serine | I, II, III | 0–8.1 GPa | GO | SIESTA | PBE | 2008 | [56] |
| D. Others | ||||||||
| 53. | Ammonium carbamate | α, β | 0–15 GPa | GO | CASTEP | PBE TS | 2022 | [57] |
| 54. | Chloroform (CHCl3) | P63, Pnma | 0–35 GPa | GO, Raman, PF | CASTEP | PBE TS | 2020 | [58] |
| 55. | Croconic acid | Pca21, Pbcm | 0–55 GPa | USPEX, GO (compression and decompression), PC, Raman, PF, BG | CASTEP | PBE TS | 2020 | [59] |
| 56. | Squaric acid | P21/m, I4m | 0–25 GPa | USPEX, GO (compression and decompression), PC, Raman, PF, BG, OP | CASTEP | PBE TS | 2020 | [59] |
| 57. | Diisopropylammonium perchlorate (DIPAP) | P1 | 0–3.3 GPa | GO, Raman | DMol3 | PBE, PBE Grimme | 2020 | [60] |
| 58. | C11N4 | g-C11N4, α-C11N4, d-C11N4, and β-C11N4 | 0 -70 GPa | GO, PC, PF, TD | VASP | PBESOL | 2019 | [61] |
| 59. | Oxalic acid | Dihydrate, α and β | (−1.0)–12.0 GPa | GO, XRD | CASTEP | PBE | 2019 | [62] |
| 60. | Sorbic acid | C2/c | 0–8 GPa | GO, PC, Raman | CASTEP | PBE | 2017 | [63] |
| 61. | Oxobenzene-bridged 1,2,3-bisdithiazolyl radical conductor (3a) | α, β, γ | 0–13 GPa | SP, MO, BG | Gaussian; Quantum ESPRESSO | (U)B3LYP; PBE | 2014 | [64] |
| 62. | Bis-1,2,3-thiaselenazolyl radical dimer | [1a]2: σ-dimer, π-dimer [1b]2: NP | 0-13.7 GPa | GO, BG | VASP | PBE | 2012 | [65] |
| 63. | Indole | HB and β | 0–25 GPa | GO, TB | CASTEP | PBE TS | 2011 | [66] |
| N° | Software/Code | Basis Set | Periodic | License Type | Ref. Method | Number of Works | Ref. in This Article |
|---|---|---|---|---|---|---|---|
| 1. | CASTEP | Plane-wave | 3D | Academic, Commercial | [67,68] | 27 | [13,21,25,26,27,29,32,34,37,39,43,44,45,47,49,54,57,58,59,62,63,66,69] |
| 2. | VASP | Plane-wave | 3D | Academic, Commercial | [70,71] | 11 | [15,16,17,20,22,28,46,50,51,61,65] |
| 3. | Quantum ESPRESSO | Plane-wave | 3D | Free, General Public License | [72,73] | 9 | [14,23,35,40,41,42,52,55,64] |
| 4. | Gaussian | Gaussian-type orbitals | Any | Commercial | [74,75] | 8 | [12,19,24,36,38,50,51,64] |
| 5. | CP2K | Hybrid Gaussian-type orbitals, plane-wave | Any | Free, General Public License | [76,77] | 4 | [30,31,32,33] |
| 6. | CRYSTAL | Gaussian-type orbitals | Any | Academic, Commercial | [78,79] | 3 | [48,53,54] |
| 7. | DMol3 | Numerically tabulated atom-centered orbitals | Any | Commercial | [80,81] | 3 | [36,37,60] |
| 8. | CPMD | Plane-wave | 3D | Academic | [82,83] | 1 | [36] |
| 9. | DFTB+ | Slater-type orbitals, numerically tabulated atom-centered orbitals | Any | Free, General Public License | [84,85] | 1 | [41] |
| 10. | SIESTA | Numerically tabulated atom-centered orbitals | 3D | Free, General Public License | [86,87] | 1 | [56] |
| 11. | WIEN2k | FP-(L)APW + lo (the full-potential (linearized) augmented plane-wave and local orbitals) | 3D | Commercial | [88,89] | 1 | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napiórkowska, E.; Milcarz, K.; Szeleszczuk, Ł. Review of Applications of Density Functional Theory (DFT) Quantum Mechanical Calculations to Study the High-Pressure Polymorphs of Organic Crystalline Materials. Int. J. Mol. Sci. 2023, 24, 14155. https://doi.org/10.3390/ijms241814155
Napiórkowska E, Milcarz K, Szeleszczuk Ł. Review of Applications of Density Functional Theory (DFT) Quantum Mechanical Calculations to Study the High-Pressure Polymorphs of Organic Crystalline Materials. International Journal of Molecular Sciences. 2023; 24(18):14155. https://doi.org/10.3390/ijms241814155
Chicago/Turabian StyleNapiórkowska, Ewa, Katarzyna Milcarz, and Łukasz Szeleszczuk. 2023. "Review of Applications of Density Functional Theory (DFT) Quantum Mechanical Calculations to Study the High-Pressure Polymorphs of Organic Crystalline Materials" International Journal of Molecular Sciences 24, no. 18: 14155. https://doi.org/10.3390/ijms241814155
APA StyleNapiórkowska, E., Milcarz, K., & Szeleszczuk, Ł. (2023). Review of Applications of Density Functional Theory (DFT) Quantum Mechanical Calculations to Study the High-Pressure Polymorphs of Organic Crystalline Materials. International Journal of Molecular Sciences, 24(18), 14155. https://doi.org/10.3390/ijms241814155