
Applications of Genome Editing Technologies in CAD Research and Therapy with a Focus on Atherosclerosis

Abstract
:1. Introduction
2. Pathophysiology of Atherosclerosis
3. Genome Editing Approaches
3.1. Zinc-Finger Nucleases
3.2. Transcription-Activator-like Effector Nucleases
3.3. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-Associated (Cas) Systems
4. Genome Editing Approaches for Coronary Artery Disease
4.1. PCSK9
4.1.1. CRISPR-Based Editing of PCSK9
4.1.2. Cas9 Variants
4.1.3. Nonviral Delivery Methods
4.2. ANGPTL3
4.3. APOC3 and Other Lipid-Related Genes
4.3.1. APOC3
4.3.2. ApoE
4.3.3. LDLR
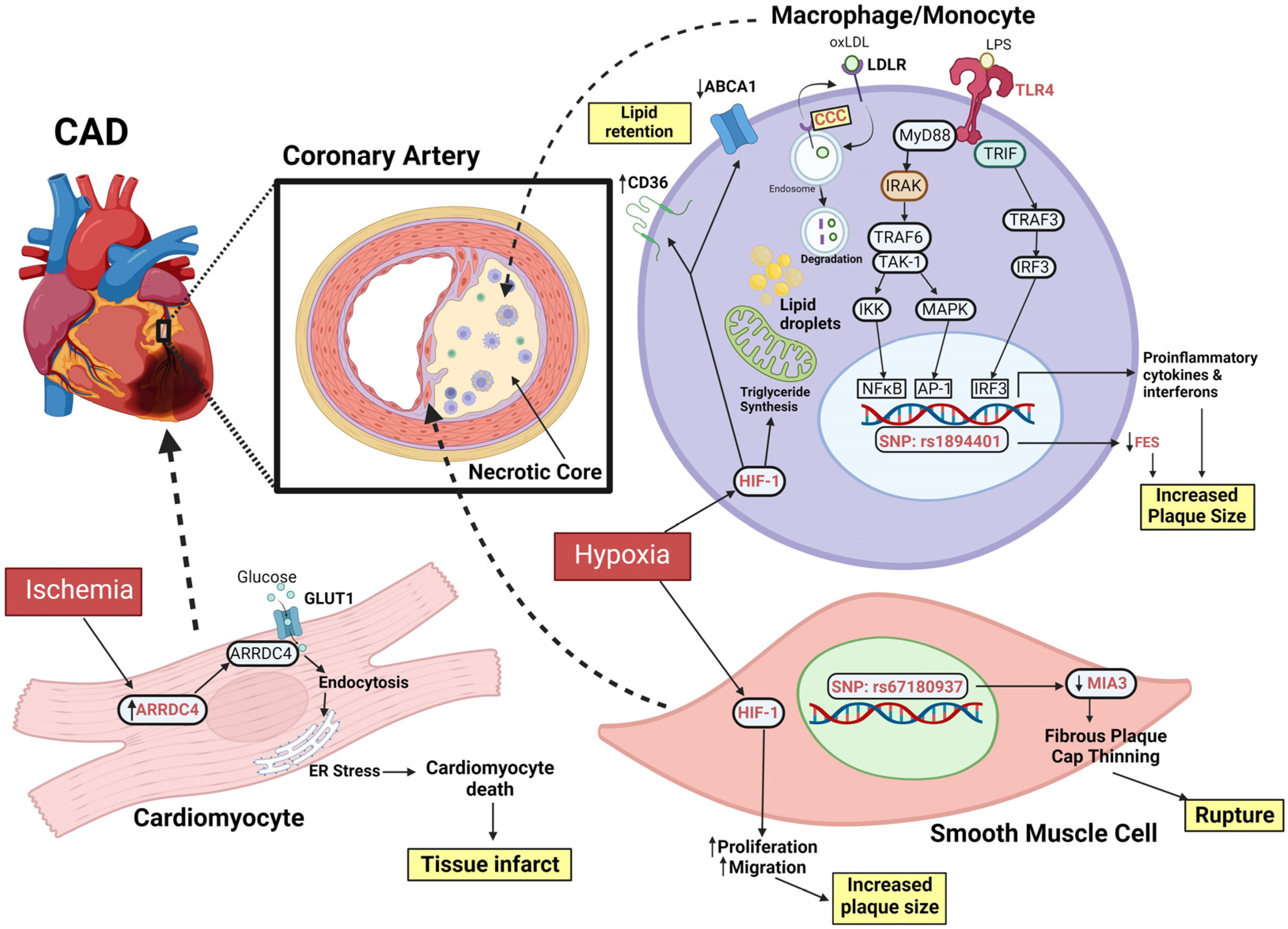
4.4. Genome Editing in the Understanding of the Complications Arising from CAD
4.4.1. CCC Complex
4.4.2. HIF1
4.4.3. Alpha-Arrestins
4.4.4. Atherosclerosis and Monocyte Activation
4.4.5. MIA3
4.4.6. CaMKIIδ
4.4.7. TLR4
5. Potential Challenges Facing Genome-Editing-Based Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| ABE | Adenine base editor |
| ALT | Alanine transaminase |
| ANGPTL4 | Angiopoietin-like 4 |
| apoAII | Apolipoprotein A-II |
| APOB | Apolipoprotein B |
| Apoc3/apoC-III | Apolipoprotein C-III |
| ApoE | Apolipoprotein E |
| ARRDC4 | Arrestin domain containing 4 |
| AST | Aspartate aminotransferase |
| BE3 | Base editor 3 |
| CAD | Coronary artery disease |
| CaMKIIδ | Calcium/calmodulin-dependent protein kinase IIδ |
| CCDC | Coiled-coil domain containing protein |
| CHO | Chinese hamster ovary |
| CIRCLE-seq | Circularization for in vitro reporting of cleavage effects by sequencing |
| COMMD | Copper metabolism gene MURR1 |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR interference |
| CRISPRmod | CRISPR modulation |
| crRNA | CRISPR RNA |
| CVD | Cardiovascular disease |
| dCas9 | Catalytically dead Cas9 |
| DNA | Deoxyribonucleic acid |
| dSaCas9 | Staphylococcus aureus Cas9 |
| DSB | Double-stranded break |
| E. coli | Escherichia coli |
| FES | FES proto-oncogene |
| gRNA | Guide RNA |
| GWAS | Genome-wide association studies |
| HBB | Haemoglobin subunit beta |
| HDR | Homology directed repair |
| HEK293 | Human embryonic kidney 293 |
| HF | Heart failure |
| HIF-1 | Hypoxia-inducible factor-1 |
| hIPSC | Human induced pluripotent stem cell |
| HMG-CoAR | 3-hydroxy-3-methylglutaryl coenzyme-A reductase |
| hMSCs | Human mesenchymal stromal cells |
| KO | Knockout |
| KRAB | Krüppel associated box domain |
| LCAT | Lecithin-cholesterol acylltransferase |
| LDL | low-density lipoprotein |
| LDLR | LDL receptor |
| LNP | Lipid nanoparticles |
| MC-3 | D-Lin-MC3-DMA |
| MI | Myocardial infarction |
| MIA3 | MIA SH3 domain ER export factor 3 |
| miR | microRNA |
| nCas9 | Nickase cas9 |
| NHEJ | Nonhomologous end joining |
| NHP | Nonhuman primate |
| NmeCas9 | Neisseria meningitidis Cas9 |
| PAM | Protospacer adjacent motif |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| RNP | Ribonucleoprotein |
| RVD | Repeat variable di-residue |
| SCNT | Somatic Cell Nuclear Transfer |
| sgRNA | Single guide RNA |
| siRNA | Small interfering RNA |
| SMC | Smooth muscle cell |
| SNP | Single nucleotide polymorphism |
| spCas9 | Streptococcus pyogenes |
| TALEN | Transcription activator-like effector nuclease |
| TLR4 | Toll-like receptor 4 |
| tracrRNA | Trans-activating CRISPR RNA |
| UCHL1 | Ubiquitin C-terminal hydrolase L1 |
| VLP | Virus-like particles |
| WT | Wild type |
| ZFN | Zinc finger nuclease |
References
- GBD 2019 Diseases and Injuries Collaborators. Global Burden of 369 Diseases and Injuries in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 18 July 2023).
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and Aetiology of Heart Failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef]
- Vedin, O.; Lam, C.S.P.; Koh, A.S.; Benson, L.; Teng, T.H.K.; Tay, W.T.; Braun, O.Ö.; Savarese, G.; Dahlström, U.; Lund, L.H. Significance of Ischemic Heart Disease in Patients with Heart Failure and Preserved, Midrange, and Reduced Ejection Fraction: A Nationwide Cohort Study. Circ. Heart Fail. 2017, 10, e003875. [Google Scholar] [CrossRef] [PubMed]
- Grech, E.D. Pathophysiology and Investigation of Coronary Artery Disease. BMJ 2003, 326, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Hashim, M.J.; Mustafa, H.; Baniyas, M.Y.; Suwaidi, S.K.B.M.A.; AlKatheeri, R.; Alblooshi, F.M.K.; Almatrooshi, M.E.A.H.; Alzaabi, M.E.H.; Darmaki, R.S.A.; et al. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 2020, 12, e9349. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury Following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, e7018393. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.; Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. Circ. Res. 2016, 118, 564–578. [Google Scholar] [CrossRef]
- Khera, A.V.; Kathiresan, S. Genetics of Coronary Artery Disease: Discovery, Biology and Clinical Translation. Nat. Rev. Genet. 2017, 18, 331–344. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease. 1. Evidence from Genetic, Epidemiologic, and Clinical Studies. A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Schunkert, H.; König, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.R.; Barbalic, M.; Gieger, C.; et al. Large-Scale Association Analysis Identifies 13 New Susceptibility Loci for Coronary Artery Disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef]
- Erdmann, J.; Kessler, T.; Munoz Venegas, L.; Schunkert, H. A Decade of Genome-Wide Association Studies for Coronary Artery Disease: The Challenges Ahead. Cardiovasc. Res. 2018, 114, 1241–1257. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Zemaitis, M.R.; Boll, J.M.; Dreyer, M.A. Peripheral Arterial Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Basatemur, G.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular Smooth Muscle Cells in Atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Grootaert, M.O.J.; Bennett, M.R. Vascular Smooth Muscle Cells in Atherosclerosis: Time for a Re-Assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Yurdagul, A.; Doran, A.C.; Cai, B.; Fredman, G.; Tabas, I.A. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front. Cardiovasc. Med. 2018, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Moons, A.H.M.; Levi, M.; Peters, R.J.G. Tissue Factor and Coronary Artery Disease. Cardiovasc. Res. 2002, 53, 313–325. [Google Scholar] [CrossRef]
- Bacigaluppi, M.; Semerano, A.; Gullotta, G.S.; Strambo, D. Insights from Thrombi Retrieved in Stroke Due to Large Vessel Occlusion. J. Cereb. Blood Flow. Metab. 2019, 39, 1433–1451. [Google Scholar] [CrossRef]
- Patil, S.; Darcourt, J.; Messina, P.; Bozsak, F.; Cognard, C.; Doyle, K. Characterising Acute Ischaemic Stroke Thrombi: Insights from Histology, Imaging and Emerging Impedance-Based Technologies. Stroke Vasc. Neurol. 2022, 7, 001038. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and Safety of More Intensive Lowering of LDL Cholesterol: A Meta-Analysis of Data from 170,000 Participants in 26 Randomised Trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Peng, Z.; Gu, H.; Wang, M.; Wang, G.; Zhang, D. Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8, 764038. [Google Scholar] [CrossRef]
- Hummelgaard, S.; Vilstrup, J.P.; Gustafsen, C.; Glerup, S.; Weyer, K. Targeting PCSK9 to Tackle Cardiovascular Disease. Pharmacol. Ther. 2023, 249, 108480. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G.; Benn, M.; Tybjærg-Hansen, A.; Kamstrup, P.R. PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. J. Clin. Endocrinol. Metab. 2016, 101, 3281–3287. [Google Scholar] [CrossRef]
- Fasano, T.; Cefalù, A.B.; Di Leo, E.; Noto, D.; Pollaccia, D.; Bocchi, L.; Valenti, V.; Bonardi, R.; Guardamagna, O.; Averna, M.; et al. A Novel Loss of Function Mutation of PCSK9 Gene in White Subjects with Low-Plasma Low-Density Lipoprotein Cholesterol. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 677–681. [Google Scholar] [CrossRef]
- Kent, S.T.; Rosenson, R.S.; Avery, C.L.; Chen, Y.-D.I.; Correa, A.; Cummings, S.R.; Cupples, L.A.; Cushman, M.; Evans, D.S.; Gudnason, V.; et al. PCSK9 Loss-of-Function Variants, Low-Density Lipoprotein Cholesterol, and Risk of Coronary Heart Disease and Stroke. Circ. Cardiovasc. Genet. 2017, 10, e001632. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL Cholesterol in Individuals of African Descent Resulting from Frequent Nonsense Mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Xhaard, C.; Lamiral, Z.; Borges-Canha, M.; Neves, J.S.; Dandine-Roulland, C.; LeFloch, E.; Deleuze, J.; Bacq-Daian, D.; Bozec, E.; et al. PCSK9 Protein and Rs562556 Polymorphism Are Associated with Arterial Plaques in Healthy Middle-Aged Population: The STANISLAS Cohort. J. Am. Heart Assoc. 2020, 9, e014758. [Google Scholar] [CrossRef]
- Deedwania, P.; Murphy, S.A.; Scheen, A.; Badariene, J.; Pineda, A.L.; Honarpour, N.; Keech, A.C.; Sever, P.S.; Pedersen, T.R.; Sabatine, M.S.; et al. Efficacy and Safety of PCSK9 Inhibition with Evolocumab in Reducing Cardiovascular Events in Patients with Metabolic Syndrome Receiving Statin Therapy: Secondary Analysis From the FOURIER Randomized Clinical Trial. JAMA Cardiol. 2021, 6, 139–147. [Google Scholar] [CrossRef]
- Raal, F.J.; Honarpour, N.; Blom, D.J.; Hovingh, G.K.; Xu, F.; Scott, R.; Wasserman, S.M.; Stein, E.A. Inhibition of PCSK9 with Evolocumab in Homozygous Familial Hypercholesterolaemia (TESLA Part B): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2015, 385, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.T.; Turner, T.; Visseren, F.L.J.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Wright, R.S.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-Term Efficacy and Safety of Inclisiran in Patients with High Cardiovascular Risk and Elevated LDL Cholesterol (ORION-3): Results from the 4-Year Open-Label Extension of the ORION-1 Trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid Restriction Enzymes: Zinc Finger Fusions to Fok I Cleavage Domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef]
- Bibikova, M.; Beumer, K.; Trautman, J.K.; Carroll, D. Enhancing Gene Targeting with Designed Zinc Finger Nucleases. Science 2003, 300, 764. [Google Scholar] [CrossRef]
- Santiago, Y.; Chan, E.; Liu, P.-Q.; Orlando, S.; Zhang, L.; Urnov, F.D.; Holmes, M.C.; Guschin, D.; Waite, A.; Miller, J.C.; et al. Targeted Gene Knockout in Mammalian Cells by Using Engineered Zinc-Finger Nucleases. Proc. Natl. Acad. Sci. USA 2008, 105, 5809–5814. [Google Scholar] [CrossRef]
- Ou, L.; DeKelver, R.C.; Rohde, M.; Tom, S.; Radeke, R.; Martin, S.J.S.; Santiago, Y.; Sproul, S.; Przybilla, M.J.; Koniar, B.L.; et al. ZFN-Mediated In Vivo Genome Editing Corrects Murine Hurler Syndrome. Mol. Ther. 2019, 27, 178–187. [Google Scholar] [CrossRef]
- Yan, H.; Niimi, M.; Matsuhisa, F.; Zhou, H.; Kitajima, S.; Chen, Y.; Wang, C.; Yang, X.; Yao, J.; Yang, D.; et al. Apolipoprotein CIII Deficiency Protects Against Atherosclerosis in Knockout Rabbits. ATVB 2020, 40, 2095–2107. [Google Scholar] [CrossRef] [PubMed]
- Sithu, S.D.; Malovichko, M.V.; Riggs, K.A.; Wickramasinghe, N.S.; Winner, M.G.; Agarwal, A.; Hamed-Berair, R.E.; Kalani, A.; Riggs, D.W.; Bhatnagar, A.; et al. Atherogenesis and Metabolic Dysregulation in LDL Receptor–Knockout Rats. JCI Insight 2017, 2, 86442. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Quan, C.; Hu, C.; Xie, B.; Du, Y.; Chen, L.; Yang, W.; Yang, L.; Chen, Q.; Shen, B.; et al. A Lipidomics Study Reveals Hepatic Lipid Signatures Associating with Deficiency of the LDL Receptor in a Rat Model. Biol. Open 2016, 5, 979–986. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE Nuclease Architecture for Efficient Genome Editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A Widely Applicable Technology for Targeted Genome Editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef]
- Sun, N.; Zhao, H. Transcription Activator-like Effector Nucleases (TALENs): A Highly Efficient and Versatile Tool for Genome Editing. Biotechnol. Bioeng. 2013, 110, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Boch, J. TALE and TALEN Genome Editing Technologies. Gene Genome Ed. 2021, 2, 100007. [Google Scholar] [CrossRef]
- Benjamin, R.; Berges, B.K.; Solis-Leal, A.; Igbinedion, O.; Strong, C.L.; Schiller, M.R. TALEN Gene Editing Takes Aim on HIV. Hum. Genet. 2016, 135, 1059–1070. [Google Scholar] [CrossRef]
- Jain, S.; Shukla, S.; Yang, C.; Zhang, M.; Fatma, Z.; Lingamaneni, M.; Abesteh, S.; Lane, S.T.; Xiong, X.; Wang, Y.; et al. TALEN Outperforms Cas9 in Editing Heterochromatin Target Sites. Nat. Commun. 2021, 12, 606. [Google Scholar] [CrossRef]
- Karakikes, I.; Termglinchan, V.; Cepeda, D.A.; Lee, J.; Diecke, S.; Hendel, A.; Itzhaki, I.; Ameen, M.; Shrestha, R.; Wu, H.; et al. A Comprehensive TALEN-Based Knockout Library for Generating Human-Induced Pluripotent Stem Cell–Based Models for Cardiovascular Diseases. Circ. Res. 2017, 120, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Moiani, A. Non-Viral DNA Delivery Associated to TALEN® Gene Editing Leads to Highly Efficient Correction of Sickle Cell Mutation in Long-Term Repopulating Hematopoietic Stem Cells. Available online: https://cellectis.com/uploads/files/2022_ESGCT_Moiani._October_14._FINAL3.pdf (accessed on 2 August 2023).
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide Sequence of the Iap Gene, Responsible for Alkaline Phosphatase Isozyme Conversion in Escherichia Coli, and Identification of the Gene Product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Terns, M.P.; Terns, R.M. CRISPR-Based Adaptive Immune Systems. Curr. Opin. Microbiol. 2011, 14, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas Systems in Bacteria and Archaea: Versatile Small RNAs for Adaptive Defense and Regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the Immune System of Bacteria and Archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA Maturation by Trans-Encoded Small RNA and Host Factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Collias, D.; Beisel, C.L. CRISPR Technologies and the Search for the PAM-Free Nuclease. Nat. Commun. 2021, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, Classification and Evolution of CRISPR-Cas Systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and Evolution of Class 2 CRISPR–Cas Systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Yang, S.; Hwang, G.-H.; Yu, J.; Bae, S. Analysis of NHEJ-Based DNA Repair after CRISPR-Mediated DNA Cleavage. Int. J. Mol. Sci. 2021, 22, 6397. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Li, X.-L.; Li, G.-H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.-W.; et al. Efficient Precise Knockin with a Double Cut HDR Donor after CRISPR/Cas9-Mediated Double-Stranded DNA Cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base Editing: Precision Chemistry on the Genome and Transcriptome of Living Cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base Editing: Advances and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA–Guided Activation of Endogenous Human Genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef]
- Forstnerič, V.; Oven, I.; Ogorevc, J.; Lainšček, D.; Praznik, A.; Lebar, T.; Jerala, R.; Horvat, S. CRISPRa-Mediated FOXP3 Gene Upregulation in Mammalian Cells. Cell Biosci. 2019, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Steinhart, Z.; Layeghi, M.; Freimer, J.W.; Bueno, R.; Nguyen, V.Q.; Blaeschke, F.; Ye, C.J.; Marson, A. CRISPR Activation and Interference Screens Decode Stimulation Responses in Primary Human T Cells. Science 2022, 375, eabj4008. [Google Scholar] [CrossRef]
- Jensen, T.I.; Mikkelsen, N.S.; Gao, Z.; Foßelteder, J.; Pabst, G.; Axelgaard, E.; Laustsen, A.; König, S.; Reinisch, A.; Bak, R.O. Targeted Regulation of Transcription in Primary Cells Using CRISPRa and CRISPRi. Genome Res. 2021, 31, 2120–2130. [Google Scholar] [CrossRef]
- Thakore, P.I.; Kwon, J.B.; Nelson, C.E.; Rouse, D.C.; Gemberling, M.P.; Oliver, M.L.; Gersbach, C.A. RNA-Guided Transcriptional Silencing in Vivo with S. Aureus CRISPR-Cas9 Repressors. Nat. Commun. 2018, 9, 1674. [Google Scholar] [CrossRef]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid Nanoparticle-Mediated Codelivery of Cas9 MRNA and Single-Guide RNA Achieves Liver-Specific in Vivo Genome Editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [Google Scholar] [CrossRef]
- Jarrett, K.E.; Lee, C.M.; Yeh, Y.-H.; Hsu, R.H.; Gupta, R.; Zhang, M.; Rodriguez, P.J.; Lee, C.S.; Gillard, B.K.; Bissig, K.-D.; et al. Somatic Genome Editing with CRISPR/Cas9 Generates and Corrects a Metabolic Disease. Sci. Rep. 2017, 7, 44624. [Google Scholar] [CrossRef]
- Zha, Y.; Lu, Y.; Zhang, T.; Yan, K.; Zhuang, W.; Liang, J.; Cheng, Y.; Wang, Y. CRISPR/Cas9-Mediated Knockout of APOC3 Stabilizes Plasma Lipids and Inhibits Atherosclerosis in Rabbits. Lipids Health Dis. 2021, 20, 180. [Google Scholar] [CrossRef]
- Guo, M.; Xu, Y.; Dong, Z.; Zhou, Z.; Cong, N.; Gao, M.; Huang, W.; Wang, Y.; Liu, G.; Xian, X. Inactivation of ApoC3 by CRISPR/Cas9 Protects Against Atherosclerosis in Hamsters. Circ. Res. 2020, 127, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Ren, X.; Wang, Y.; Li, Z.; Zhao, L.; Zhang, M.; Li, C.; Zhang, Z.; Chen, L.; Li, X.; et al. Apolipoprotein E Deficiency Accelerates Atherosclerosis Development in Miniature Pigs. Dis. Models Mech. 2018, 11, dmm036632. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hua, Z.; Xiao, H.; Cheng, Y.; Xu, K.; Gao, Q.; Xia, Y.; Liu, Y.; Zhang, X.; Zheng, X.; et al. CRISPR/Cas9-Mediated ApoE -/- and LDLR -/- Double Gene Knockout in Pigs Elevates Serum LDL-C and TC Levels. Oncotarget 2017, 8, 37751–37760. [Google Scholar] [CrossRef] [PubMed]
- Fedoseienko, A.; Wijers, M.; Wolters, J.C.; Dekker, D.; Smit, M.; Huijkman, N.; Kloosterhuis, N.; Klug, H.; Schepers, A.; Willems van Dijk, K.; et al. The COMMD Family Regulates Plasma LDL Levels and Attenuates Atherosclerosis Through Stabilizing the CCC Complex in Endosomal LDLR Trafficking. Circ. Res. 2018, 122, 1648–1660. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shi, H.; Zhao, M.; Zhang, X.; Huang, W.; Wang, Y.; Zheng, L.; Xian, X.; Liu, G. Loss of LCAT Activity in the Golden Syrian Hamster Elicits Pro-Atherogenic Dyslipidemia and Enhanced Atherosclerosis. Metabolism 2018, 83, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.-T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9–Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef]
- Lu, R.; Yuan, T.; Wang, Y.; Zhang, T.; Yuan, Y.; Wu, D.; Zhou, M.; He, Z.; Lu, Y.; Chen, Y.; et al. Spontaneous Severe Hypercholesterolemia and Atherosclerosis Lesions in Rabbits with Deficiency of Low-Density Lipoprotein Receptor (LDLR) on Exon 7. EBioMedicine 2018, 36, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Zhong, Y.; Wang, Y.; Zhang, T.; Lu, R.; Zhou, M.; Lu, Y.; Yan, K.; Chen, Y.; Hu, Z.; et al. Generation of Hyperlipidemic Rabbit Models Using Multiple SgRNAs Targeted CRISPR/Cas9 Gene Editing System. Lipids Health Dis. 2019, 18, 69. [Google Scholar] [CrossRef]
- Park, H.; Kim, D.; Cho, B.; Byun, J.; Kim, Y.S.; Ahn, Y.; Hur, J.; Oh, Y. In Vivo Therapeutic Genome Editing via CRISPR/Cas9 Magnetoplexes for Myocardial Infarction. Biomaterials 2021, 281, 121327. [Google Scholar] [CrossRef]
- Wang, X.; Raghavan, A.; Chen, T.; Qiao, L.; Zhang, Y.; Ding, Q.; Musunuru, K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 783–786. [Google Scholar] [CrossRef]
- Carreras, A.; Pane, L.S.; Nitsch, R.; Madeyski-Bengtson, K.; Porritt, M.; Akcakaya, P.; Taheri-Ghahfarokhi, A.; Ericson, E.; Bjursell, M.; Perez-Alcazar, M.; et al. In Vivo Genome and Base Editing of a Human PCSK9 Knock-in Hypercholesterolemic Mouse Model. BMC Biol. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, J.; Liu, Y.; Jin, X.; Zhong, X.; Mo, L.; Wang, Q.; Deng, H.; Yang, Y. In Vivo PCSK9 Gene Editing Using an All-in-One Self-Cleavage AAV-CRISPR System. Mol. Ther.-Methods Clin. Dev. 2021, 20, 652–659. [Google Scholar] [CrossRef]
- Ding, Q.; Strong, A.; Patel, K.M.; Ng, S.-L.; Gosis, B.S.; Regan, S.N.; Cowan, C.A.; Rader, D.J.; Musunuru, K. Permanent Alteration of PCSK9 With In Vivo CRISPR-Cas9 Genome Editing. Circ. Res. 2014, 115, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Xie, Y.; Wang, P.; Deng, S.; Qin, A.; Zhang, J.; Yu, X.; Zheng, W.; Jiang, X. Triple-Targeting Delivery of CRISPR/Cas9 To Reduce the Risk of Cardiovascular Diseases. Angew. Chem. Int. Ed. 2019, 58, 12404–12408. [Google Scholar] [CrossRef] [PubMed]
- Ibraheim, R.; Song, C.-Q.; Mir, A.; Amrani, N.; Xue, W.; Sontheimer, E.J. All-in-One Adeno-Associated Virus Delivery and Genome Editing by Neisseria Meningitidis Cas9 in Vivo. Genome Biol. 2018, 19, 137. [Google Scholar] [CrossRef]
- Jarrett, K.E.; Lee, C.M.; De Giorgi, M.; Hurley, A.; Gillard, B.K.; Doerfler, A.M.; Li, A.; Pownall, H.J.; Henry, J.; Pownall, H.J.; et al. Somatic Editing of Ldlr With Adeno-Associated Viral-CRISPR Is an Efficient Tool for Atherosclerosis Research. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1997–2006. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In Vivo Genome Editing Using Staphylococcus Aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered Virus-like Particles for Efficient in Vivo Delivery of Therapeutic Proteins. Cell 2022, 185, 250–265.e16. [Google Scholar] [CrossRef]
- Zuo, Y.; Zhang, C.; Zhou, Y.; Li, H.; Xiao, W.; Herzog, R.W.; Xu, J.; Zhang, J.; Chen, Y.E.; Han, R. Liver-Specific in Vivo Base Editing of Angptl3 via AAV Delivery Efficiently Lowers Blood Lipid Levels in Mice. Cell Biosci. 2023, 13, 109. [Google Scholar] [CrossRef]
- Lebek, S.; Chemello, F.; Caravia, X.M.; Tan, W.; Li, H.; Chen, K.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Ablation of CaMKIIδ Oxidation by CRISPR-Cas9 Base Editing as a Therapy for Cardiac Disease. Science 2023, 379, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhi, S.; Liu, W.; Wen, J.; Hu, S.; Cao, T.; Sun, H.; Li, Y.; Huang, L.; Liu, Y.; et al. Development of Highly Efficient Dual-AAV Split Adenosine Base Editor for In Vivo Gene Therapy. Small Methods 2020, 4, 2000309. [Google Scholar] [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, W.; Nguyen, G.N.; Zhang, C.; Zeng, C.; Yan, J.; Du, S.; Hou, X.; Li, W.; Jiang, J.; et al. Functionalized Lipid-like Nanoparticles for in Vivo MRNA Delivery and Base Editing. Sci. Adv. 2020, 6, eabc2315. [Google Scholar] [CrossRef]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In Vivo Adenine Base Editing of PCSK9 in Macaques Reduces LDL Cholesterol Levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Evitt, N.H.; Lv, W.; Musunuru, K. Reduced Blood Lipid Levels with In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation 2018, 137, 975–977. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Wang, X.; Musunuru, K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1741–1747. [Google Scholar] [CrossRef]
- Wang, L.; Smith, J.; Breton, C.; Clark, P.; Zhang, J.; Ying, L.; Che, Y.; Lape, J.; Bell, P.; Calcedo, R.; et al. Meganuclease Targeting of PCSK9 in Macaque Liver Leads to Stable Reduction in Serum Cholesterol. Nat. Biotechnol. 2018, 36, 717–725. [Google Scholar] [CrossRef]
- Wang, L.; Breton, C.; Warzecha, C.C.; Bell, P.; Yan, H.; He, Z.; White, J.; Zhu, Y.; Li, M.; Buza, E.L.; et al. Long-Term Stable Reduction of Low-Density Lipoprotein in Nonhuman Primates Following In Vivo Genome Editing of PCSK9. Mol. Ther. 2021, 29, 2019–2029. [Google Scholar] [CrossRef]
- Wei, S.; Zhang, Y.; Su, L.; He, K.; Wang, Q.; Zhang, Y.; Yang, D.; Yang, Y.; Ma, S. Apolipoprotein E-Deficient Rats Develop Atherosclerotic Plaques in Partially Ligated Carotid Arteries. Atherosclerosis 2015, 243, 589–592. [Google Scholar] [CrossRef]
- Koike, T.; Koike, Y.; Yang, D.; Guo, Y.; Rom, O.; Song, J.; Xu, J.; Chen, Y.; Wang, Y.; Zhu, T.; et al. Human Apolipoprotein A-II Reduces Atherosclerosis in Knock-in Rabbits. Atherosclerosis 2021, 316, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Conway, A.; Mendel, M.; Kim, K.; McGovern, K.; Boyko, A.; Zhang, L.; Miller, J.C.; DeKelver, R.C.; Paschon, D.E.; Mui, B.L.; et al. Non-Viral Delivery of Zinc Finger Nuclease MRNA Enables Highly Efficient In Vivo Genome Editing of Multiple Therapeutic Gene Targets. Mol. Ther. 2019, 27, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Hopewell, J.C.; Malik, R.; Valdés-Márquez, E.; Worrall, B.B.; Collins, R. METASTROKE Collaboration of the ISGC Differential Effects of PCSK9 Variants on Risk of Coronary Disease and Ischaemic Stroke. Eur. Heart J. 2018, 39, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Small, A.M.; Huffman, J.E.; Klarin, D.; Lynch, J.A.; Assimes, T.; DuVall, S.; Sun, Y.V.; Shere, L.; Natarajan, P.; Gaziano, M.; et al. PCSK9 Loss of Function Is Protective against Extra-Coronary Atherosclerotic Cardiovascular Disease in a Large Multi-Ethnic Cohort. PLoS ONE 2020, 15, e0239752. [Google Scholar] [CrossRef] [PubMed]
- Amrani, N.; Gao, X.D.; Liu, P.; Edraki, A.; Mir, A.; Ibraheim, R.; Gupta, A.; Sasaki, K.E.; Wu, T.; Donohoue, P.D.; et al. NmeCas9 Is an Intrinsically High-Fidelity Genome-Editing Platform. Genome Biol. 2018, 19, 214. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.; Furmanak, T.; Avitto, A.N.; Smith, M.K.; Latshaw, C.; Yan, H.; Greig, J.A.; Wilson, J.M. Increasing the Specificity of AAV-Based Gene Editing through Self-Targeting and Short-Promoter Strategies. Mol. Ther. 2021, 29, 1047–1056. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Shimizugawa, T.; Ono, M.; Shimamura, M.; Yoshida, K.; Ando, Y.; Koishi, R.; Ueda, K.; Inaba, T.; Minekura, H.; Kohama, T.; et al. ANGPTL3 Decreases Very Low Density Lipoprotein Triglyceride Clearance by Inhibition of Lipoprotein Lipase*. J. Biol. Chem. 2002, 277, 33742–33748. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef]
- Khera, A.; Lee, R.; Rohde, E.; Jayaram, H.; Kathiresan, S.; Bellinger, A. An in Vivo CRISPR Base Editing Therapy to Inactivate the ANGPTL3 Gene: Nomination of a Development Candidate for VERVE-201. Eur. Heart J. 2022, 43, ehac544.3087. [Google Scholar] [CrossRef]
- Lee, R.; Denizio, J.; Dutta, C.; Hsu, H.-T.; Garrity, R.; Pacheco, A.; Rohde, E.; Jayaram, H.; Kathiresan, S.; Bellinger, A.; et al. Preclinical Data Supporting Potential Efficacy of Verve-201—An Investigational Crispr Base Editing Medicine Targeting Angptl3—In Primary Human Cells, Mice, and Non-Human Primates. J. Am. Coll. Cardiol. 2023, 81, 1115. [Google Scholar] [CrossRef]
- Giammanco, A.; Spina, R.; Cefalù, A.B.; Averna, M. APOC-III: A Gatekeeper in Controlling Triglyceride Metabolism. Curr. Atheroscler. Rep. 2023, 25, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Corder, C.; Mueller, G.; Centurion, H.; Hallum, G.; Fesmire, J.; McConathy, W.D.; Alaupovic, P. Triglyceride Enriched Lipoprotein Particles Correlate with the Severity of Coronary Artery Disease. Atherosclerosis 1996, 122, 105–115. [Google Scholar] [CrossRef] [PubMed]
- The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute. Loss-of-Function Mutations in APOC3, Triglycerides, and Coronary Disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Karwatowska-Prokopczuk, E.; Amour, E.S.; Ballantyne, C.M.; Shapiro, M.D.; Moriarty, P.M.; Baum, S.J.; Hurh, E.; Bartlett, V.J.; Kingsbury, J.; et al. Apolipoprotein C-III Reduction in Subjects with Moderate Hypertriglyceridaemia and at High Cardiovascular Risk. Eur. Heart J. 2022, 43, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guo, J.; Zhang, L.; Miao, G.; Lai, P.; Zhang, W.; Liu, L.; Hou, X.; Wang, Y.; Huang, W.; et al. Targeting ApoC3 Paradoxically Aggravates Atherosclerosis in Hamsters With Severe Refractory Hypercholesterolemia. Front. Cardiovasc. Med. 2022, 9, 840358. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and Function in Lipid Metabolism, Neurobiology, and Alzheimer’s Diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Bouchareychas, L.; Raffai, R.L. Apolipoprotein E and Atherosclerosis: From Lipoprotein Metabolism to MicroRNA Control of Inflammation. J. Cardiovasc. Dev. Dis. 2018, 5, 30. [Google Scholar] [CrossRef]
- Jeon, H.; Blacklow, S.C. Structure and Physiologic Function of the Low-Density Lipoprotein Receptor. Annu. Rev. Biochem. 2005, 74, 535–562. [Google Scholar] [CrossRef]
- Sun, X.M.; Patel, D.D.; Webb, J.C.; Knight, B.L.; Fan, L.M.; Cai, H.J.; Soutar, A.K. Familial Hypercholesterolemia in China. Identification of Mutations in the LDL-Receptor Gene That Result in a Receptor-Negative Phenotype. Arterioscler. Thromb. A J. Vasc. Biol. 1994, 14, 85–94. [Google Scholar] [CrossRef]
- Singla, A.; Fedoseienko, A.; Giridharan, S.S.P.; Overlee, B.L.; Lopez, A.; Jia, D.; Song, J.; Huff-Hardy, K.; Weisman, L.; Burstein, E.; et al. Endosomal PI(3)P Regulation by the COMMD/CCDC22/CCDC93 (CCC) Complex Controls Membrane Protein Recycling. Nat. Commun. 2019, 10, 4271. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, P.; Billadeau, D.D.; Favier, R.; Rong, S.; Dekker, D.; Fedoseienko, A.; Fieten, H.; Wijers, M.; Levels, J.H.; Huijkman, N.; et al. CCC- and WASH-Mediated Endosomal Sorting of LDLR Is Required for Normal Clearance of Circulating LDL. Nat. Commun. 2016, 7, 10961. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early Expression of Angiogenesis Factors in Acute Myocardial Ischemia and Infarction. N. Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-G.; Hausenloy, D.J. Hypoxia-Inducible Factor as a Therapeutic Target for Cardioprotection. Pharmacol. Ther. 2012, 136, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Geng, B.; Wang, X.; Park, K.H.; Lee, K.E.; Kim, J.; Chen, P.; Zhou, X.; Tan, T.; Yang, C.; Zou, X.; et al. UCHL1 Protects against Ischemic Heart Injury via Activating HIF-1α Signal Pathway. Redox Biol. 2022, 52, 102295. [Google Scholar] [CrossRef] [PubMed]
- Hovsepian, J.; Albanèse, V.; Becuwe, M.; Ivashov, V.; Teis, D.; Léon, S. The Yeast Arrestin-Related Protein Bul1 Is a Novel Actor of Glucose-Induced Endocytosis. MBoC 2018, 29, 1012–1020. [Google Scholar] [CrossRef]
- Nakayama, Y.; Mukai, N.; Kreitzer, G.; Patwari, P.; Yoshioka, J. Interaction of ARRDC4 with GLUT1 Mediates Metabolic Stress in the Ischemic Heart. Circ. Res. 2022, 131, 510–527. [Google Scholar] [CrossRef]
- Karamanavi, E.; McVey, D.G.; van der Laan, S.W.; Stanczyk, P.J.; Morris, G.E.; Wang, Y.; Yang, W.; Chan, K.; Poston, R.N.; Luo, J.; et al. The FES Gene at the 15q26 Coronary-Artery-Disease Locus Inhibits Atherosclerosis. Circ. Res. 2022, 131, 1004–1017. [Google Scholar] [CrossRef]
- Luo, C.; Wang, F.; Ren, X.; Ke, T.; Xu, C.; Tang, B.; Qin, S.; Yao, Y.; Chen, Q.; Wang, Q.K. Identification of a Molecular Signaling Gene-Gene Regulatory Network between GWAS Susceptibility Genes ADTRP and MIA3/TANGO1 for Coronary Artery Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1640–1653. [Google Scholar] [CrossRef]
- Aherrahrou, R.; Guo, L.; Nagraj, V.P.; Aguhob, A.; Hinkle, J.; Chen, L.; Yuhl Soh, J.; Lue, D.; Alencar, G.F.; Boltjes, A.; et al. Genetic Regulation of Atherosclerosis-Relevant Phenotypes in Human Vascular Smooth Muscle Cells. Circ. Res. 2020, 127, 1552–1565. [Google Scholar] [CrossRef]
- Duran, J.; Nickel, L.; Estrada, M.; Backs, J.; van den Hoogenhof, M.M.G. CaMKIIδ Splice Variants in the Healthy and Diseased Heart. Front. Cell Dev. Biol. 2021, 9, 644630. [Google Scholar] [CrossRef]
- Paz-García, M.; Povo-Retana, A.; Jaén, R.I.; Prieto, P.; Peraza, D.A.; Zaragoza, C.; Hernandez-Jimenez, M.; Pineiro, D.; Regadera, J.; García-Bermejo, M.L.; et al. Beneficial Effect of TLR4 Blockade by a Specific Aptamer Antagonist after Acute Myocardial Infarction. Biomed. Pharmacother. 2023, 158, 114214. [Google Scholar] [CrossRef] [PubMed]
- Schary, Y.; Rotem, I.; Caller, T.; Lewis, N.; Shaihov-Teper, O.; Brzezinski, R.Y.; Lendengolts, D.; Raanani, E.; Sternik, L.; Naftali-Shani, N.; et al. CRISPR-Cas9 Editing of TLR4 to Improve the Outcome of Cardiac Cell Therapy. Sci. Rep. 2023, 13, 4481. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Farbas, C.F., III. ZFN, TALEN and CRISPR/Cas-based Methods for Genome Engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Nain, V. TALENs—An Indespensable Tool in the Era of CRISPR: A Mini Review. J. Genet. Eng. Biotechnol. 2021, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.; Yu, T. CRISPR/Cas9 Therapeutics: Progress and Prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef]
- Zhu, L.J.; Holmes, B.R.; Aronin, N.; Brodsky, M.H. CRISPRseek: A Bioconductor Package to Identify Target-Specific Guide RNAs for CRISPR-Cas9 Genome-Editing Systems. PLoS ONE 2014, 9, e108424. [Google Scholar] [CrossRef]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A Web-Based Tool for Identifying and Validating CRISPR/Cas Off-Target Sites. Mol. Ther.-Nucleic Acids 2014, 3, e214. [Google Scholar] [CrossRef]
- Rasul, M.F.; Hussen, B.M.; Salihi, A.; Ismael, B.S.; Jalal, P.J.; Zanichelli, A.; Jamali, E.; Banlahmad, A.; Ghafouri-Fard, S.; Basiri, A.; et al. Strategies to Overcome the Main Challenges of the Use of CRISPR/Cas9 as a Replacement for Cancer Therapy. Mol. Cancer 2022, 21, 64. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Technology | General Mechanism | Strengths | Weaknesses |
|---|---|---|---|
| ZFNs | Zinc finger domains conjugated to a FokI endonuclease domain recognises target sequence. Dimerisation of FokI domain allowed for activity and cleavage of target sequence. Each zinc finger binding domain recognises 3 nucleotides. Recognition sites are usually at least 18 base pairs long. | Easily delivered using viral and nonviral delivery vectors. | Protein engineering required to generate different ZFNs. Not as specific compared to TALENs. |
| TALENs | TALEs conjugated to a FokI endonuclease domain recognises target sequence. Dimerisation of FokI domain allowed for activity and cleavage of target sequence. Each TAL repeat unit recognises 1 nucleotide. Recognition sites are usually at least 14 base pairs long. | More specific compared to ZFNs. Can target heterochromatin with greater efficiency compared to CRISPR/Cas systems. Easier to engineer for specific sequence targeting as compared to ZFNs | Hard to deliver with viral vectors due to large size of TALENs. Possibility of off-target editing remains. |
| CRISPR/Cas | Guide RNAs direct Cas protein to the target site for cleavage. Guide sequences are generally 20 base pairs long. | Easy to multiplex. Easily cloned and synthesised. Easy to modify to target novel sequences as long as a PAM site is present. | Typically used spCas9 protein is relatively large, difficult to deliver with viral vectors (e.g., AAVs), but possible to use smaller orthologs. Possibility of off-target editing. |
| Gene Editor | Delivery Vector | Target Gene | Model | Model Phenotype | Reference |
|---|---|---|---|---|---|
| CRISPR/Cas9 | LNPs | Angptl3 | 6–80-week-old female C57BL/6 mice | 65.2% reduction in serum ANGPTL3 protein, lower LDL cholesterol and triglyceride levels. | [81] |
| CRISPR/Cas9 | AAV8 | Apob | 6–9-week old Cas9 transgenic male mice | Mice with both Ldlr and Apob KO showed rapid drop in plasma cholesterol. Near complete loss of both Apob and LDLR protein. No atherosclerotic lesions. | [82] |
| CRISPR/Cas9 | Microinjection and embryo transfer | Apoc3 | New Zealand White rabbits | Chow diet: KO rabbits had 50% lower triglyceride levels and increased plasma lipoprotein lipase levels. High fat diet: No change in plasma triglycerides, total cholesterol, and LDL cholesterol levels. Mild atherosclerotic lesions. | [83] |
| CRISPR/Cas9 | not mentioned | Apoc3 | Syrian golden hamsters | Reduced triglyceride and total cholesterol in blood, marked increase in HDL cholesterol. Fewer atherosclerotic lesions in both thoracic and abdominal arteries compared to WT. | [84] |
| CRISPR/Cas9 | Somatic Cell Nuclear Transfer (SCNT) | ApoE | Bama miniature pigs | Normal diet: KO pigs have moderately increased plasma cholesterol levels. High-fat, high-cholesterol diet: severe hypercholesterolemia, human-like atherosclerotic lesions in aorta and coronary arteries. | [85] |
| CRISPR/Cas9 | SCNT | ApoE, LDLR | Bama minipigs | Significant elevation in LDL cholesterol, total cholesterol, and apolipoprotein B. | [86] |
| CRISPR/Cas9 | Adenovirus | Ccdc22 | Liver-specific Cas9-expressing C57BL/6J mice | 70% reduction in CCDC22 levels, decreased expression of all COMMD proteins except COMMD6, ~35% increased plasma total cholesterol. | [87] |
| CRISPR/Cas9 | mRNA | LCAT | Golden Syrian hamster | Extremely low HDL in plasma, hypertriglyceridemia. Proatherogenic dyslipidaemia. | [88] |
| CRISPR/Cas9 | AAV8 | Ldlr | P1 and P2 LdlrE208X mutant mice | Significant reduction in total cholesterol, triglycerides, and LDL cholesterol in serum. Smaller atherosclerotic plaques in aorta. | [89] |
| CRISPR/Cas9 | AAV8 | Ldlr | 6–9-week-old Cas9 transgenic male mice | Significantly higher plasma cholesterol. Visible atherosclerotic lesions. Near complete loss in LDLR protein expression. | [82] |
| CRISPR/Cas9 | microinjection of zygote | LDLR | New Zealand White rabbits | Spontaneous development of hypercholesterolemia and atherosclerosis on normal chow diet. | [90] |
| CRISPR/Cas9 | Microinjection and embryo transfer | LDLR, apoE | New Zealand White rabbits | Normal chow diet: 10× higher cholesterol levels compared to WT, aortic, and coronary atherosclerosis. | [91] |
| CRISPR/Cas9 | AAV8 | LdlrE208X | Not stated | Severe atherosclerotic phenotype after high-fat diet. | [89] |
| CRISPR/Cas9 | Magnetoplexes | miR34a | 8-week-old ICR mice that underwent MI | Decreased miR34a expression, reduced collagen fibril formation, increased proliferation and cardiomyocyte number. | [92] |
| CRISPR/Cas9 | Adenovirus | Pcsk9 | FRG KO mouse model, engrafted with primary human hepatocytes | 52% reduction in human PCSK9 levels. Twofold increase in mouse Pcsk9 levels. No change in total cholesterol levels. | [93] |
| CRISPR/Cas9 | Adenovirus | Pcsk9 | 28-week-old hPCSK9-KI mouse model | Decreased Pcsk9 protein and mRNA levels in liver. Significant reduction in total plasma cholesterol and LDL cholesterol levels. | [94] |
| CRISPR/Cas9 | AAV8 | Pcsk9 | 4- to 6-week-old C57/BL6J male mice | ~80% decrease in serum Pcsk9. ~35% decrease in total cholesterol after 24 weeks. | [95] |
| CRISPR/Cas9 | Adenovirus | Pcsk9 | 5-week-old female C57BL/6 mice | 35–40% reduction in total plasma cholesterol. Substantially lower Pcsk9 levels. | [96] |
| CRISPR/Cas9 | gold nano clusters | Pcsk9 | 6 weeks old C57BL6/J female mice | Decrease in serum LDL cholesterol, reduced serum Pcsk9 level. | [97] |
| NmeCas9 | AAV8 | Pcsk9 | 12- to 16-week-old female C57BL/6 mice | Lower cholesterol levels. Decreased Pcsk9 levels. | [98] |
| SaCas9 | AAV8 | Ldlr | 6 weeks old C57BL6/J (both sexes) | Decrease in LDLR protein expression. Hypercholesterolemia. Atherosclerotic lesion formation (more significant for male mice). | [99] |
| SaCas9 | AAV8 | Pcsk9 | 5–6-week-old male C57/BL6 mice | ~95% decrease in serum Pcsk9. ~40% decrease in total cholesterol one week post administration, sustained throughout four-week course, | [100] |
| ABE8.8m base editor | LNPs | Pcsk9 | Cynomolgus monkeys | 81% reduction in serum Pcsk9. 65% reduction in serum LDL cholesterol. | [101] |
| ABE8e base editor | Virus like particles | Pcsk9 | 6- to 7-week-old adult C57BL/6J mice | ~78% reduction in serum Pcsk9 levels 1 week post injection. | [102] |
| AncBE4max | AAV9 | Angptl3 | 6-week-old B6 mice | ~58% decrease in serum triglyceride levels. ~61% decrease in total cholesterol. ~88% decrease in ANGPTL3 protein. | [103] |
| CRISPR/Cas9 ABE | AAV9 | CaMKIIδ | 12-week-old male C57Bl6 mice | Similar levels of fractional shortening comparable to sham. LV end-diastolic dilation not observed. | [104] |
| CRISPR/Cas9 ABE | AAV8 | Pcsk9 | 8-week-old female mice | ~50% decrease in Pcsk9 protein level, lower VLDL/LDL levels 6–8 weeks post injection. | [105] |
| CRISPR/Cas9 ABE | LNPs | Pcsk9 | Cynomolgus monkey | Up to 83% lower blood Pcsk9 protein and 69% lower low-density lipoprotein cholesterol up to 476 days after dosing. | [106] |
| CRISPR/Cas9 ABE | Lipid-like nanomaterials | Pcsk9 | Balb/c mice | Reduction in serum Pcsk9 levels. | [107] |
| CRISPR/Cas9 ABE | AAV8 | Pcsk9 | 5-week-old male C57BL/6J mice | Decrease in plasma Pcsk9 and LDL levels. | [108] |
| CRISPR/Cas9 ABE | LNPs | Pcsk9 | Male C57BL/6J mice | 95% reduction in plasma Pcsk9. 58% reduction in LDL cholesterol. | [108] |
| CRISPR/Cas9 ABE | LNPs | Pcsk9 | Cynomolgus macaques | 32% reduction in plasma Pcsk9. 14% reduction in LDL cholesterol. | [108] |
| CRISPR/Cas9 BE3 | Adenovirus | Angptl3 | 5-week-old C57BL/6J male mice | 49% reduction in plasma ANGPTL3. 31% reduction in plasma triglycerides. 19% reduction in cholesterol after 7 days. | [109] |
| CRISPR/Cas9 BE3 | Adenovirus | Angptl3 | 5-week-old male hyperlipidaemic Ldlr KO mice | 56% reduction in triglycerides. 51% reduction in cholesterol after 14 days. | [109] |
| CRISPR/Cas9 BE3 | Adenovirus | Pcsk9 | 5-week-old C57BL/6J mice | >50% reduction in Pcsk9 protein levels. ~30% reduction in plasma cholesterol levels. | [110] |
| CRISPR/Cas9 BE3 | Adenovirus | Pcsk9 | 10-week-old hPCSK9-KI mouse model | Significant reductions in levels of circulating human and mouse Pcsk9 protein. Significant reductions in plasma total cholesterol. | [94] |
| dSaCas9KRA | AAV8 | Pcsk9 | 6–8-week-old C57Bl/6 male mice | Reduction in serum Pcsk9 and cholesterol levels. | [80] |
| I-CreI-based meganuclease first generation (M1PCSK9) | AAV8 | Pcsk9 | Nonhuman primate. Rhesus macaques | Up to 84% decrease in serum Pcsk9, up to 60% decrease in serum LDL. | [111] |
| I-CreI-based meganuclease second generation (M2PCSK9) | AAV8 | Pcsk9 | Nonhuman primate. Rhesus macaques | ~63% reduction in Pcsk9 protein. ~40% reduction in serum LDL levels. | [112] |
| M2PCSK9 | AAV8 | Pcsk9 | Nonhuman primate. Rhesus macaques | Up to 62% reduction in serum Pcsk9, up to 39% reduction in serum LDL. | [111] |
| TALEN | ApoE | 6–8-week-old male rats | High cholesterol diet: typical dyslipidaemia, not observed in WT. No obvious atherosclerotic lesions on aorta and aortic root. Partial ligation of carotid arteries resulted in formation of plaques in KO rats. | [113] | |
| TALEN | microinjection and embryo transfer | human ApoAII | New Zealand White rabbits | Knock-in rabbits had lower atherosclerotic burden, lower plasma triglycerides, and higher plasma HDL levels. | [114] |
| ZFN | Microinjection | Apoc3 | Embryos | Normal diet: KO rabbits had significantly lower plasma levels of triglycerides but unchanged levels of total cholesterol and HDL (compared to WT). Cholesterol-rich diet: KO rabbits had significantly lower levels of plasma total cholesterol and triglycerides than WT. | [44] |
| ZFN | Lipid nanoparticle | Pcsk9 | 8- to 10-week-old C57BL/6 mice | >90% reduction in Pcsk9protein levels in plasma. | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mak, M.C.E.; Gurung, R.; Foo, R.S.Y. Applications of Genome Editing Technologies in CAD Research and Therapy with a Focus on Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 14057. https://doi.org/10.3390/ijms241814057
Mak MCE, Gurung R, Foo RSY. Applications of Genome Editing Technologies in CAD Research and Therapy with a Focus on Atherosclerosis. International Journal of Molecular Sciences. 2023; 24(18):14057. https://doi.org/10.3390/ijms241814057
Chicago/Turabian StyleMak, Michelle C. E., Rijan Gurung, and Roger S. Y. Foo. 2023. "Applications of Genome Editing Technologies in CAD Research and Therapy with a Focus on Atherosclerosis" International Journal of Molecular Sciences 24, no. 18: 14057. https://doi.org/10.3390/ijms241814057
APA StyleMak, M. C. E., Gurung, R., & Foo, R. S. Y. (2023). Applications of Genome Editing Technologies in CAD Research and Therapy with a Focus on Atherosclerosis. International Journal of Molecular Sciences, 24(18), 14057. https://doi.org/10.3390/ijms241814057