
Peculiarities of the Spatial and Electronic Structure of 2-Aryl-1,2,3-Triazol-5-Carboxylic Acids and Their Salts on the Basis of Spectral Studies and DFT Calculations




Abstract
:1. Introduction
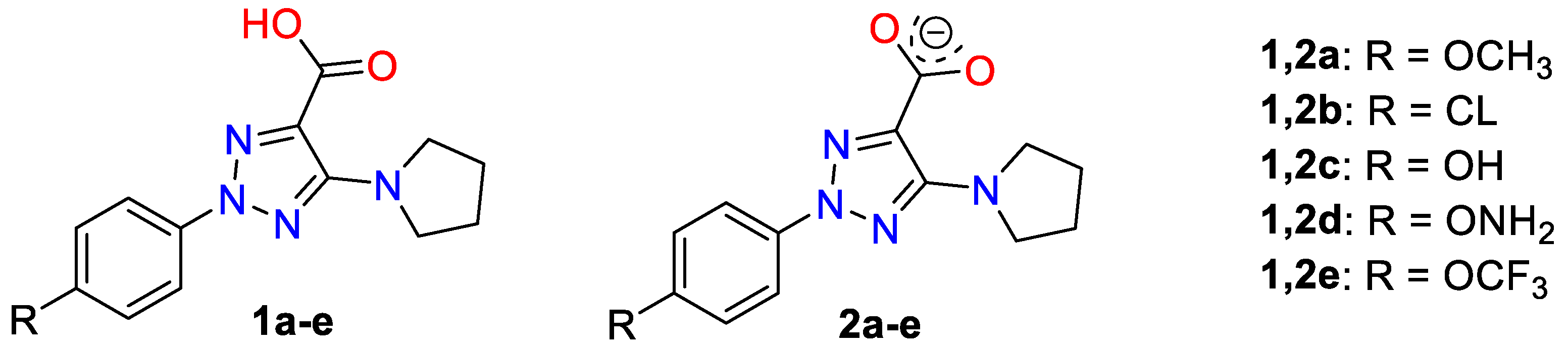
2. Results and Discussion
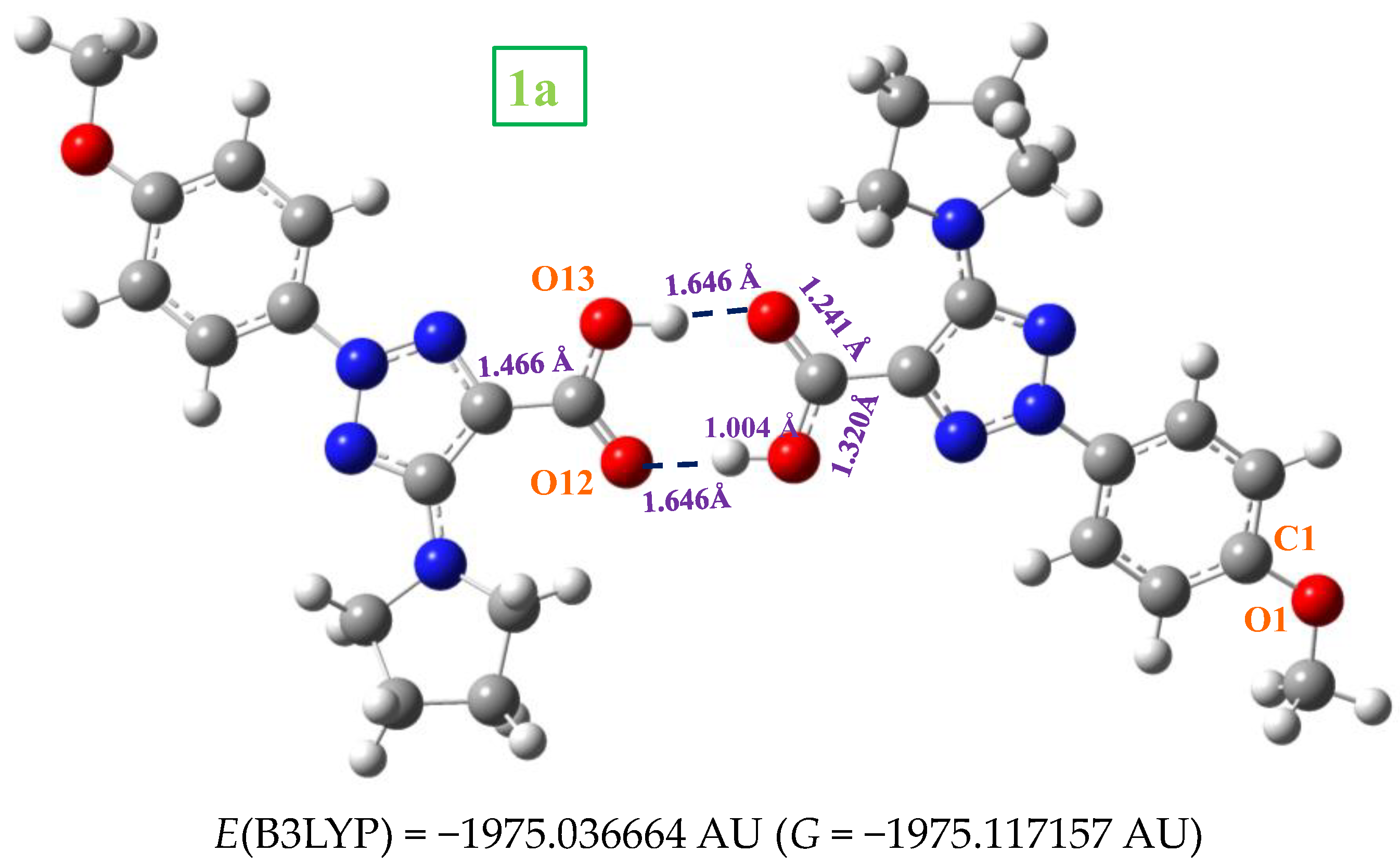
2.1. Molecular Geometries in the Monomer Form of 1a and 2a
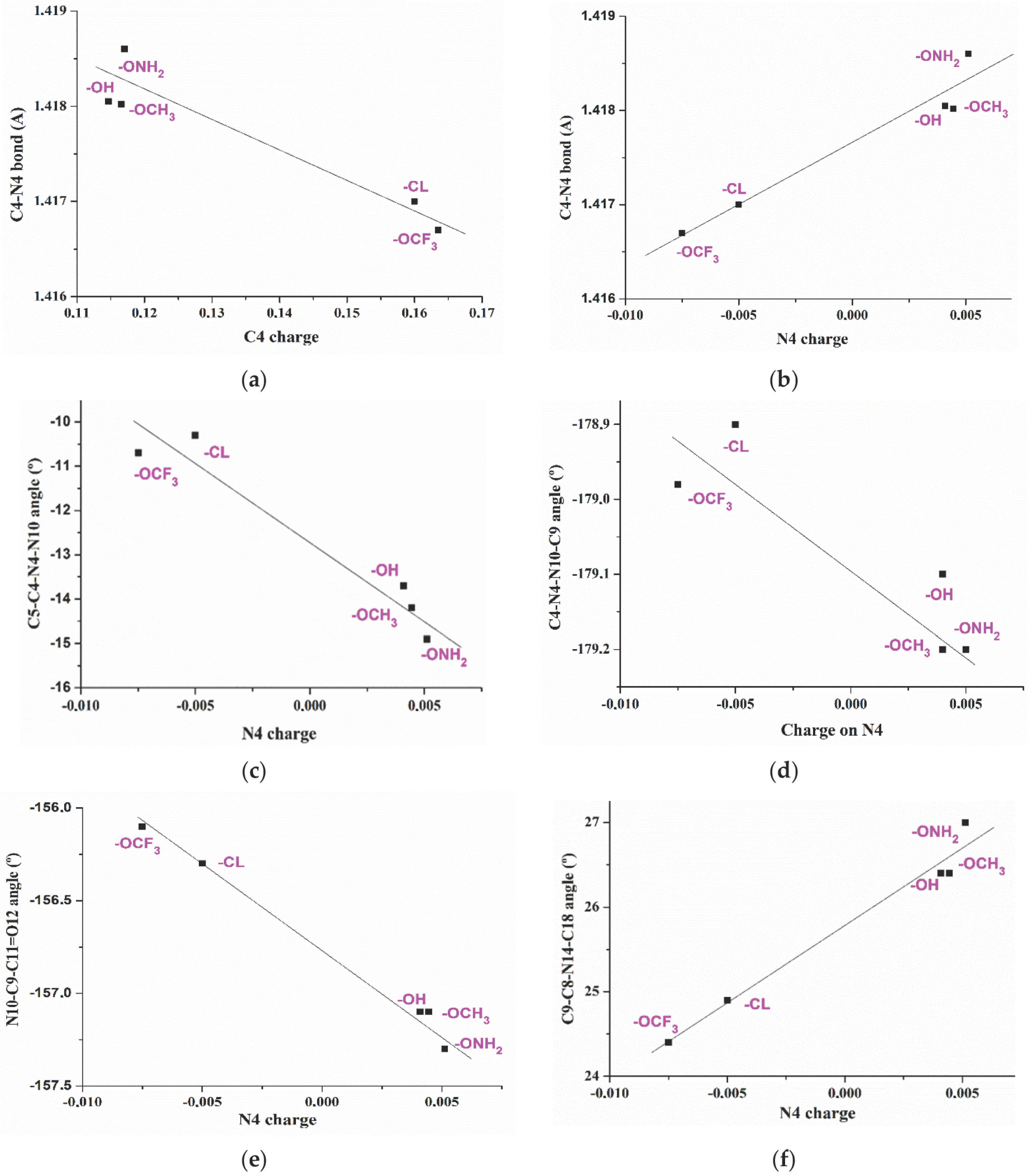
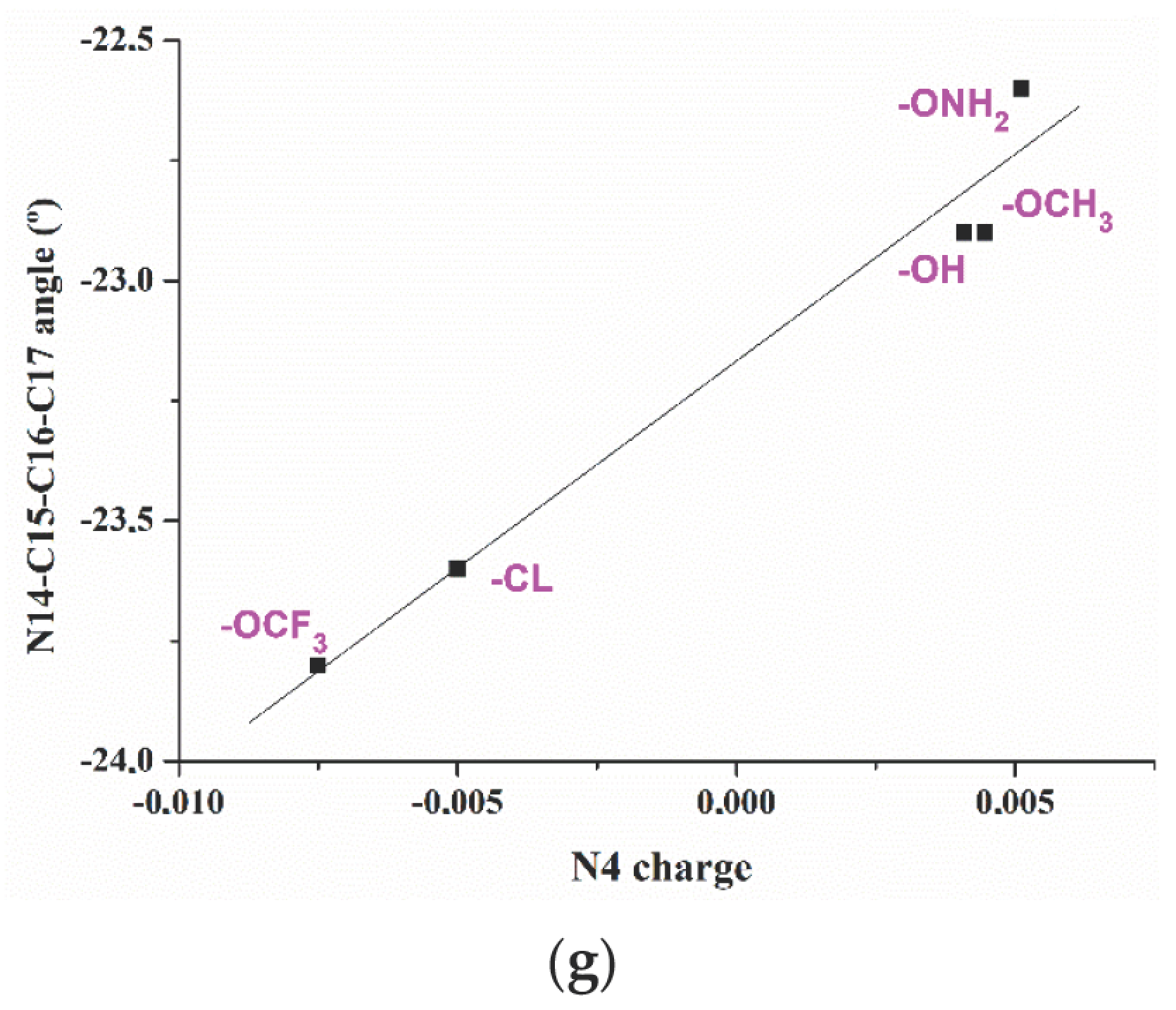
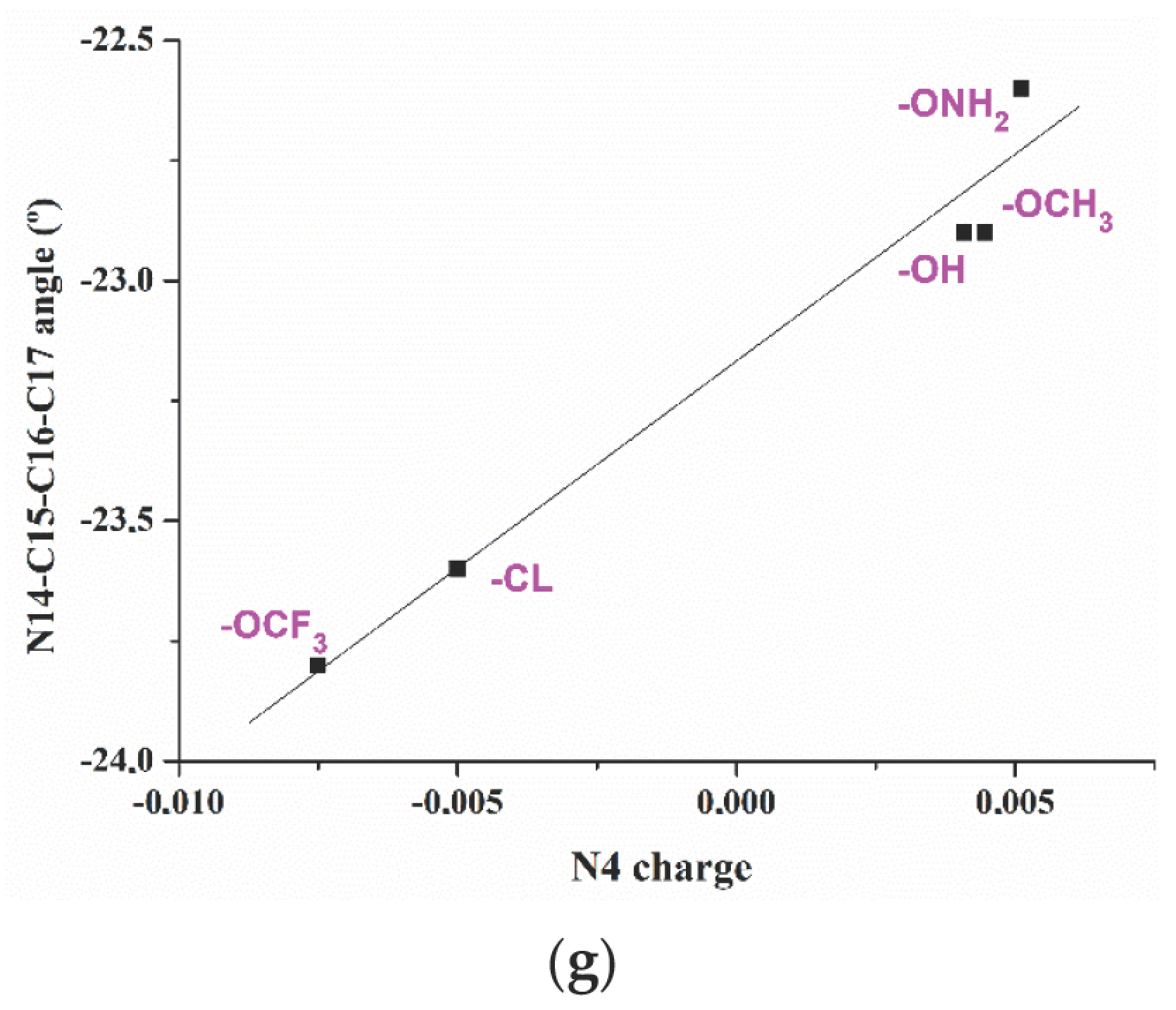
2.2. Relationships Established between the Molecular Parameters
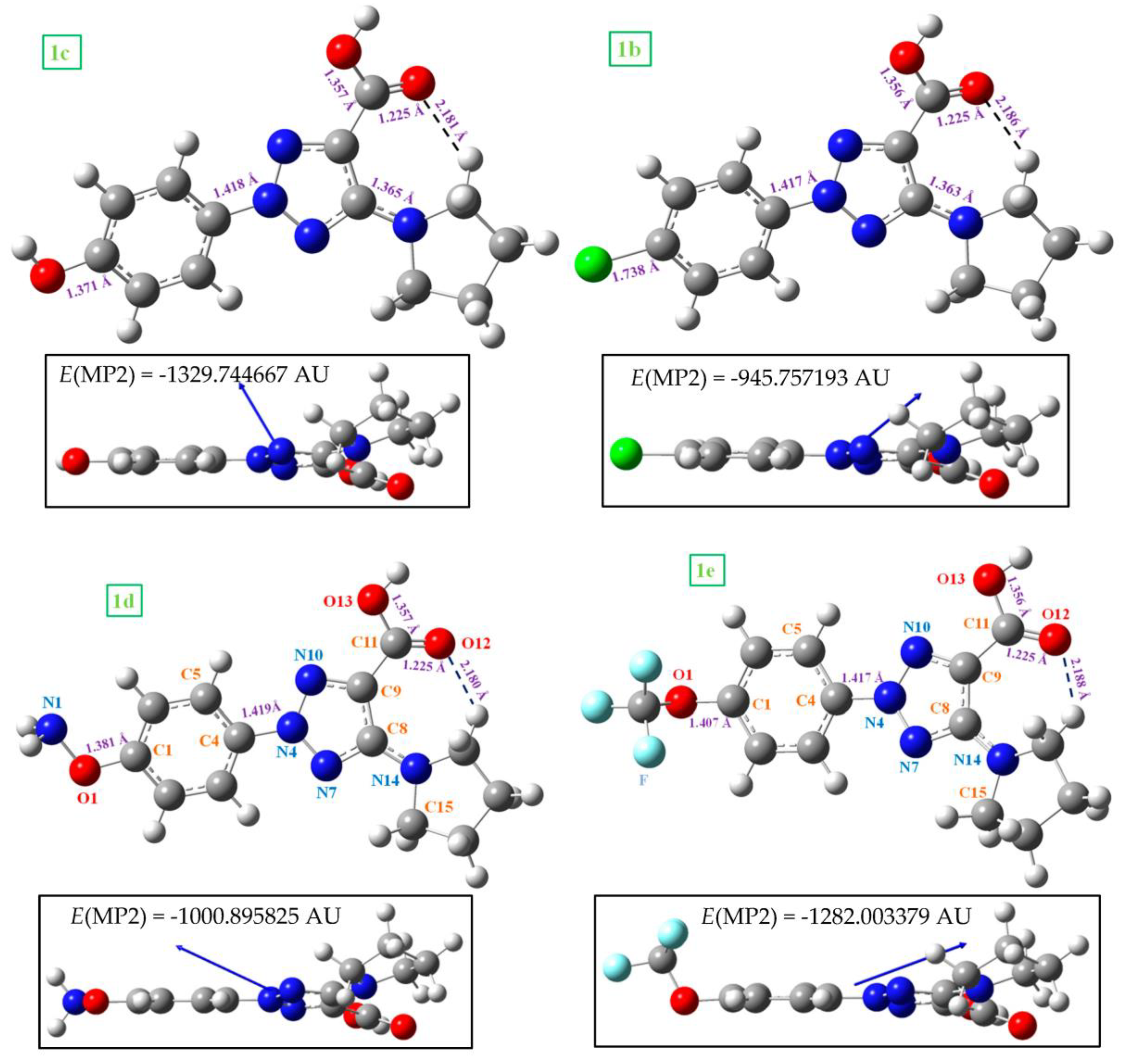
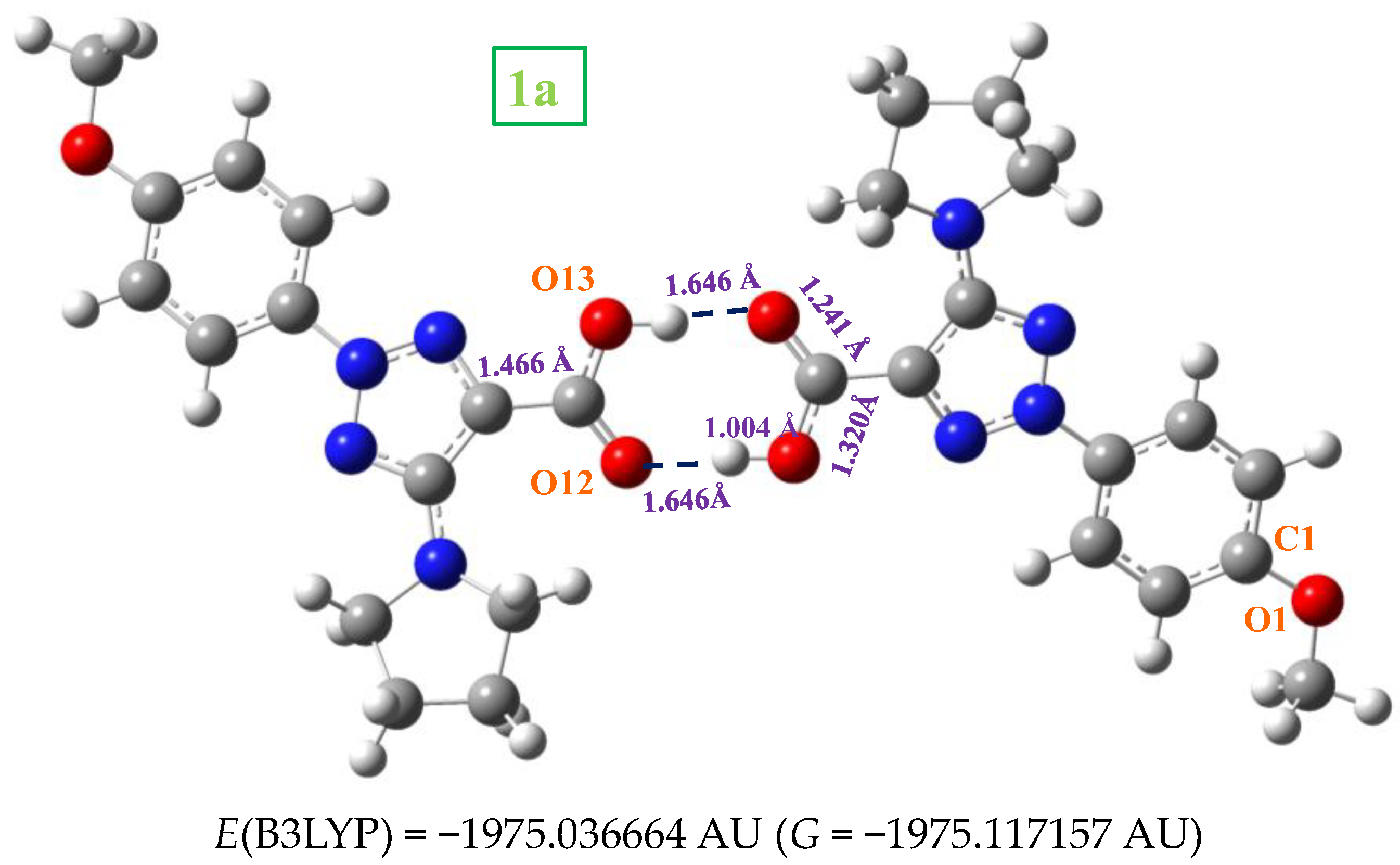
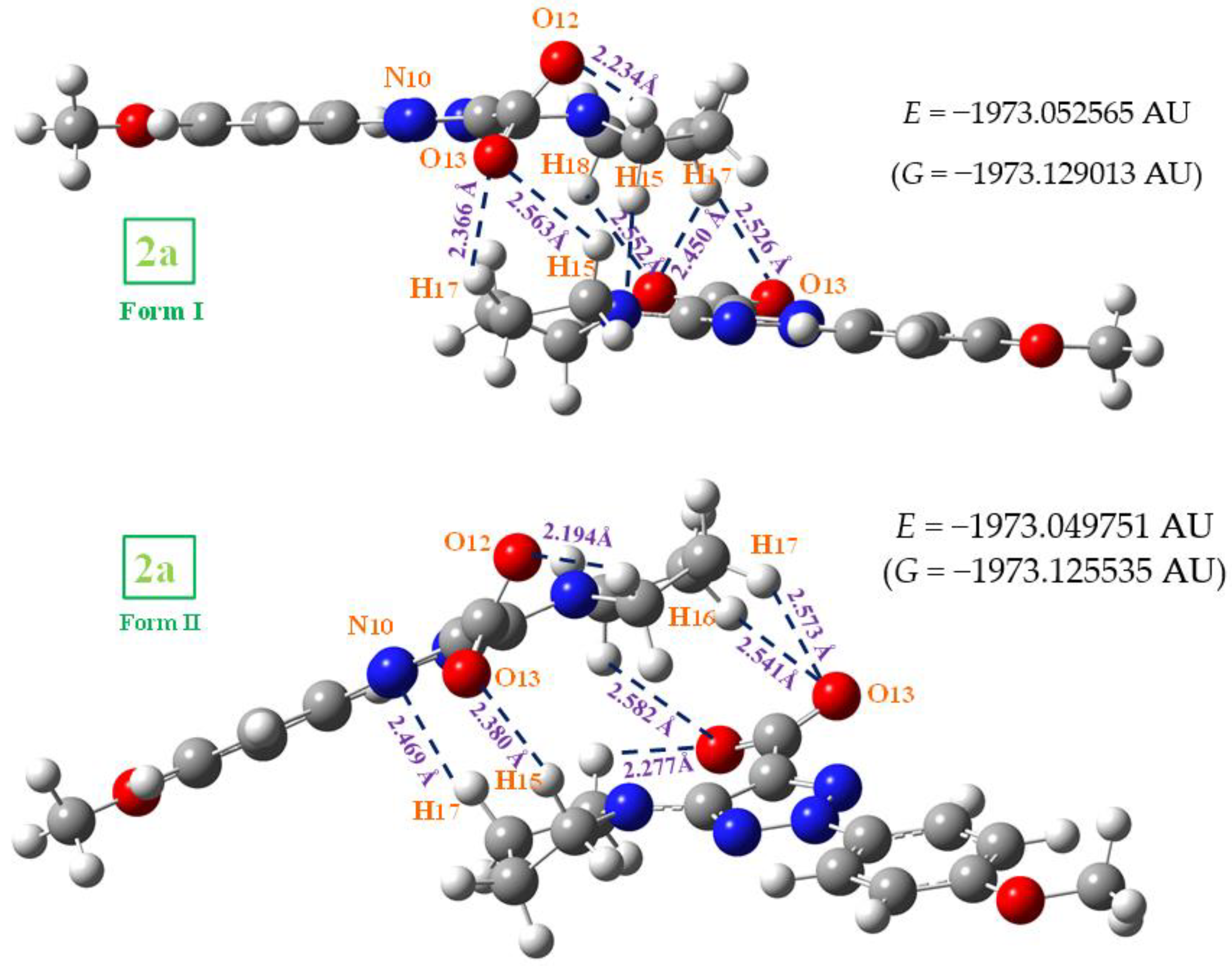
2.3. Molecular Geometries in the Dimer Forms
2.4. Molecular Properties
2.5. Scaling the Wavenumbers
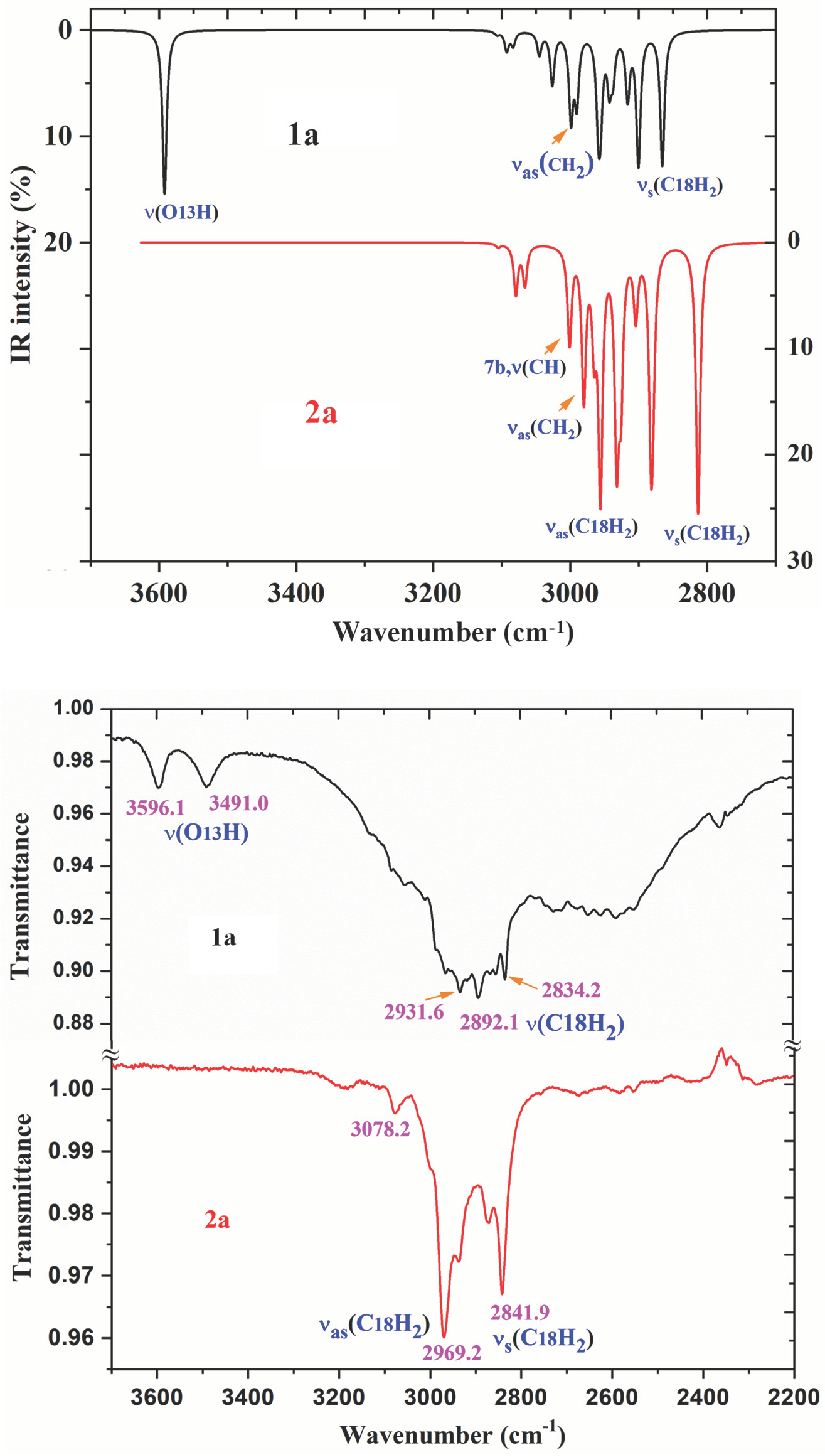
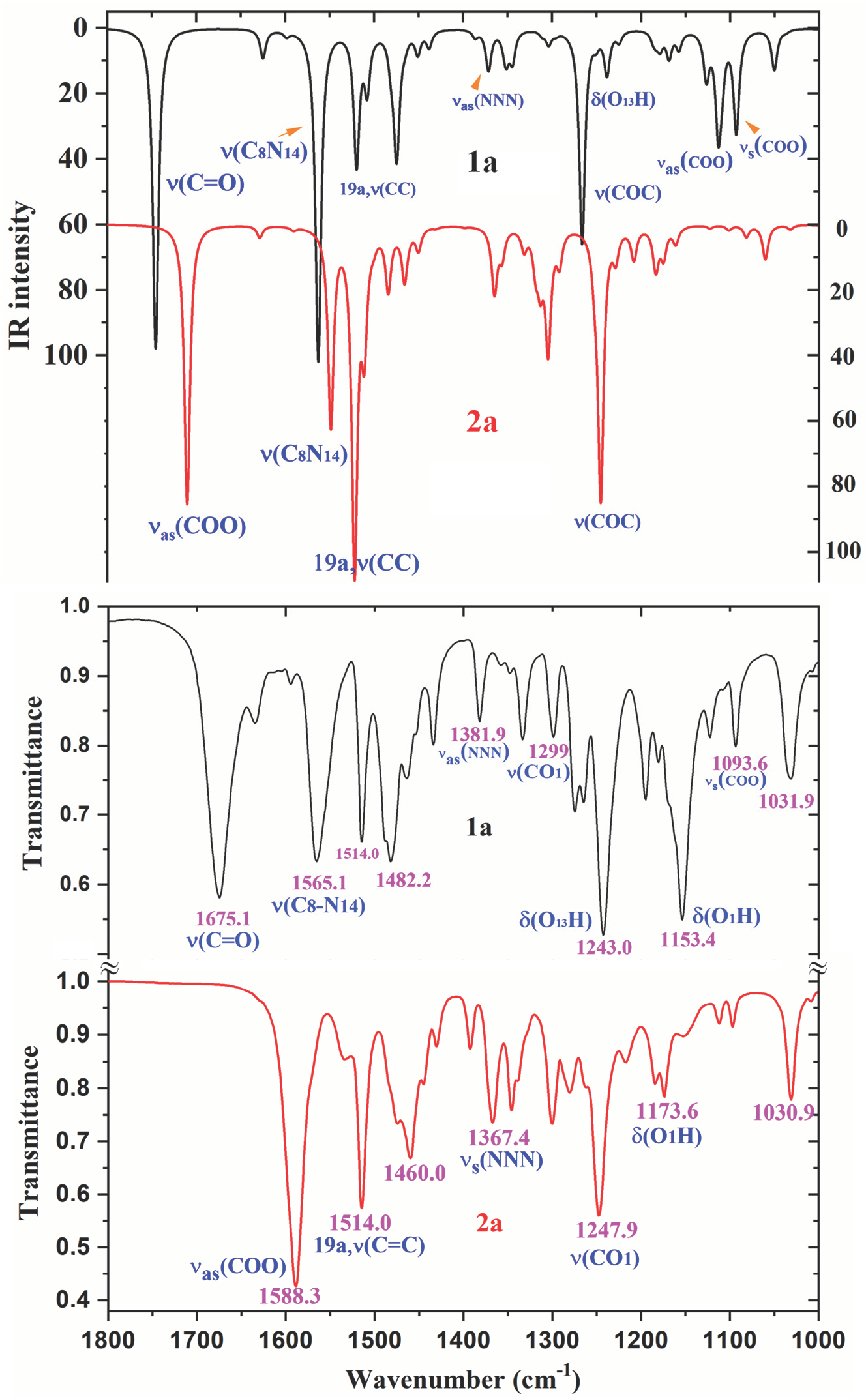
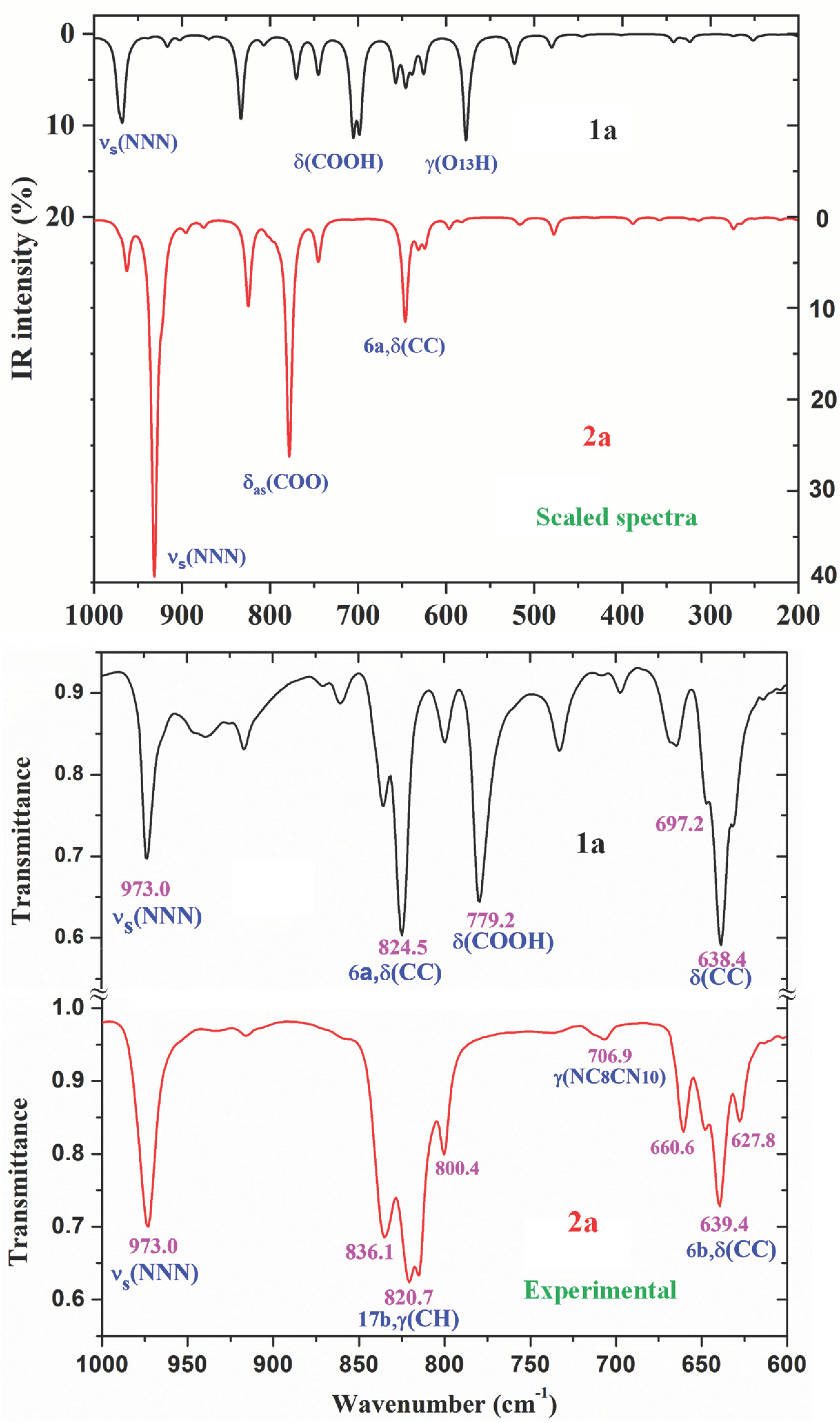
2.6. Vibrational Analysis of 1a and 2a
2.6.1. The Carboxylic COOH Group Modes in Molecule 1a

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Mode | A | S | TLSE | PSE | IR | Raman | Characterization | |
|---|---|---|---|---|---|---|---|---|---|
| νcal | νscal | νscal | |||||||
| COOH | ν(O-H) δ(O-H) γ(O-H) | 3762 3161 1269 591 | 15 100 13 11 | 17 0 2 1 | 3607 3035 1236 576 | 3592 3035 1238 578 | 3596.1 w 3491.0 w 1243.1 vs | 3504.2 vw 1243.0 w 570.9 w | ν(O-H) (100) Dimer: ν(O-H) out-of-phase δ(O-H) (52) + ν(CN)(25) + γas(CH) pyrrol (16) γ(O-H) (81) |
| ν(C=O) | 1743 1702 | 80 0 | 0 70 | 1687 1648 | 1695 1655 | 1675.1 vs | 1643.2 m | Dimer: ν(C=O) out-of-phase Dimer: ν(C=O) in-phase | |
| ν(C-O) | 1140 1119 | 29 30 | 0 3 | 1114 1094 | 1113 1093 | 1122.5 m 1093.6 m | 1120.6 w 1096.5 w | νas(COO) (33) + νs(NNN)(31) + 15,δ(CH)(28) νs(COO) (45) + δ(NNN) (38) | |
| δ(C=O) | 714 796 777 | 9 15 0 | 0 0 8 | 699 782 763 | 699 780 761 | 697.2 w 779.2 vs | 696.3 w 768.0 m | δ(COOH) (46) + γ(triazole) (38) Dimer: δ(COOH) out-of-phase + ν(CC) Dimer: δ(COOH) in-phase | |
| γ(C=O) | 721 | 9 | 1 | 706 | 705 | 707.8 vvw | 705.9 w | γ(COOH) (62) + γ(NC8C) (21) + γ(CN14) (16) | |
| COO | νas νs δas γs | 1759 1337 795 808 | 83 35 25 1 | 2 4 1 1 | 1702 1301 786 798 | 1710 1304 778 790 | 1588.3 vs 1299.9 m | 1629.7 vw 1307.6 vw 774.4 w 783.1 vw | νas(COO) (96) νs(COO)(34) + ν(triazole)(32) + γ(CH)pyrrol(18) δas(COO) (58) + δas(C-H) in pyrrolidine (15) γs(CCOO) (55) + γ(C8C) (30) + 6a, δ(CC) (27) |
2.6.2. The Carboxylate COO Group Modes in 2a
2.6.3. The Triazole Ring Modes
2.6.4. The Aryl Ring Modes
2.6.5. The Methoxy OCH3 Modes
3. Materials and Methods
Computational Details
4. Summary and Conclusions
- (1)
- By rotation on the C-O1 bond length, the conformers differ less than 1 kJ/mol, while by rotation on the C9-C11 bond it is about 10 kJ/mol. This feature means a large flexibility of the substituents bonded to the triazole ring and variability of its spatial arrangements.
- (2)
- The ionization of neutral carboxylic acid and formation of the anionic form demonstrated a larger effect on the triazole ring structure and its charges than the electronic nature of different substituents on the aryl ring.
- (3)
- The effect of five substituents in the para-position of the aryl ring on the molecular structure of the triazole and on its atomic charge distribution was determined and several relationships were established.
- (4)
- The FT-IR and FT-Raman spectra in the solid state of 1a and 2a were recorded, and an accurate assignment of all bands observed was carried out for the first time. For this task, the calculated wavenumbers were improved by two main scaling procedures, leading the PSE to the best results, with errors less than 3%.
- (5)
- The scaled wavenumbers in the acid 1a dimer and stacking forms of triazole 2a were in better accordance with the experimental bands than those with the monomer, which confirms our simplified optimized system for the crystal unit cell of the solid state.
- (6)
- The large red shift of the ν(C=O) mode to 1675.1 cm−1 indicates that strong H-bonds in the dimer form appear in the solid state through this group. These features mean that in the solid state, free and H-bonded COOH groups exist simultaneously.
- (7)
- A comparison of the scaled and experimental wavenumbers confirms these features for the solid state, with free and H-bonded COOH groups in 1a, and stacking forms in 2a.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mahboob Alam, M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. 2022, 355, 2100158. [Google Scholar] [CrossRef]
- Slavova, K.I.; Todorov, L.T.; Belskaya, N.P.; Alcolea Palafox, M.; Kostova, I.P. Developments in the Application of 1,2,3-Triazoles in Cancer Treatment. Recent Patents Anti-Cancer Drug Discov. 2020, 15, 92–112. [Google Scholar] [CrossRef]
- Safronov, N.E.; Kostova, I.P.; Alcolea Palafox, M.; Belskaya, N.P. Combined NMR Spectroscopy and Quantum-Chemical Calculations in Fluorescent 1,2,3-Triazole-4-Carboxylic Acids Fine Structures Analysis. Int. J. Mol. Sci. 2023, 24, 8947. [Google Scholar] [CrossRef] [PubMed]
- Hrimla, M.; Oubella, A.; Laamari, M.R.; Bahsis, L.; Ghaleb, A.; Auhmani, A.; Morjani, H.; Julve Olcina, M.; Stiriba, S.E.; Itto, M.Y.A. Click Synthesis, Anticancer Activity, and Molecular Docking Investigation of some Functional 1,2,3-triazole Derivatives. Biointerface Res. Appl. Chem. 2021, 12, 7633–7667. [Google Scholar] [CrossRef]
- Poonia, N.; Kumar, A.; Kumar, V.; Yadav, M.; Lal, K. Recent Progress in 1H-1,2,3-triazoles as Potential Antifungal Agents. Curr. Top. Med. Chem. 2021, 21, 2109–2133. [Google Scholar] [CrossRef]
- Liang, T.; Sun, X.; Li, W.; Hou, G.; Gao, F. 1,2,3-Triazole-Containing Compounds as Anti–Lung Cancer Agents: Current Developments, Mechanisms of Action, and Structure–Activity Relationship. Front. Pharmacol. 2021, 12, 661173. [Google Scholar] [CrossRef]
- Dong, G.; Jiang, Y.; Zhang, F.; Zhu, F.; Liu, J.; Xu, Z. Recent updates on 1,2,3-, 1,2,4-, and 1,3,5-triazine hybrids (2017–present): The anticancer activity, structure–activity relationships, and mechanisms of action. Arch. Pharm. 2022, 356, 2200479. [Google Scholar] [CrossRef]
- Wu, X.; Wang, J.; Xia, S.; Cheng, S.; Shi, Y. 1,2,3-Triazole Derivatives with Anti-breast Cancer Potential. Curr. Top. Med. Chem. 2022, 22, 1406–1425. [Google Scholar] [CrossRef]
- Kumar Trivedi, M.; Tallapragada, R.M.R.; Branton, A.; Trivedi, D.; Nayak, G.; Mishra, R.K.; Jana, S. Characterization of Physical, Spectral and Thermal Properties of Biofield Treated 1,2,4-Triazole. J. Mol. Pharm. Org. Process. Res. 2015, 3, 1000128. [Google Scholar] [CrossRef]
- Alcolea Palafox, M.; Gil, M.; Núñez, J.L.; Rastogi, V.K.; Mittal, L.; Sharma, R. Scaling factors for the prediction of vibrational spectra. II. The aniline molecule and several derivatives. Int. J. Quantum Chem. 2005, 103, 394–421. [Google Scholar] [CrossRef]
- Alcolea Palafox, M.; Núñez, J.L.; Gil, M. Theoretical Quantum Chemical Study of Benzoic Acid: Geometrical Parameters and Vibrational Wavenumbers. Int. J. Quantum Chem. 2002, 89, 1–24. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991; Chapter 5. [Google Scholar]
- Alcolea Palafox, M. Effect of the Sulphur Atom on S2 and S4 Positions of the Uracil Ring in different DNA:RNA Hybrid Microhelixes with three Nucleotide Base Pairs. Biopolymers 2019, 110, e23247. [Google Scholar] [CrossRef] [PubMed]
- Alcolea Palafox, M.; Iza, N.; Gil, M. The hydration effect on the uracil frequencies: An experimental and quantum chemical study. J. Mol. Struct. 2002, 585, 69–92. [Google Scholar] [CrossRef]
- Alcolea Palafox, M. Scaling factors for the prediction of vibrational spectra. I. Benzene molecule. Int. J. Quantum Chem. 2000, 77, 661–684. [Google Scholar] [CrossRef]
- Alcolea Palafox, M. DFT computations on vibrational spectra: Scaling procedures to improve the wavenumbers. In Density Functional Theory: Advances in Applications; Ramasami, P., Ed.; Walter de Gruyter, Inc.: Boston, MA, USA, 2019; pp. 147–191, Chapter 10. [Google Scholar]
- Varsányi, G. Assignment for Vibrational Spectra of Seven Hundred Benzene Derivatives; Adam Hilder: London, UK, 1974; Volumes 1 and 2. [Google Scholar]
- Boczar, M.; Szczeponek, K.; Wójcik, M.J.; Paluszkiewicz, C. Theoretical modeling of infrared spectra of benzoic acid and its deuterated derivative. J. Mol. Struct. 2004, 700, 39–48. [Google Scholar] [CrossRef]
- Alcolea Palafox, M.; Gil, M.; Núñez, J.L. Spectroscopy of p-methoxybenzoic acid: An AM1 and ab initio study. Appl. Spectrosc. 1994, 48, 27–36. [Google Scholar] [CrossRef]
- Reva, I.D.; Stepanian, S.G. An infrared study on matrix-isolated benzoic acid. J. Mol. Struct. 1995, 349, 337–340. [Google Scholar] [CrossRef]
- Stepanian, S.G.; Reva, I.D.; Radchenko, E.D.; Sheina, G.G. Infrared spectra of benzoic acid monomers and dimers in argon matrix. Vibrat. Spectrosc. 1996, 11, 123–133. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies, 3rd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- George, S. Infrared and Raman Characteristic Group Wavenumbers, Tables and Charts, 3rd ed.; Wiley: Chichester, UK, 2001. [Google Scholar]
- Gavlik, K.D.; Lesogorova, S.G.; Sukhorukova, E.S.; Subbotina, J.O.; Slepukhin, P.A.; Benassi, E.; Belskaya, N.P. Synthesis of 2-Aryl-1,2,3-triazoles by Oxidative Cyclization of 2-(Arylazo)ethene-1,1-diamines: A One-Pot Approach. Eur. J. Org. Chem. 2016, 15, 2700–2710. [Google Scholar] [CrossRef]
- Safronov, N.E.; Fomin, T.O.; Minin, A.S.; Todorov, L.; Kostova, I.; Benassi, E.; Belskaya, N.P. 5-Amino-2-aryl-1,2,3-triazol-4-carboxylic acids: Synthesis, photophysical properties, and application prospects. Dye. Pigm. 2020, 178, 108343. [Google Scholar] [CrossRef]
- Seminario, J.M.; Politzer, P. (Eds.) Modern Density Functional Theory: A Tool for Chemistry; Elsevier: Amsterdam, The Netherlands, 1995; Volume 2. [Google Scholar]
- Szafran, M.; Katrusiak, A.; Koput, J.; Dega-Szafran, Z. X-ray, MP2 and DFT studies of the structure, vibrational and NMR spectra of homarine. J. Molec. Struct. 2007, 846, 1–12. [Google Scholar] [CrossRef]
- Rasheed, T.; Ahmad, S. Computational studies of vibrational spectra and molecular properties of 6-methyluracil using HF, DFT and MP2 methods. Ind. J. Phys. 2011, 85, 239–260. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J. Mol. Struct. 1988, 169, 41–62. [Google Scholar] [CrossRef]
- Szafran, M.; Ostrowska, K.; Katrusiak, A.; Dega-Szafran, Z. Spectral and structural studies of dimethylphenyl betaine hydrate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 128, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, V.; Devi, L.; Subbalakshmi, R.; Rani, T.; Mohan, S. Synthesis, vibrational, NMR, quantum chemical and structure-activity relation studies of 2-hydroxy-4-methoxyacetophenone. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 130, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, A.G.; Yurenko, Y.P.; Zhurakivsky, R.O.; van Mourik, T.; Hovorun, D.M. Complete conformational space of the potential HIV-1 reverse transcriptase inhibitors d4U and d4C. A quantum chemical study. Phys. Chem. Chem. Phys. 2012, 14, 6787–6795. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Gorg, L.; Zhikol, O.A.; Leszczynski, J. Conformational Analysis of Canonical 2-Deoxyribonucleotides. 2. Purine Nucleotides. J. Biomol. Struct. Dyn. 2004, 22, 227–243. [Google Scholar] [CrossRef]
- Brovarets’, O.O.; Hovorun, D.M. Can tautomerization of the A·T Watson–Crick base pair via double proton transfer provoke point mutations during DNA replication? A comprehensive QM and QTAIM analysis. J. Biomol. Struct. Dyn. 2014, 32, 127–154. [Google Scholar] [CrossRef]
- Koyambo-Konzapa, S.-J.; Kongbonga, G.Y.M.; Premkumar, R.; Ramlina Vamhindi Nsangou, M.; Franklin Benial, A.M. Spectroscopic, quantum chemical, molecular docking and molecular dynamics investigations of hydroxylic indole-3-pyruvic acid: A potent candidate for nonlinear optical applications and Alzheimer’s drug. J. Biomol. Struct. Dyn. 2022, 40, 10651–10664. [Google Scholar] [CrossRef]
- Nisha, S.V.D.; Hubert Joe, I. Quantum chemical computation and spectroscopic investigation on antiviral drug Acyclovir:-In-silico and in-vitro analysis. J. Molec. Struct. 2021, 1233, 130033. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Applications and validations of the Minnesota density functionals. Chem. Phys. Lett. 2011, 502, 1–13. [Google Scholar] [CrossRef]
- van Mourik, T. Comment on ‘To stack or not to stack: Performance of a new density functional for the uracil and thymine dimers’. Chem. Phys. Lett. 2009, 473, 206–208. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Boys, S.F.; Bernardi, F. Calculation of small molecular interactions by differences of separate total energies. Some procedures with reduced errors. Molec. Phys. 1970, 19, 553. [Google Scholar] [CrossRef]


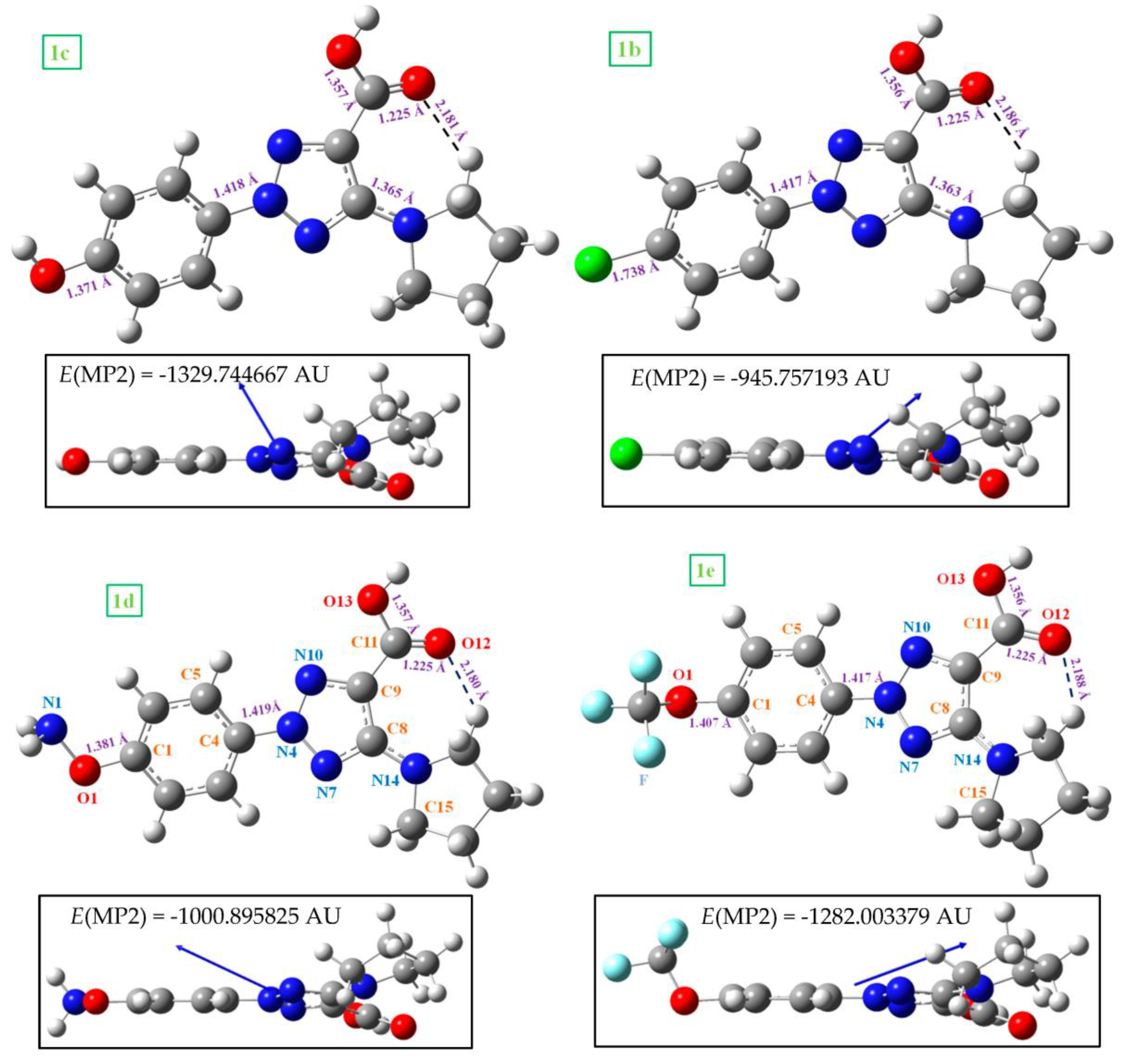



| Parameters | 0 | −ΧΛ | −OH | 0 | 0 | |||
|---|---|---|---|---|---|---|---|---|
| B3LYP | MP2 | MP2 | MP2 | MP2 | MP2 | |||
| 1a | 2a | 1a | 2a | 1b | 1c | 1d | 1e | |
| r(C4−N4) | 1.421 | 1.398 | 1.418 | 1.401 | 1.417 | 1.418 | 1.419 | 1.417 |
| r(N4−N7) | 1.349 | 1.357 | 1.340 | 1.348 | 1.340 | 1.340 | 1.34 | 1.34 |
| r(N4−N10) | 1.311 | 1.343 | 1.334 | 1.350 | 1.334 | 1.334 | 1.334 | 1.334 |
| r(C8−C9) | 1.440 | 1.448 | 1.422 | 1.427 | 1.423 | 1.422 | 1.422 | 1.424 |
| r(C9−N10) | 1.347 | 1.329 | 1.358 | 1.350 | 1.357 | 1.358 | 1.358 | 1.356 |
| r(C9−C11) | 1.465 | 1.549 | 1.466 | 1.541 | 1.467 | 1.466 | 1.466 | 1.468 |
| O12…H18 | 2.152 | 1.980 | 2.181 | 2.030 | 2.186 | 2.181 | 2.18 | 2.188 |
| ∠(C4−N4−N10) | 122.5 | 122.7 | 121.7 | 122.0 | 121.7 | 121.7 | 121.8 | 121.7 |
| ∠N−N−N) | 115.9 | 115.0 | 116.7 | 116.1 | 116.8 | 116.7 | 116.7 | 116.8 |
| ∠(N10−C9−C11) | 119.7 | 121.0 | 119.2 | 120.7 | 119.2 | 119.2 | 119.3 | 119.2 |
| ∠(C9−C8−N14) | 133.2 | 131.0 | 132.6 | 131.0 | 132.7 | 132.6 | 132.6 | 132.7 |
| ∠(C9−C11=O12) | 126.1 | 113.9 | 125.7 | 113.6 | 125.6 | 125.7 | 125.7 | 125.6 |
| ∠(C9−C11−O13) | 112.4 | 115.7 | 111.9 | 115.3 | 111.8 | 111.9 | 111.9 | 111.8 |
| ∠(O=C=O) | 121.4 | 130.3 | 122.4 | 131.0 | 122.6 | 122.4 | 122.4 | 122.6 |
| ∠(C5−C4−N4−N10) | −1.6 | −1.1 | −14.2 | −3.0 | −10.3 | −13.7 | −14.9 | −10.7 |
| ∠(N4−N10−C9−C11) | 175.6 | 175.3 | 173.6 | 176.1 | 173.7 | 173.6 | 173.6 | 173.7 |
| ∠(N10−N4−N7−C8) | −0.4 | −1.1 | −0.9 | −1.4 | −0.8 | −0.9 | −0.9 | −0.8 |
| ∠(N10−C9−C=O12) | −165.9 | −145.7 | −157.1 | −149.3 | −156.3 | −157.1 | −157.3 | −156.1 |
| ∠(N10−C9−C−O13) | 13 | 32.8 | 21.9 | 29.8 | 22.6 | 21.9 | 21.7 | 22.9 |
| ∠(C8−C9−C=O12) | 9.6 | 29.1 | 15.0 | 26.5 | 16.0 | 15.0 | 14.7 | 16.4 |
| ∠(C11−C9−C8−N14) | 4.8 | 4.4 | 8.0 | 2.5 | 8.0 | 8.0 | 8.1 | 8 |
| ∠(C9−C8−N14−C18) | 19.7 | 22.2 | 26.4 | 35.6 | 24.9 | 26.4 | 27 | 24.4 |
| ∠(C8−N14−C15−C16) | −163.0 | −169.6 | −151.8 | −163.7 | −152.0 | −151.8 | −151.7 | −152.0 |
| ∠(N14−C15−C16−C17) | −21.5 | −14.0 | −22.9 | −5.1 | −23.6 | −22.9 | −22.6 | −23.8 |
| −OCH3 | −CL | −OH | −ONH2 | −OCF3 | ||
|---|---|---|---|---|---|---|
| Atom | 1a | 2a | 1b | 1c | 1d | 1e |
| O1 | −0.606 | −0.615 | −0.012 * | −0.756 | −0.472 | −0.651 |
| C1 | 0.378 | 0.332 | −0.052 | 0.385 | 0.368 | 0.288 |
| C4 | 0.117 | 0.167 | 0.160 | 0.115 | 0.117 | 0.163 |
| N4 | 0.004 | −0.064 | −0.005 | 0.004 | 0.005 | −0.007 |
| N7 | −0.380 | −0.398 | −0.381 | −0.380 | −0.380 | −0.381 |
| C8 | 0.458 | 0.409 | 0.462 | 0.458 | 0.457 | 0.462 |
| C9 | −0.099 | 0.019 | −0.091 | −0.099 | −0.101 | −0.089 |
| N10 | −0.188 | −0.240 | −0.183 | −0.188 | −0.185 | −0.182 |
| C11 | 0.979 | 0.956 | 0.979 | 0.979 | 0.979 | 0.978 |
| =O12 | −0.724 | −0.889 | −0.720 | −0.724 | −0.725 | −0.719 |
| O13 | −0.776 | −0.843 | −0.775 | −0.776 | −0.776 | −0.775 |
| N14 | −0.561 | −0.558 | −0.560 | −0.561 | −0.561 | −0.560 |
| C18 | −0.211 | −0.219 | −0.211 | −0.211 | −0.211 | −0.211 |
| H18 | 0.263 | 0.297 | 0.263 | 0.263 | 0.263 | 0.263 |
| Form | Molecular Properties | 1a | 2a | |
|---|---|---|---|---|
| monomer | Rotational constants (GHz): A | 0.616 | 0.647 | |
| B | 0.142 | 0.140 | ||
| C | 0.116 | 0.116 | ||
| Cv (cal/mol·K) | 70.04 | 69.04 | ||
| S (cal/mol·K) | 144.58 | 144.01 | ||
| Dipole moment (Debye) | 1.632 | 12.775 | ||
| dimer | Rotational constants (GHz): A | 0.114 | (0.169) | |
| B | 0.024 | (0.034) | ||
| C | 0.021 | (0.032) | ||
| Cv (cal/mol·K) | 143.7 | (141.8) | ||
| S (cal/mol·K) | 252.9 | (241.5) | ||
| Dipole moment (Debye) | 0.751 | (9.734) | ||
| νcal 1a | TLSE | PSE | A | S | Experimental 1a † | Characterization of 1a * | |
|---|---|---|---|---|---|---|---|
| νscal | νscal | IR | Raman | ||||
| 1670 1643 1605 1561 1515 1513 1476 1422 1407 1389 1298 1282 1269 1215 1207 1154 1075 995 990 825 672 652 640 591 | 1618 1592 1556 1514 1470 1468 1433 1382 1368 1351 1264 1249 1236 1185 1177 1127 1052 980 975 810 657 637 625 576 | 1625 1599 1562 1520 1476 1474 1438 1386 1372 1354 1266 1251 1238 1186 1178 1127 1050 973 968 807 657 638 626 578 | 9 2 100 40 22 17 5 2 12 2 64 3 13 3 5 14 12 5 7 1 4 3 4 11 | 100 6 17 48 6 10 1 57 46 0 0 2 2 0 1 0 0 9 11 0 0 1 1 1 | 1634.6 w 1594.0 vw 1565.1 vs 1514.0 vs 1482.2 vs 1463.9 s 1434.0 m 1381.9 m 1357.8 vw 1274.9 s 1265.2 s 1243.1 vs 1194.8 s 1153.4 vs 973.0 s 824.5vs 664.4 m 638.4 vs 614.3 vvw | 1615.2 vs 1594.0 m 1561.2 m 1511.1 s 1485.1 w 1472.5 w 1420.5 m 1383.8 s 1362.6 vw 1357.8 vw 1262.0 m 1246.9 w 1243.0 w 1201.6 vw 1175.5 sh 1140.8 w 972.1 s 965.3 m 813.0 w 662.5 vw 639.2 m 631.7 m 570.9 w | 8a, ν(C=C) (96) 8b, ν(C=C) (97) ν(C8-N14) (65) + νs(N7CC) (20) 19a, ν(CC) (87) + δs(CH) in pyrrolidine (11) δs(C-H) out-of-phase pyrrolidine (83) δs(C-H) out-of-phase pyrrolidine (75) 19b, ν(CC,CH) in aryl (72) νs(NNN) (35) + νs(C4N) (28) + ν(COO)(25) ν(C4N) (28) + ν(NNN) (22) + δ(COO) (18) ν(C9N) (32) +δ(COO) (25) + δs(pyrrolidine) (23) ν(C-O1) (65) + 14, ν(CC) in aryl (22) γas(C-H) in pyrrolidine (78) ν(NN)(53) + γas(C-H) in pyrrolidine (33) γas(C-H) in pyrrolidine (88) γas(C-H) in pyrrolidine (82) + δ(O13H) (14) νs(COOH) (45) + δas(CH) in pyrrolidine (42) ν(O1-CH3) (83) νas(NNN) (42)+δ(CN14) (24) + γ(CC) pyrrolid (22) νs(NNN) (38) + 12, δ(CC) (34) + δ(CC,CN) (24) 10a, γ(C-H) in aryl (97) γs(triazole) (78) + γ(O13-H) (15) 6b, δ(CC) (42) + δ(triazole) (24) Γ(triazole) (34) + 6b, δ(CC) in aryl (33) γ(O13-H) (81) |
| νcal 2a | TLSE | PSE | A | S | Experimental 2a† | Characterization of 2a * | |
| νscal | νscal | IR | Raman | ||||
| 1674 1634 1591 1563 1514 1505 1435 1400 1391 1276 1213 1203 1085 994 953 722 638 610 | 1622 1583 1543 1516 1469 1461 1394 1361 1352 1243 1183 1174 1061 975 936 716 636 610 | 1629 1590 1549 1522 1475 1466 1399 1365 1356 1245 1184 1174 1060 972 932 706 624 597 | 3 1 58 100 1 15 0 19 8 79 8 8 10 1 32 0 2 1 | 100 2 12 23 2 13 20 7 0 1 0 1 4 2 6 0 0 0 | 1588.3 vs 1533.3 w 1514.0 vs 1474.5 m 1460.0 s 1392.5 w 1367.4 m 1346.2 m 1247.9 vs 1184.2 m 1173.6 m 973.0 m 933.4 vvw 706.9 vw 627.8 w 613.3 vvw | 1610.4 vs 1592.1 m 1535.2 w 1515.9 vs 1476.4 m 1466.7 m 1393.4 vs 1372.2 vs 1356.8 w 1251.7 vw 1175.5 m 1063.7 w 975.0 vs 927.7 vw 707.9 w 627.8 w 607.6 w | 8a, ν(C=C) (95) 8b, ν(C=C) (89) ν(C8-N14) (72) + νs(N7CC) (15) 19a, ν(CC) (76) + δs(CH) in pyrrolidine (18) δs(C-H) out-of-phase in pyrrolidine (92) νs(C8C9N) (62) + δs(CH2) (16) + ν(NN) (15) ν(C4N) (41) + ν(NNN)(25) + 19a, ν(CC) (15) γs(C-H) in-phase in pyrrolidine (87) νs(NNN) (38) + γs(C-H)pyrrolidine(30) + ν(C8C) (25) 7a,ν(COC) (62) + δ(CH) aryl (25) + γas(CH3) (11) δ(triazole) (73) + γ(CC,CN) in pyrrolidine (16) γas(C-H) in pyrrolidine (68) + δ(triazole) (15) νas(COC) (62) + 15, δ(CH)(17) + γs(CH3) (15) νas(NNN) (32) + δ(CN14) (29)+δ(CC) pyrrolidi (25) νs(NNN) (44) + ν(C8C) (20) + 10b, γ(CH) (18) γ(NC8CN10) (53) + γs(COO) (38) Γ(triazole) (38) + δ(CC) in aryl (25) γs(NNN) (55) + γ(COO) (18) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcolea Palafox, M.; Belskaya, N.P.; Kostova, I.P. Peculiarities of the Spatial and Electronic Structure of 2-Aryl-1,2,3-Triazol-5-Carboxylic Acids and Their Salts on the Basis of Spectral Studies and DFT Calculations. Int. J. Mol. Sci. 2023, 24, 14001. https://doi.org/10.3390/ijms241814001
Alcolea Palafox M, Belskaya NP, Kostova IP. Peculiarities of the Spatial and Electronic Structure of 2-Aryl-1,2,3-Triazol-5-Carboxylic Acids and Their Salts on the Basis of Spectral Studies and DFT Calculations. International Journal of Molecular Sciences. 2023; 24(18):14001. https://doi.org/10.3390/ijms241814001
Chicago/Turabian StyleAlcolea Palafox, Mauricio, Nataliya P. Belskaya, and Irena P. Kostova. 2023. "Peculiarities of the Spatial and Electronic Structure of 2-Aryl-1,2,3-Triazol-5-Carboxylic Acids and Their Salts on the Basis of Spectral Studies and DFT Calculations" International Journal of Molecular Sciences 24, no. 18: 14001. https://doi.org/10.3390/ijms241814001
APA StyleAlcolea Palafox, M., Belskaya, N. P., & Kostova, I. P. (2023). Peculiarities of the Spatial and Electronic Structure of 2-Aryl-1,2,3-Triazol-5-Carboxylic Acids and Their Salts on the Basis of Spectral Studies and DFT Calculations. International Journal of Molecular Sciences, 24(18), 14001. https://doi.org/10.3390/ijms241814001