
Foveal Hypoplasia in CRB1-Related Retinopathies

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
2.1. Demographics
2.2. Clinical and FH Classification
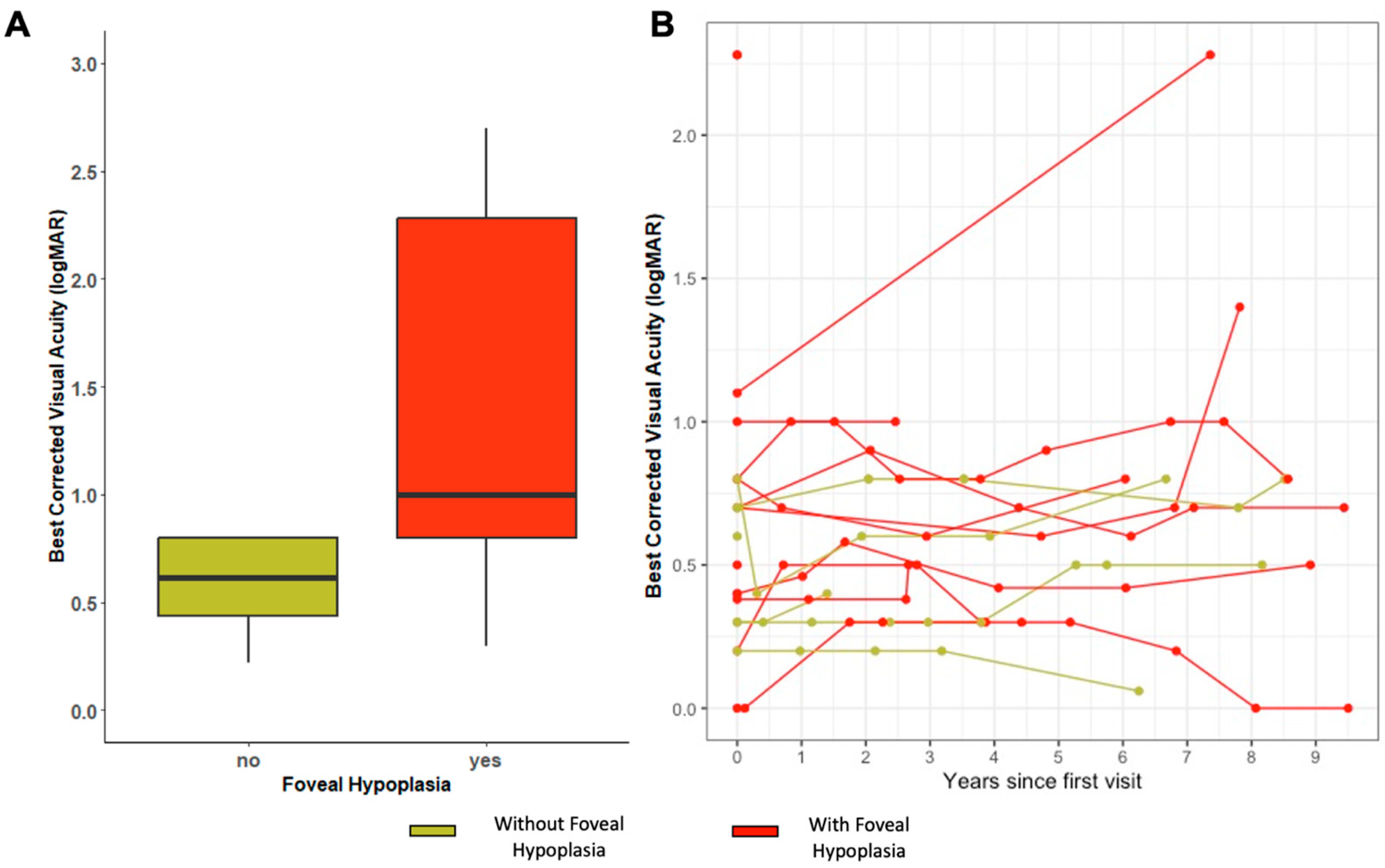
2.3. Visual Acuity and Refraction
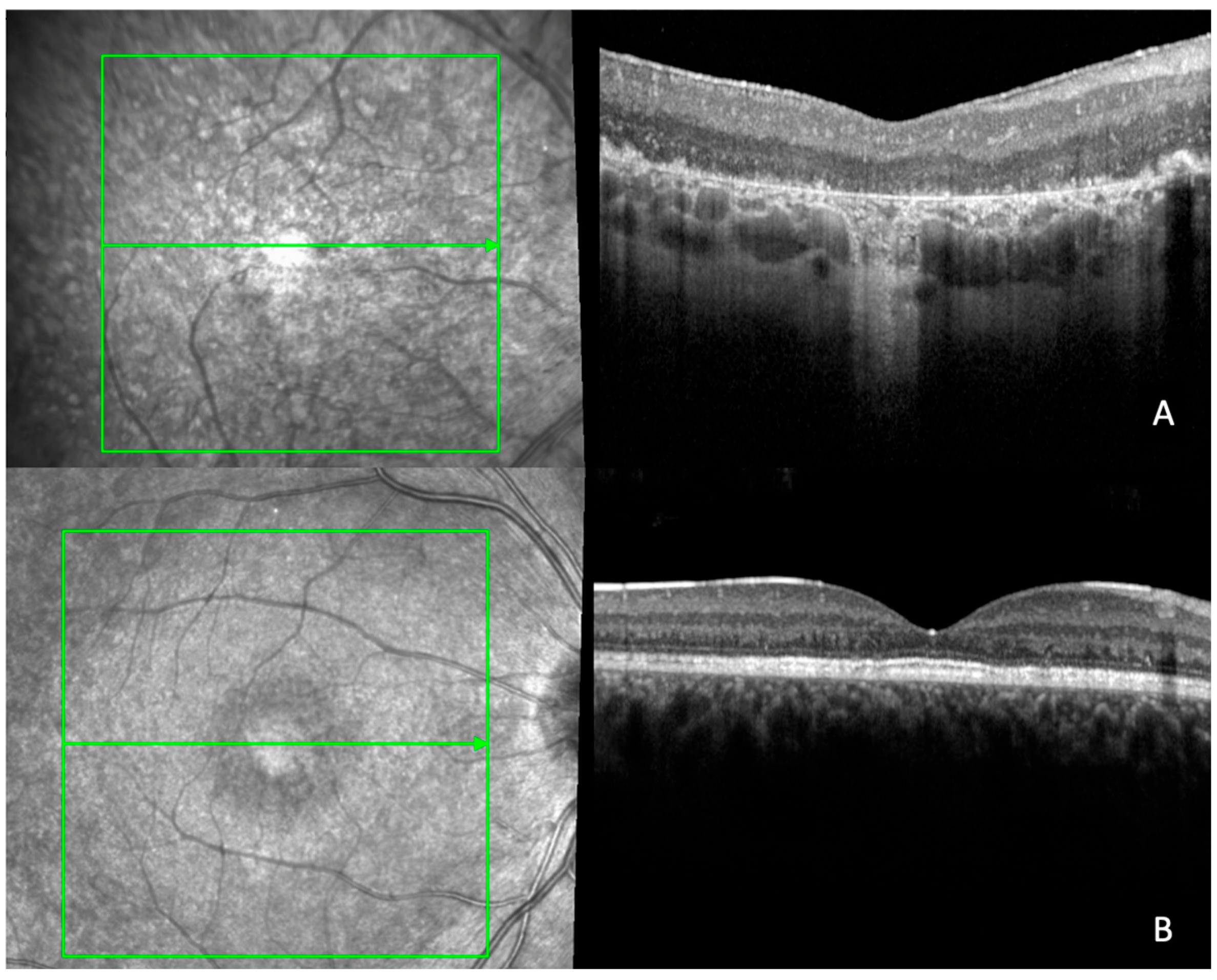
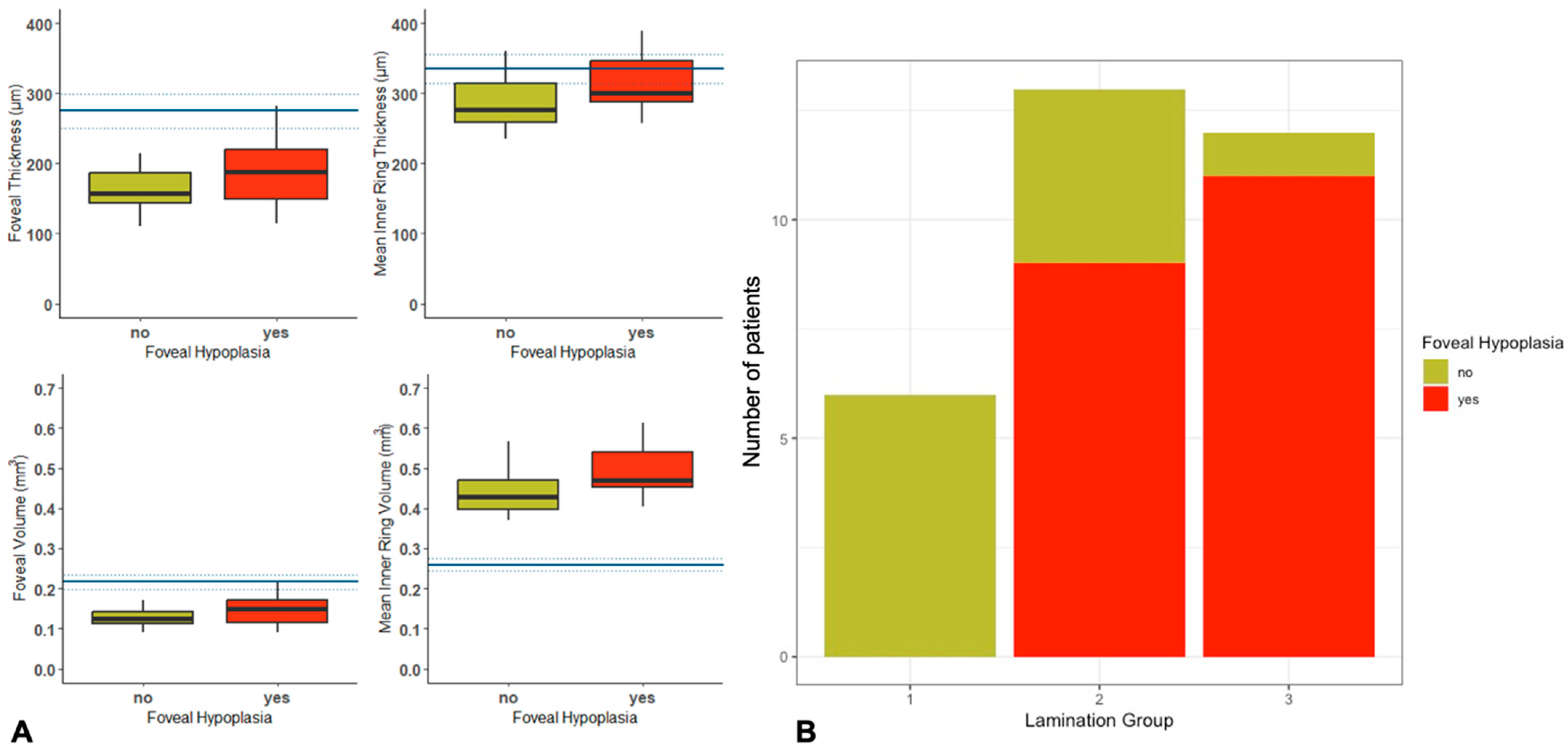
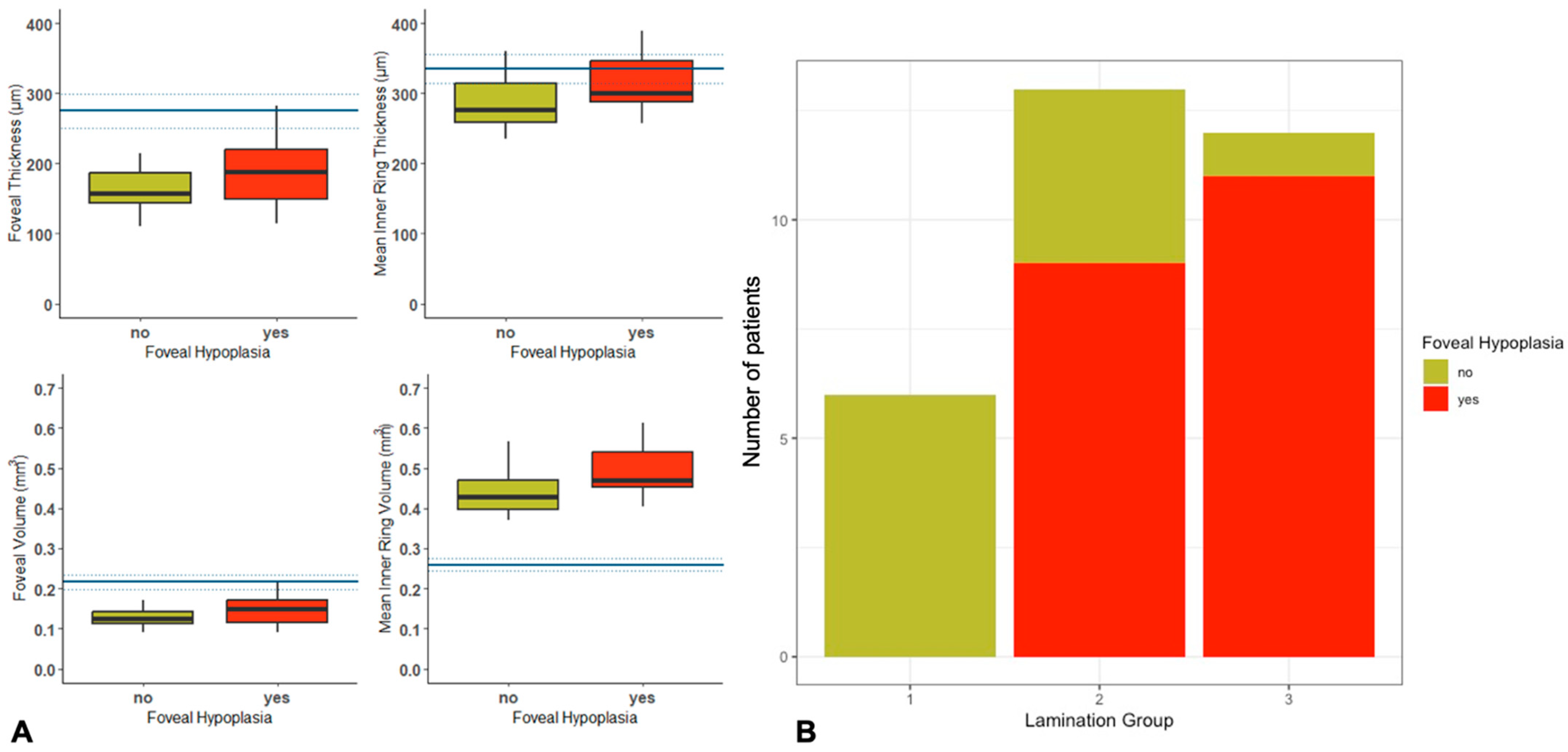
2.4. Quantitative and Qualitative OCT Imaging
2.5. Molecular Characteristics
3. Discussion
Limitations
4. Materials and Methods
4.1. Subjects
4.2. Genetics
4.3. Clinical
4.4. Retinal Imaging
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nguyen, X.-T.; Talib, M.; van Schooneveld, M.J.; Wijnholds, J.; van Genderen, M.M.; Schalij-Delfos, N.E.; Klaver, C.C.; Talsma, H.E.; Fiocco, M.; Florijn, R.J.; et al. CRB1-Associated Retinal Dystrophies: A Prospective Natural History Study in Anticipation of Future Clinical Trials. Am. J. Ophthalmol. 2022, 234, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Talib, M.; Van Cauwenbergh, C.; De Zaeytijd, J.; Van Wynsberghe, D.; De Baere, E.; Boon, C.J.F.; Leroy, B.P. CRB1-associated retinal dystrophies in a Belgian cohort: Genetic characteristics and long-term clinical follow-up. Br. J. Ophthalmol. 2022, 106, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.D.; Georgiou, M.; Alswaiti, Y.; Kabbani, J.; Fujinami, K.; Fujinami-Yokokawa, Y.; Khoda, S.; Mahroo, O.A.; Robson, A.G.; Webster, A.R.; et al. CRB1-Associated Retinal Dystrophies: Genetics, Clinical Characteristics, and Natural History. Am. J. Ophthalmol. 2023, 246, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Pianta, M.J.; Sumaroka, A.; Schwartz, S.B.; Smilko, E.E.; Milam, A.H.; Sheffield, V.C.; Stone, E.M. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 2003, 12, 1073–1078. [Google Scholar] [CrossRef]
- Henderson, R.H.; Mackay, D.S.; Li, Z.; Moradi, P.; Sergouniotis, P.; Russell-Eggitt, I.; Thompson, D.A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br. J. Ophthalmol. 2011, 95, 811–817. [Google Scholar] [CrossRef]
- Owen, N.; Toms, M.; Tian, Y.; Toualbi, L.; Richardson, R.; Young, R.; Tracey-White, D.; Dhami, P.; Beck, S.; Moosajee, M. Loss of the crumbs cell polarity complex disrupts epigenetic transcriptional control and cell cycle progression in the developing retina. J. Pathol. 2023, 259, 441–454. [Google Scholar] [CrossRef]
- Khan, K.N.; Robson, A.; Mahroo, O.A.R.; Arno, G.; Inglehearn, C.F.; Armengol, M.; Waseem, N.; Holder, G.E.; Carss, K.J.; Raymond, L.F.; et al. A clinical and molecular characterisation of CRB1-associated maculopathy. Eur. J. Hum. Genet. 2018, 26, 687–694. [Google Scholar] [CrossRef]
- Rufai, S.R.; Thomas, M.G.; Purohit, R.; Bunce, C.; Lee, H.; Proudlock, F.A.; Gottlob, I. Can Structural Grading of Foveal Hypoplasia Predict Future Vision in Infantile Nystagmus?: A Longitudinal Study. Ophthalmology 2020, 127, 492–500. [Google Scholar] [CrossRef]
- Thomas, M.G.; Papageorgiou, E.; Kuht, H.J.; Gottlob, I. Normal and abnormal foveal development. Br. J. Ophthalmol. 2022, 106, 593–599. [Google Scholar] [CrossRef]
- Dubis, A.M.; Costakos, D.M.; Subramaniam, C.D.; Godara, P.; Wirostko, W.J.; Carroll, J.; Provis, J.M. Evaluation of normal human foveal development using optical coherence tomography and histologic examination. Arch. Ophthalmol. 2012, 130, 1291–1300. [Google Scholar] [CrossRef]
- Schiff, E.R.; Tailor, V.K.; Chan, H.W.; Theodorou, M.; Webster, A.R.; Moosajee, M. Novel Biallelic Variants and Phenotypic Features in Patients with SLC38A8-Related Foveal Hypoplasia. Int. J. Mol. Sci. 2021, 22, 1130. [Google Scholar] [CrossRef]
- Kit, V.; Cunha, D.L.; Hagag, A.M.; Moosajee, M. Longitudinal genotype-phenotype analysis in 86 patients with PAX6-related aniridia. JCI Insight 2021, 6, e148406. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Kumar, A.; Mohammad, S.; Proudlock, F.A.; Engle, E.C.; Andrews, C.; Chan, W.-M.; Thomas, S.; Gottlob, I. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography: A predictor of visual acuity? Ophthalmology 2011, 118, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Poulter, J.A.; Al-Araimi, M.; Conte, I.; van Genderen, M.M.; Sheridan, E.; Carr, I.M.; Parry, D.A.; Shires, M.; Carrella, S.; Bradbury, J.; et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am. J. Hum. Genet. 2013, 93, 1143–1150. [Google Scholar] [CrossRef]
- Chan, H.W.; Schiff, E.R.; Tailor, V.K.; Malka, S.; Neveu, M.M.; Theodorou, M.; Moosajee, M. Prospective Study of the Phenotypic and Mutational Spectrum of Ocular Albinism and Oculocutaneous Albinism. Genes 2021, 12, 508. [Google Scholar] [CrossRef]
- Kuht, H.J.; Maconachie, G.D.; Han, J.; Kessel, L.; van Genderen, M.M.; McLean, R.J.; Hisaund, M.; Tu, Z.; Hertle, R.W.; Gronskov, K.; et al. Genotypic and Phenotypic Spectrum of Foveal Hypoplasia: A Multicenter Study. Ophthalmology 2022, 129, 708–718. [Google Scholar] [CrossRef]
- Mayer, A.K.; Mahajnah, M.; Thomas, M.G.; Cohen, Y.; Habib, A.; Schulze, M.; Maconachie, G.D.; AlMoallem, B.; De Baere, E.; Lorenz, B.; et al. Homozygous stop mutation in AHR causes autosomal recessive foveal hypoplasia and infantile nystagmus. Brain 2019, 142, 1528–1534. [Google Scholar] [CrossRef]
- Khan, K.N.; Islam, F.; Moore, A.T.; Michaelides, M. Clinical and Genetic Features of Choroideremia in Childhood. Ophthalmology 2016, 123, 2158–2165. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Han, G.; Serrano, L.W.; Fuerst, N.M.; Charlson, E.S.; Pearson, D.J.; Chung, D.C.; Traband, A.; Pan, W.; Ying, G.-S.; et al. Natural History of the Central Structural Abnormalities in Choroideremia: A Prospective Cross-Sectional Study. Ophthalmology 2017, 124, 359–373. [Google Scholar] [CrossRef]
- Ehrenberg, M.; Pierce, E.A.; Cox, G.F.; Fulton, A.B. CRB1: One gene, many phenotypes. Semin. Ophthalmol. 2013, 28, 397–405. [Google Scholar] [CrossRef]
- Boon, N.; Wijnholds, J.; Pellissier, L. Research Models and Gene Augmentation Therapy for CRB1 Retinal Dystrophies. Front. Neurosci. 2020, 14, 860. [Google Scholar] [CrossRef]
- Quinn, M.; Pellissier, L.; Wijnholds, J. The CRB1 Complex: Following the Trail of Crumbs to a Feasible Gene Therapy Strategy. Front. Neurosci. 2017, 11, 175. [Google Scholar] [CrossRef]
- Pellissier, L.; Quinn, M.; Alves, C.H.; Vos, R.M.; Klooster, J.; Flannery, J.G.; Heimel, J.A.; Wijnholds, J. Gene therapy into photoreceptors and Müller glial cells restores retinal structure and function in CRB1 retinitis pigmentosa mouse models. Hum. Mol. Genet. 2015, 24, 3104–3118. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.L.; Arno, G.; Corton, M.; Moosajee, M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes 2019, 10, 1050. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Dai, S.; Gao, T.; Hu, J.; Fang, Y.; Zheng, R.; Jin, C.; Zhang, B. Nystagmus-related FRMD7 gene influences the maturation and complexities of neuronal processes in human neurons. Brain Behav. 2019, 9, e01473. [Google Scholar] [CrossRef]
- Bedoukian, E.C.M.; Zhu, X.; Serrano, L.W.B.; Scoles, D.; Aleman, T.S. NMNAT1-Associated Cone–Rod Dystrophy: Evidence for a Spectrum of Foveal Maldevelopment. Retin. Cases Brief Rep. 2022, 16, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, K.A.; Battista, R.; Keep, R.B.; Capasso, J.E.; Levin, A.V. Autosomal-dominant Leber Congenital Amaurosis Caused by a Heterozygous CRX Mutation in a Father and Son. Ophthalmic Genet. 2013, 36, 156–159. [Google Scholar] [CrossRef]
- Kousal, B.; Dudakova, L.; Gaillyova, R.; Hejtmankova, M.; Diblik, P.; Michaelides, M.; Liskova, P. Phenotypic features of CRB1-associated early-onset severe retinal dystrophy and the different molecular approaches to identifying the disease-causing variants. Graefe’s Arch. Clin. Exp. Ophthalmol. 2016, 254, 1833–1839. [Google Scholar] [CrossRef]
- Boon, N.; Lu, X.; Andriessen, C.A.; Moustakas, I.; Buck, T.M.; Freund, C.; Arendzen, C.H.; Böhringer, S.; Boon, C.J.; Mei, H.; et al. AAV-mediated gene augmentation therapy of CRB1 patient-derived retinal organoids restores the histological and transcriptional retinal phenotype. Stem Cell Rep. 2023, 18, 1123–1137. [Google Scholar] [CrossRef]
- Zenteno, J.C.; Buentello-Volante, B.; Ayala-Ramirez, R.; Villanueva-Mendoza, C. Homozygosity mapping identifies the Crumbs homologue 1 (Crb1) gene as responsible for a recessive syndrome of retinitis pigmentosa and nanophthalmos. Am. J. Med. Genet. Part A 2011, 155, 1001–1006. [Google Scholar] [CrossRef]
- Nguyen, X.-T.-A.; Talib, M.; van Schooneveld, M.J.; Wijnholds, J.; van Genderen, M.M.; Florijn, R.J.; ten Brink, J.; Cremers, F.P.; Meester, M.A.; Klaver, C.; et al. A two-year prospective natural history study in patients with CRB1-associated retinal dystrophies: Establishing clinical endpoints for future gene therapy trials. In Acta Ophthalmologica; Wiley: Hoboken, NJ, USA, 2020. [Google Scholar]
- Mohammad, S.; Gottlob, I.; Kumar, A.; Thomas, M.; Degg, C.; Sheth, V.; Proudlock, F.A. The functional significance of foveal abnormalities in albinism measured using spectral-domain optical coherence tomography. Ophthalmology 2011, 118, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Méjécase, C.; Malka, S.; Guan, Z.; Slater, A.; Arno, G.; Moosajee, M. Practical guide to genetic screening for inherited eye diseases. Ther. Adv. Ophthalmol. 2020, 12, 2515841420954592. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Duker, J.S.; Ko, T.H.; Fujimoto, J.G.; Schuman, J.S. Normal macular thickness measurements in healthy eyes using Stratus optical coherence tomography. Arch. Ophthalmol. 2006, 124, 193–198. [Google Scholar] [CrossRef]
- Grover, S.; Murthy, R.K.; Brar, V.S.; Chalam, K.V. Normative data for macular thickness by high-definition spectral-domain optical coherence tomography (spectralis). Am. J. Ophthalmol. 2009, 148, 266–271. [Google Scholar] [CrossRef]
- Murthy, R.K.; Diaz, M.; Chalam, K.V.; Grover, S. Normative data for macular volume with high-definition spectral-domain optical coherence tomography (Spectralis). Eur. J. Ophthalmol. 2015, 25, 546–551. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Family Number | Subject | Gender | Ethnicity | Age | Phenotype | Zygosity | Variant 1 cDNA Variant 1 Protein | Variant 1 cDNA Variant 1 Protein | FH |
|---|---|---|---|---|---|---|---|---|---|
| 3760 | 01 | M | Asian–Pakistani | 37 | RP | Homozygous | c.2536G>A p.Gly846Arg | 1a | |
| 16291 | 02 | M | White | 31 | EOSRD/LCA | Compound Heterozygous | c.2290C>T p.Arg764Cys | c.2401A>T p.Lys801Ter | 1a |
| 20059 | 03 | M | Unknown | 53 | EOSRD/LCA | Homozygous | c.1831T>C p.Ser611Pro | 1b | |
| 24835 | 04 | M | White | 24 | EOSRD/LCA | Compound Heterozygous | c.2129A>T p.Glu710Val | c.3988del p.Glu1330Serfs*11 | 1b |
| 24839 | 05 | M | Unknown | 47 | MD | Compound Heterozygous | c.498_506del p.Ile167_Gly169del | c.584G>Tp.Ile167_Gly169del | 1a |
| 19331 | 06 | F | Unknown | 23 | EOSRD/LCA | Compound Heterozygous | c.2843G>A p.Cys948Tyr | c.1712A>C p.Glu571Ala | 1a |
| 17595 | 07 | M | Unknown | 19 | EOSRD/LCA | Compound Heterozygous | c.2043T>A p.Cys681Ter | c.2843G>A p.Cys948Tyr | 1b |
| 1826 | 08 | M | White | 52 | EOSRD/LCA | Compound Heterozygous | c.2129A>T p.Glu710Val | c.2843G>A p.Cys948Tyr | 1a |
| 2824 | 09 | M | White | 24 | EOSRD/LCA | Compound Heterozygous | c.2129A>T p.Glu710Val | c.2234C>T p.Thr745Met | 1a |
| 2824 | 10 | F | White | 17 | EOSRD/LCA | Compound Heterozygous | c.455G>A p.Cys152Tyr | c.3014A>T p.Asp1005Val | 1a |
| 20745 | 11 | M | Black | 19 | EOSRD/LCA | Compound Heterozygous | c.988+1G>T N/A | c.1183G>T p.Glu395Ter | 1a |
| 30437 | 12 | M | Unknown | 9 | EOSRD/LCA | Compound Heterozygous | c.2843G>A p.Cys948Tyr | c.1712A>C p.Glu571Ala | 1b |
| 24605 | 13 | M | Unknown | 12 | MD | Compound Heterozygous | c.2234C>T p.Thr745Met | c.2506C>A p.Pro836Thr | 1a |
| 26081 | 14 | F | Black | 38 | MD | Compound Heterozygous | c.2506C>A p.Pro836Thr | Del exon 6 | 1a |
| 3642 | 15 | M | Unknown | 36 | EOSRD/LCA | Homozygous | c.750T>G p.Cys250Trp | 1b | |
| 4441 | 16 | M | White | 40 | RP | Homozygous | c.2639A>G p.Asn880Ser | 1a | |
| 11201 | 17 | M | White | 76 | MD | Compound Heterozygous | c.253T>C p.Cys85Arg | c.4009_4015del p.Ala1337Thrfs*2 | 1a |
| 20409 | 18 | M | Unknown | 36 | MD | Compound Heterozygous | c.584G>T | c.2843G>A p.Cys948Tyr | 1a |
| 4441 | 19 | M | White | 61 | CORD | Homozygous | c.2639A>Gp.N880S | 1a | |
| 4441 | 20 | M | White | 28 | MD | Compound Heterozygous | c.498_506del p.Ile167_Gly169del | c.2688T>A p.Cys896Ter | 1a |
| Z413096 | 21 | F | Unknown | 11 | MD | Homozygous | c.2506C>Ap.Pro836Thr | No | |
| Z889804 | 22 | F | Unknown | 13 | CORD | Compound Heterozygous | c.498_506del p.Ile167_Gly169del | c.4005 + 1G>A N/A | No |
| 29543 | 23 | F | Unknown | 42 | MD | Compound Heterozygous | c.2234C>T p.Thr745Met | c.1690G>A p.Asp564Asn | No |
| 16429 | 24 | M | White | 33 | EOSRD/LCA | Homozygous | c.2843G>Ap.Cys948Tyr | No | |
| 28566 | 25 | F | Unknown | 11 | MD | Compound Heterozygous | c.498_506del p.Ile167_Gly169del | c.2843G>A p.Cys948Tyr | No |
| 17311 | 26 | M | Unknown | 40 | MD | Compound Heterozygous | c.498_506del p.Ile167_Gly169del | c.1431delG | No |
| 19403 | 27 | F | White | 39 | EOSRD/LCA | Compound Heterozygous | c.4006-1G>T N/A | c.2308G>A p.Gly770Ser | No |
| 20736 | 28 | F | White | 16 | EOSRD/LCA | Compound Heterozygous | c.2548G>A p.Gly850Ser | c.4006-10A>G | No |
| 4441 | 29 | F | White | 46 | CORD | Homozygous | c.2639A>Gp.Asn880Ser | No | |
| 22771 | 30 | M | White | 38 | MD | Compound Heterozygous | c.498_506del p.Ile167_Gly169del | c.4142C>G p.Pro1381Arg | No |
| 24139 | 31 | M | Other | 17 | MD | Compound Heterozygous | c.498_506del p.Ile167_Gly169del | c.2308G>T p.Gly770Cys | No |
| FH Mean (SD) n = 20 (65%) | No FH n = 11 (35%) | p-Value | |
|---|---|---|---|
| Foveal volume | 0.15 (SD ± 0.04) | 0.13 (SD ± 0.03) | 0.190 |
| Foveal thickness | 193.89 (SD ± 54.21) | 161.13 (SD ± 36.18) | 0.183 |
| Inner ring thickness | 312.26 (SD ± 38.86) | 288.64 (SD ± 43.55) | 0.318 |
| Inner ring volume | 0.49 (SD ± 0.06) | 0.44 (SD ± 0.06) | 0.104 |
| Superior inner volume | 0.51 (SD ± 0.07) | 0.46 (SD ± 0.06) | 0.162 |
| Superior inner thickness | 319.28 (SD ± 40.14) | 292.88 (SD ± 39.62) | 0.193 |
| Nasal inner volume | 0.49 (SD ± 0.07) | 0.45 (SD ± 0.07) | 0.109 |
| Nasal inner thickness | 312.44 (SD ± 44.56) | 294.63 (SD ± 58.04) | 0.395 |
| Inferior inner volume | 0.50 (SD ± 0.07) | 0.45 (SD ± 0.07) | 0.138 |
| Inferior inner thickness | 319.28 (SD ± 40.14) | 286.88 (SD ± 43.78) | 0.118 |
| Temporal inner volume | 0.47 (SD ± 0.05) | 0.41 (SD ± 0.61) | 0.048 |
| Temporal inner thickness | 296.22 (SD ± 32.84) | 265.57 (SD ± 38.34) | 0.096 |
| Change in Foveal Thickness (μm/Year) | Foveal Volume (mm3/Year) | Inner ring Thickness (μm/Year) | Inner Ring Volume (mm3/Year) | |
|---|---|---|---|---|
| All patients | −2.36 | −0.002 | 0.645549 | −0.002 |
| With FH | −3.45 | −0.003 | −0.483 | −0.0003 |
| Without FH | −1.26 | −0.001 | −0.808 | −0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Martinez, A.C.; Higgins, B.E.; Tailor-Hamblin, V.; Malka, S.; Cheloni, R.; Collins, A.M.; Bladen, J.; Henderson, R.; Moosajee, M. Foveal Hypoplasia in CRB1-Related Retinopathies. Int. J. Mol. Sci. 2023, 24, 13932. https://doi.org/10.3390/ijms241813932
Rodriguez-Martinez AC, Higgins BE, Tailor-Hamblin V, Malka S, Cheloni R, Collins AM, Bladen J, Henderson R, Moosajee M. Foveal Hypoplasia in CRB1-Related Retinopathies. International Journal of Molecular Sciences. 2023; 24(18):13932. https://doi.org/10.3390/ijms241813932
Chicago/Turabian StyleRodriguez-Martinez, Ana Catalina, Bethany Elora Higgins, Vijay Tailor-Hamblin, Samantha Malka, Riccardo Cheloni, Alexander Mark Collins, John Bladen, Robert Henderson, and Mariya Moosajee. 2023. "Foveal Hypoplasia in CRB1-Related Retinopathies" International Journal of Molecular Sciences 24, no. 18: 13932. https://doi.org/10.3390/ijms241813932
APA StyleRodriguez-Martinez, A. C., Higgins, B. E., Tailor-Hamblin, V., Malka, S., Cheloni, R., Collins, A. M., Bladen, J., Henderson, R., & Moosajee, M. (2023). Foveal Hypoplasia in CRB1-Related Retinopathies. International Journal of Molecular Sciences, 24(18), 13932. https://doi.org/10.3390/ijms241813932