
A Transcriptomic Analysis of Smoking-Induced Gene Expression Alterations in Coronary Artery Disease Patients

 ,
, 

Abstract
1. Background
2. Results
3. Discussion
3.1. Link of Upregulated Genes to CAD and/or Smoking
3.2. Pathways of the Downregulated Genes
3.3. Limitations
4. Materials and Methods
4.1. Study Design and Population
4.2. Blood Sample Collection
4.3. RNA Isolation, Library Preparation, and Sequencing
4.4. Cotinine Assay
4.5. Data Collected
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAD | Coronary artery disease |
| CHD | Coronary heart disease |
| CX | Circumflex artery |
| DEGs | Differentially expressed genes |
| FC | Fold change |
| FDR | False discovery rate |
| GO | Gene Ontology |
| Igs | Immunoglobulins |
| LAD | Left anterior descending artery |
| LM | Left main coronary artery |
| ND | Nicotine dependence |
| NGS | Next-generation sequencing |
| OS | Oxidative stress |
| RCA | Right coronary artery |
| RIN | RNA integrity number |
| SD | Standard deviation |
References
- CDC. Coronary Artery Disease (CAD). Coronary Artery Disease, This Process Is Called Atherosclerosis. Available online: https://www.cdc.gov/heartdisease/coronary_ad.htm#:~:text=Print- (accessed on 23 May 2023).
- Libby, P.; Theroux, P. Pathophysiology of Coronary Artery Disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef]
- Malakar, A.K.; Choudhury, D.; Halder, B.; Paul, P.; Uddin, A.; Chakraborty, S. A Review on Coronary Artery Disease, Its Risk Factors, and Therapeutics. J. Cell. Physiol. 2019, 234, 16812–16823. [Google Scholar] [CrossRef] [PubMed]
- Kamceva, G.; Arsova-Sarafinovska, Z.; Ruskovska, T.; Zdravkovska, M.; Kamceva-Panova, L.; Stikova, E. Cigarette Smoking and Oxidative Stress in Patients with Coronary Artery Disease. Open Access Maced. J. Med. Sci. 2016, 4, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Caliri, A.W.; Tommasi, S.; Besaratinia, A. Relationships among Smoking, Oxidative Stress, Inflammation, Macromolecular Damage, and Cancer. Mutat. Res.—Rev. Mutat. Res. 2021, 787, 108365. [Google Scholar] [CrossRef] [PubMed]
- Amit, V.K.; Sekar, K. Genetics of Coronary Artery Disease: Discovery, Biology and Clinical Translation. Nat. Rev. Genet. 2017, 18, 331–344. [Google Scholar]
- Ding, N.; Sang, Y.; Chen, J.; Ballew, S.H.; Kalbaugh, C.A.; Salameh, M.J.; Blaha, M.J.; Allison, M.; Heiss, G.; Selvin, E.; et al. Cigarette Smoking, Smoking Cessation, and Long-Term Risk of 3 Major Atherosclerotic Diseases. J. Am. Coll. Cardiol. 2019, 74, 498–507. [Google Scholar] [CrossRef]
- McEvoy, J.W.; Blaha, M.J.; DeFilippis, A.P.; Lima, J.A.; Bluemke, D.A.; Hundley, W.G.; Min, J.K.; Shaw, L.J.; Lloyd-Jones, D.M.; Barr, R.G.; et al. Cigarette Smoking and Cardiovascular Events: Role of Inflammation and Subclinical Atherosclerosis: The Multi-Ethnic Study of Atherosclerosis (MESA). Arter. Thromb. Vasc. Biol. 2015, 35, 700–709. [Google Scholar] [CrossRef]
- OECD. European Observatory on Health Systems and Policies. State of Health in the EU—Hungary: Country Health Profile; OECD: Paris, France, 2021; Volume 2021, p. 4. [Google Scholar]
- Nasr, N.; Soltész, B.; Sándor, J.; Ádány, R.; Fiatal, S. Comparison of Genetic Susceptibility to Coronary Heart Disease in the Hungarian Populations: Risk Prediction Models for Coronary Heart Disease. Genes 2023, 14, 1033. [Google Scholar] [CrossRef]
- Porsch, F.; Mallat, Z.; Binder, C.J. Humoral Immunity in Atherosclerosis and Myocardial Infarction: From B Cells to Antibodies. Cardiovasc. Res. 2021, 117, 2544–2562. [Google Scholar] [CrossRef]
- Hafiz, M.; Abdullah, N.; Othman, Z.; Mohd, H.; Suri, S.; Khairuddin, A.; Yusof, M.; Jamal, R.; Rashid, A.; Rahman, A. Peripheral Blood Gene Expression Profile of Atherosclerotic Coronary Artery Disease in Patients of Different Ethnicity in Malaysia. J. Cardiol. 2012, 60, 192–203. [Google Scholar] [CrossRef][Green Version]
- Li, J.; Liu, S.; Cao, G.; Sun, Y.; Chen, W.; Dong, F.; Xu, J.; Zhang, C.; Zhang, W. Nicotine Induces Endothelial Dysfunction and Promotes Atherosclerosis via GTPCH1. J. Cell. Mol. Med. 2018, 22, 5406–5417. [Google Scholar] [CrossRef]
- Banks, E.; Joshy, G.; Korda, R.J.; Stavreski, B.; Soga, K.; Egger, S.; Day, C.; Clarke, N.E.; Lewington, S.; Lopez, A.D. Tobacco Smoking and Risk of 36 Cardiovascular Disease Subtypes: Fatal and Non-Fatal Outcomes in a Large Prospective Australian Study. BMC Med. 2019, 17, 128. [Google Scholar] [CrossRef]
- Khoramdad, M.; Leila, A.V. Association between Passive Smoking and Cardiovascular Disease: A Systematic Review and Meta-Analysis. Int. Union Biochem. Mol. Biol. 2019, 72, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Salehi, N.; Janjani, P.; Tadbiri, H.; Rozbahani, M.; Jalilian, M. Effect of Cigarette Smoking on Coronary Arteries and Pattern and Severity of Coronary Artery Disease: A Review. J. Int. Med. Res. 2021, 49, 03000605211059893. [Google Scholar] [CrossRef]
- Tolstrup, J.S.; Hvidtfeldt, U.A.; Flachs, E.M.; Spiegelman, D.; Heitmann, B.L.; Bälter, K.; Goldbourt, U.; Hallmans, G.; Knekt, P.; Liu, S.; et al. Smoking and Risk of Coronary Heart Disease in Younger, Middle-Aged, and Older Adults. Am. J. Public Health 2014, 104, 96–102. [Google Scholar] [CrossRef]
- Tian, X.; Chen, S.; Zuo, Y.; Zhang, Y.; Zhang, X.; Xu, Q.; Luo, Y.; Wu, S.; Wang, A. Association of Lipid, Inflammatory, and Metabolic Biomarkers with Age at Onset for Incident Cardiovascular Disease. BMC Med. 2022, 20, 383. [Google Scholar] [CrossRef] [PubMed]
- Dugani, S.B.; Moorthy, M.V.; Li, C.; Demler, O.V.; Alsheikh-Ali, A.A.; Ridker, P.M.; Glynn, R.J.; Mora, S. Association of Lipid, Inflammatory, and Metabolic Biomarkers with Age at Onset for Incident Coronary Heart Disease in Women. JAMA Cardiol. 2021, 6, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Jamee, A.; Abed, Y.; Jalambo, M.O. Gender Difference and Characteristics Attributed to Coronary Artery Disease in Gaza-Palestine. Glob. J. Health Sci. 2013, 5, 51–56. [Google Scholar] [CrossRef]
- Sayed, A.I. Gender Differences in Coronary Artery Disease, Clinical Characteristics, and Angiographic Features in the Jazan Region, Saudi Arabia. Cureus 2022, 14, e30239. [Google Scholar] [CrossRef]
- Schroeder, H.W.J.; Cavacini, L. Structure and Function of Immunoglobulins. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S41–S52. [Google Scholar] [CrossRef]
- Tarbiah, N.; Todd, I.; Tighe, P.J.; Fairclough, L.C. Cigarette Smoking Differentially Affects Immunoglobulin Class Levels in Serum and Saliva: An Investigation and Review. Basic Clin. Pharmacol. Toxicol. 2019, 125, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Núñez, E.; Fuster, V.; Gómez-Serrano, M.; Valdivielso, J.M.; Fernández-Alvira, J.M.; Martínez-López, D.; Rodríguez, J.M.; Bonzon-Kulichenko, E.; Calvo, E.; Alfayate, A.; et al. Unbiased Plasma Proteomics Discovery of Biomarkers for Improved Detection of Subclinical Atherosclerosis. eBioMedicine 2022, 76, 103874. [Google Scholar] [CrossRef] [PubMed]
- Piaggeschi, G.; Rolla, S.; Rossi, N.; Brusa, D.; Naccarati, A.; Couvreur, S.; Spector, T.D.; Roederer, M.; Mangino, M.; Cordero, F.; et al. Immune Trait Shifts in Association with Tobacco Smoking: A Study in Healthy Women. Front. Immunol. 2021, 12, 637974. [Google Scholar] [CrossRef] [PubMed]
- Ewald, D.A.; Malajian, D.; Krueger, J.G.; Workman, C.T.; Wang, T.; Tian, S.; Litman, T.; Guttman-yassky, E.; Suárez-fariñas, M. Meta-Analysis Derived Atopic Dermatitis (MADAD) Transcriptome Defines a Robust AD Signature Highlighting the Involvement of Atherosclerosis and Lipid Metabolism Pathways. BMC Med. Genom. 2015, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-F.; Huang, Y.; Lu, S.-F.; Hong, H.; Xu, S.-J.; Xie, J.-S.; Wu, Z.-Y.; Tang, Y.; Xu, H.-X.; Fu, S.-P.; et al. Comparative Study of Gene Expression Profiles Rooted in Acute Myocardial Infarction and Ischemic/Reperfusion Rat Models. Am. J. Cardiovasc. Dis. 2020, 10, 84–100. [Google Scholar] [PubMed]
- Vink, J.M.; Jansen, R.; Brooks, A.; Willemsen, G.; van Grootheest, G.; de Geus, E.; Smit, J.H.; Penninx, B.W.; Boomsma, D.I. Differential Gene Expression Patterns between Smokers and Non-Smokers: Cause or Consequence? Addict. Biol. 2017, 22, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Haase, T.; Müller, C.; Stoffers, B.; Kirn, P.; Waldenberger, M.; Kaiser, F.J.; Karakas, M.; Kim, S.V.; Voss, S.; Wild, P.S.; et al. G Protein-Coupled Receptor 15 Expression Is Associated with Myocardial Infarction. Int. J. Mol. Sci. 2023, 24, 180. [Google Scholar] [CrossRef]
- Andersen, A.M.; Lei, M.K.; Beach, S.R.H.; Philibert, R.A.; Sinha, S.; Colgan, J.D. Cigarette and Cannabis Smoking Effects on GPR15+ Helper T Cell Levels in Peripheral Blood: Relationships with Epigenetic Biomarkers. Genes 2020, 11, 149. [Google Scholar] [CrossRef]
- Grievink, H.W.; Smit, V.; Huisman, B.W.; Gal, P.; Yavuz, Y.; Klerks, C.; Binder, C.J.; Bot, I.; Kuiper, J.; Foks, A.C.; et al. Cardiovascular Risk Factors: The Effects of Ageing and Smoking on the Immune System, an Observational Clinical Study. Front. Immunol. 2022, 13, 968815. [Google Scholar] [CrossRef]
- Wang, C.; Song, C.; Liu, Q.; Zhang, R.; Fu, R.; Wang, H.; Yin, D.; Song, W.; Zhang, H.; Dou, K. Gene Expression Analysis Suggests Immunological Changes of Peripheral Blood Monocytes in the Progression of Patients with Coronary Artery Disease. Front. Genet. 2021, 12, 641117. [Google Scholar] [CrossRef]
- Ma, C.; Lu, T.; He, Y.; Guo, D.; Duan, L.; Jia, R.; Cai, D.; Gao, T.; Chen, Z.; Xue, B.; et al. Comprehensive Analysis of Autophagy-Related Gene Expression Profiles Identified Five Gene Biomarkers Associated with Immune Infiltration and Advanced Plaques in Carotid Atherosclerosis. Orphanet J. Rare Dis. 2023, 18, 66. [Google Scholar] [CrossRef]
- Xia, Y.; Brewer, A.; Bell, J.T. DNA Methylation Signatures of Incident Coronary Heart Disease: Findings from Epigenome-Wide Association Studies. Clin. Epigenetics 2021, 13, 186. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Palmer, R. The Influence of Tobacco Smoking on Adhesion Molecule Profiles. Tob. Induc. Dis. 2002, 1, 7–25. [Google Scholar] [CrossRef]
- Agha, G.; Mendelson, M.M.; Ward-Caviness, C.K.; Joehanes, R.; Huan, T.X.; Gondalia, R.; Salfati, E.; Brody, J.A.; Fiorito, G.; Bressler, J.; et al. Blood Leukocyte DNA Methylation Predicts Risk of Future Myocardial Infarction and Coronary Heart Disease. Circulation 2019, 140, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Petsophonsakul, P.; Burgmaier, M.; Willems, B.; Heeneman, S.; Stadler, N.; Gremse, F.; Reith, S.; Burgmaier, K.; Kahles, F.; Marx, N.; et al. Nicotine Promotes Vascular Calcification via Intracellular Ca2+-Mediated, Nox5-Induced Oxidative Stress, and Extracellular Vesicle Release in Vascular Smooth Muscle Cells. Cardiovasc. Res. 2022, 118, 2196–2210. [Google Scholar] [CrossRef]
- Li, Y.; Fu, W.; Geng, Z.; Song, Y.; Yang, X.; He, T.; Wu, J.; Wang, B. A Pan-Cancer Analysis of the Oncogenic Role of Ribonucleotide Reductase Subunit M2 in Human Tumors. PeerJ 2022, 10, e14432. [Google Scholar] [CrossRef] [PubMed]
- Slizhikova, D.K.; Zinovyeva, M.V.; Kuzmin, D.V.; Snezhkov, E.V.; Shakhparonov, M.I.; Dmitriev, R.I.; Antipova, N.V.; Zavalova, L.L.; Sverdlov, E.D. Decreased Expression of the Human Immunoglobulin J-Chain Gene in Squamous Cell Cancer and Adenocarcinoma of the Lungs. Mol. Biol. 2007, 41, 594–600. [Google Scholar] [CrossRef]
- Marengo-Rowe, A.J. Structure-Function Relations of Human Hemoglobins. Proc. Baylor Univ. Med. Cent. 2006, 19, 239–245. [Google Scholar] [CrossRef]
- Thriveni, R.; Manshi, P.; Ramesh, D.V.; Rachel, B.; Byatnal, A.; Kempwade, P. Effects of Smoking on Hemoglobin and Erythrocytes Sedimentation Rate and Its Association with ABO Blood Groups. J. Indian Acad. Oral Med. Radiol. 2020, 32, 103–106. [Google Scholar] [CrossRef]
- Rietbrock, N.; Kunkel, S.; Worner, W.; Eyer, P. Oxygen-Dissociation Kinetics in the Blood of Smokers and Non-Smokers: Interaction between Oxygen and Carbon Monoxide at the Hemoglobin Molecule. Naunyn-Schmiedebergs Arch. Pharmacol. 1992, 345, 123–128. [Google Scholar] [CrossRef]
- Remigante, A.; Spinelli, S.; Pusch, M.; Sarikas, A.; Morabito, R.; Marino, A.; Dossena, S. Role of SLC4 and SLC26 Solute Carriers during Oxidative Stress. Acta Physiol. 2022, 235, e13796. [Google Scholar] [CrossRef]
- Revin, V.V.; Gromova, N.V.; Revina, E.S.; Samonova, A.Y.; Tychkov, A.Y.; Bochkareva, S.S.; Moskovkin, A.A.; Kuzmenko, T.P. The Influence of Oxidative Stress and Natural Antioxidants on Morphometric Parameters of Red Blood Cells, the Hemoglobin Oxygen Binding Capacity, and the Activity of Antioxidant Enzymes. Biomed. Res. Int. 2019, 2019, 2109269. [Google Scholar] [CrossRef]
- Cox, T.C.; Sadlon, T.J.; Schwarz, Q.P.; Matthews, C.S.; Wise, P.D.; Cox, L.L.; Bottomley, S.S.; May, B.K. The Major Splice Variant of Human 5-Aminolevulinate Synthase-2 Contributes Significantly to Erythroid Heme Biosynthesis. Int. J. Biochem. Cell Biol. 2004, 36, 281–295. [Google Scholar] [CrossRef]
- Guauque-Olarte, S.; Gaudreault, N.; Piché, M.È.; Fournier, D.; Mauriège, P.; Mathieu, P.; Bossé, Y. The Transcriptome of Human Epicardial, Mediastinal and Subcutaneous Adipose Tissues in Men with Coronary Artery Disease. PLoS ONE 2011, 6, e19908. [Google Scholar] [CrossRef]
- Beccacece, L.; Abondio, P.; Bini, C.; Pelotti, S. The Link between Prostanoids and Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 4193. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, R.A.; Zeller, T.; Rotival, M.; Wild, P.S.; Munzel, T.; Lackner, K.J.; Weidmann, H.; Ninio, E.; Tregouet, D.-A.; Cambien, F.; et al. Graphical Modeling of Gene Expression in Monocytes Suggests Molecular Mechanisms Explaining Increased Atherosclerosis in Smokers. PLoS ONE 2013, 8, e50888. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Jacob, P. Metabolism of Nicotine to Cotinine Studied by a Dual Stable Isotope Method. Clin. Pharmacol. Ther. 1994, 56, 483–493. [Google Scholar] [CrossRef]
- Heatherton, T.F.; Kozlowski, L.T.; Frecker, R.C.; Fagerström, K.O. The Fagerström Test for Nicotine Dependence: A Revision of the Fagerström Tolerance Questionnaire. Br. J. Addict. 1991, 86, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Sample Size for Microarray Experiments. Available online: https://bioinformatics.mdanderson.org/MicroarraySampleSize/ (accessed on 15 September 2020).
- Bioanalyzer. Agilent 2100 Bioanalyzer System: 2100 Expert Software User’s Guide; Agile Technologies Inc.: Waldbronn, Germany, 2020; Volume 2000–2020, pp. 1–252. [Google Scholar]
- Hukkanen, J.; Jacob, P.; Benowitz, N.L. Metabolism and Disposition Kinetics of Nicotine. Pharmacol. Rev. 2005, 57, 79–115. [Google Scholar] [CrossRef]
- Garcia-Garcia, H.M.; Eugène, P.; McFadden, A.F.; Mehran, R.; Stone, G.W.; Spertus, J.; Onuma, Y.; Morel, M.; van Es, G.-A.; Zuckerman, B.; et al. Standardized End Point Definitions for Coronary Intervention Trials. Circulation 2018, 137, 2635–2650. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]



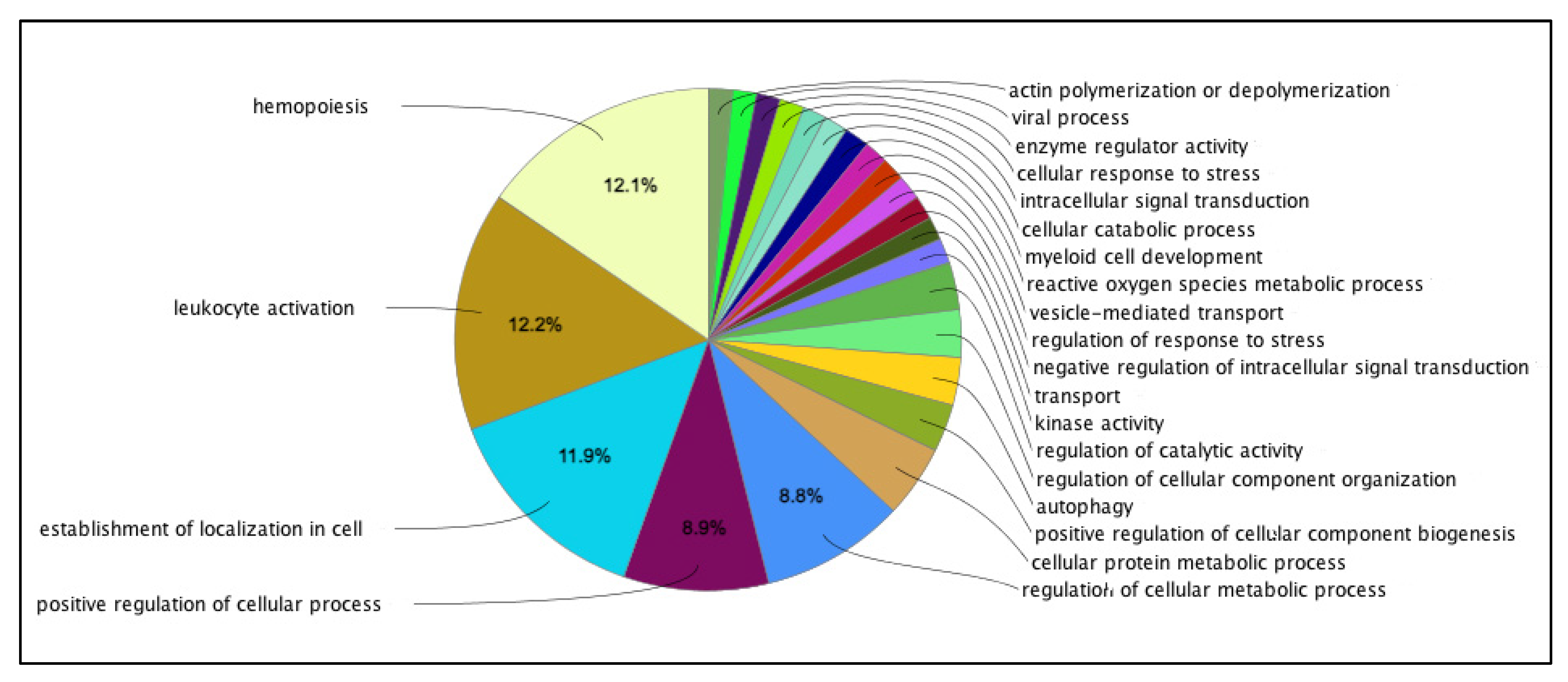{kind=link}
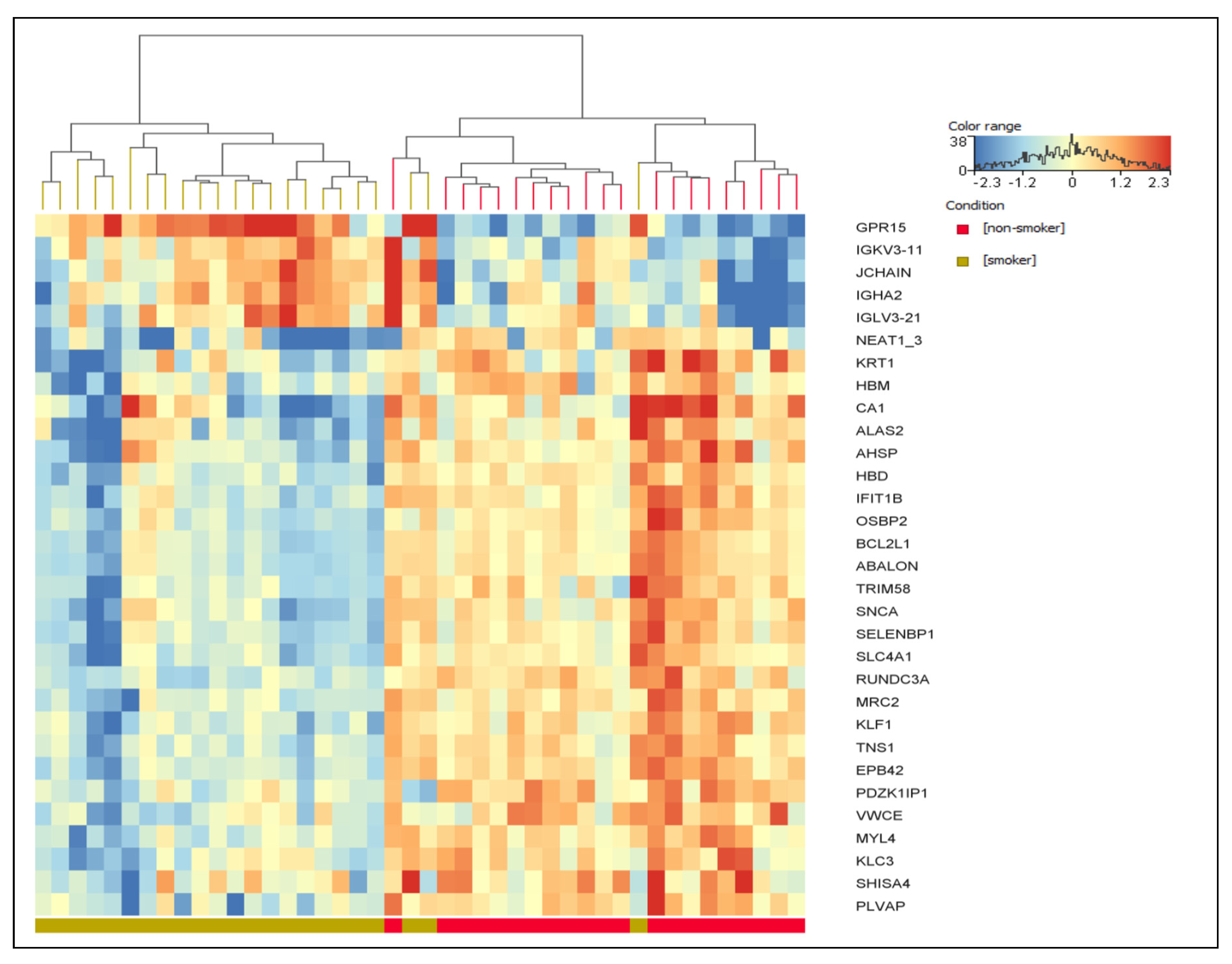{kind=link}
| Variables | Total | Smoking Status | p-Value | |
|---|---|---|---|---|
| Smokers | Nonsmokers | |||
| Gender, n (%) | ||||
| Male | 34 (57.4) | 23 (74.2) | 12 (40.0) | 0.007 |
| Female | 26 (42.6) | 8 (25.8) | 18 (60.0) | |
| Age, median (range) years | 67 (28–88) | 60 (28–75) | 75 (54–88) | <0.001 |
| Cholesterol (mmol/L), mean (SD) | 5.0 (1.4) | 5.2 (1.5) | 4.8 (1.4) | NS |
| Triglyceride (mmol/L), mean (SD) | 1.6 (2.0) | 1.9 (2.7) | 1.4 (0.7) | NS |
| HDL-C (mmol/L), mean (SD) | 1.3 (0.6) | 1.4 (0.7) | 1.3 (0.3) | NS |
| LDL-C (mmol/L), mean (SD) | 3.0 (1.2) | 3.1 (1.3) | 2.9 (1.2) | NS |
| Family history of CAD, n (%) | 20 (32.8) | 14 (45.2) | 6 (20.0) | 0.04 |
| Anticoagulation medication, n (%) | 61 (100) | 31 (100) | 30 (100) | NS |
| BMI, mean (SD) | 26.0 (3.1) | 25.3 (3.4) | 26.8 (2.5) | NS |
| Blood pressure (mmHg), mean (SD) | ||||
| Systolic | 125.9 (22.4) | 120.8 (21.9) | 131.3 (22.0) | NS |
| Diastolic | 73.3 (12.0) | 71.9 (13.5) | 74.8 (10.4) | NS |
| Plasma cotinine ng/mL, mean (SD) | 0.3 (0.1) | 0.4 (0.2) | 0.2 (0.1) | NS |
| Angiography findings, n (%) | ||||
| >2 arteries stenosis | 45 (73.8) | 20 (64.5) | 25 (83.3) | NS |
| Number of affected arteries, median (range) | 2 (1–3) | 2 (1–3) | 2 (1–3) | NS |
| LAD | 50 (82.0) | 24 (77.4) | 26 (86.6) | NS |
| LM | 10 (16.4) | 6 (19.4) | 4 (13.3) | NS |
| CX | 34 (55.7) | 13 (42.0) | 21 (70.0) | 0.03 |
| RCA | 35 (57.4) | 17 (54.8) | 18 (60.0) | NS |
| No. | Gene Symbol | FC | p-Value * | Description |
|---|---|---|---|---|
| Upregulated genes | ||||
| 1 | GPR15 | 7.5 | 1.4 × 10−9 | G protein-coupled receptor 15 [Source:HGNC Symbol;Acc:HGNC:4469] |
| 2 | IGHA2 | 2.6 | 0.034 | immunoglobulin heavy constant alpha 2 (A2m marker) [Source:HGNC Symbol;Acc:HGNC:5479] |
| 3 | IGLV3-21 | 2.4 | 0.030 | immunoglobulin lambda variable 3–21 [Source:HGNC Symbol;Acc:HGNC:5905] |
| 4 | IGKV3-11 | 2.2 | 0.006 | immunoglobulin kappa variable 3–11 [Source:HGNC Symbol;Acc:HGNC:5815] |
| 5 | JCHAIN | 2.1 | 0.037 | joining chain of multimeric IgA and IgM [Source:HGNC Symbol;Acc:HGNC:5713] |
| 6 | IGLC1 | 2.0 | 0.044 | immunoglobulin lambda constant 1 [Source:HGNC Symbol;Acc:HGNC:5855] |
| 7 | IGLL5 | 1.9 | 0.046 | immunoglobulin lambda like polypeptide 5 [Source:HGNC Symbol;Acc:HGNC:38476] |
| 8 | LEF1 | 1.7 | 0.001 | lymphoid enhancer binding Factor 1 [Source:HGNC Symbol;Acc:HGNC:6551] |
| 9 | CCR7 | 1.6 | 0.030 | C-C motif chemokine receptor 7 [Source:HGNC Symbol;Acc:HGNC:1608] |
| 10 | CD28 | 1.6 | 0.002 | CD28 molecule [Source:HGNC Symbol;Acc:HGNC:1653] |
| Downregulated genes | ||||
| 1 | KRT1 | −2.9 | 0.006 | keratin 1 [Source:HGNC Symbol;Acc:HGNC:6412] |
| 2 | NEAT1_3 | −2.7 | 0.005 | Nuclear enriched abundant transcript 1 conserved region 3 [Source:RFAM;Acc:RF01957] |
| 3 | PDZK1IP1 | −2.6 | 0.044 | PDZK1 interacting protein 1 [Source:HGNC Symbol;Acc:HGNC:16887] |
| 4 | PLVAP | −2.6 | 8.1 × 10−6 | plasmalemma vesicle associated protein [Source:HGNC Symbol;Acc:HGNC:13635] |
| 5 | CA1 | −2.4 | 7.2 × 10−4 | carbonic anhydrase 1 [Source:HGNC Symbol;Acc:HGNC:1368] |
| 6 | VWCE | −2.4 | 0.036 | von Willebrand factor C and EGF domains [Source:HGNC Symbol;Acc:HGNC:26487] |
| 7 | KLF1 | −2.4 | 8.1 × 10−4 | Kruppel-like factor 1 [Source:HGNC Symbol;Acc:HGNC:6345] |
| 8 | HBM | −2.3 | 2.3 × 10−4 | haemoglobin subunit mu [Source:HGNC Symbol;Acc:HGNC:4826] |
| 9 | AHSP | −2.3 | 0.016 | alpha haemoglobin stabilizing protein [Source:HGNC Symbol;Acc:HGNC:18075] |
| 10 | EPB42 | −2.2 | 0.006 | erythrocyte membrane protein band 4.2 [Source:HGNC Symbol;Acc:HGNC:3381] |
| No. | Gene Symbol | FC | p-Value * | Description |
|---|---|---|---|---|
| Upregulated genes | ||||
| 1 | GPR15 | 7.5 | 1.4 × 10−9 | G protein-coupled receptor 15 [Source:HGNC Symbol;Acc:HGNC:4469] |
| 2 | IGHA2 | 2.6 | 2.8 × 10−2 | immunoglobulin heavy constant alpha 2 (A2m marker) [Source:HGNC Symbol;Acc:HGNC:5479] |
| 3 | IGKV3-11 | 2.2 | 6.2 × 10−3 | immunoglobulin kappa variable 3–11 [Source:HGNC Symbol;Acc:HGNC:5815] |
| 4 | IGLV3-21 | 2.4 | 3.0 × 10−2 | immunoglobulin lambda variable 3–21 [Source:HGNC Symbol;Acc:HGNC:5905] |
| 5 | JCHAIN | 2.1 | 3.8 × 10−2 | joining chain of multimeric IgA and IgM [Source:HGNC Symbol;Acc:HGNC:5713] |
| Downregulated genes | ||||
| 1 | ABALON | −2.1 | 8.6 × 10−4 | apoptotic BCL2L1-antisense long noncoding RNA [Source:HGNC Symbol;Acc:HGNC:49667] |
| 2 | AHSP | −2.3 | 6.1 × 10−3 | alpha haemoglobin stabilizing protein [Source:HGNC Symbol;Acc:HGNC:18075] |
| 3 | ALAS2 | −2.2 | 1.6 × 10−2 | 5’-aminolevulinate synthase 2 [Source:HGNC Symbol;Acc:HGNC:397] |
| 4 | BCL2L1 | −2.0 | 1.0 × 10−3 | BCL2 like 1 [Source:HGNC Symbol;Acc:HGNC:992] |
| 5 | CA1 | −2.4 | 3.6 × 10−2 | carbonic anhydrase 1 [Source:HGNC Symbol;Acc:HGNC:1368] |
| 6 | EPB42 | −2.2 | 3.5 × 10−4 | erythrocyte membrane protein band 4.2 [Source:HGNC Symbol;Acc:HGNC:3381] |
| 7 | HBD | −2.0 | 1.7 × 10−3 | haemoglobin subunit delta [Source:HGNC Symbol;Acc:HGNC:4829] |
| 8 | HBM | −2.3 | 1.6 × 10−2 | haemoglobin subunit mu [Source:HGNC Symbol;Acc:HGNC:4826] |
| 9 | IFIT1B | −2.2 | 1.5 × 10−3 | interferon induced protein with tetratricopeptide repeats 1B [Source:HGNC Symbol;Acc:HGNC:23442] |
| 10 | KLC3 | −2.2 | 1.8 × 10−3 | kinesin light chain 3 [Source:HGNC Symbol;Acc:HGNC:20717] |
| 11 | KLF1 | −2.4 | 2.4 × 10−4 | Kruppel-like factor 1 [Source:HGNC Symbol;Acc:HGNC:6345] |
| 12 | KRT1 | −2.9 | 5.8 × 10−3 | keratin 1 [Source:HGNC Symbol;Acc:HGNC:6412] |
| 13 | MRC2 | −2.2 | 3.6 × 10−4 | mannose receptor C type 2 [Source:HGNC Symbol;Acc:HGNC:16875] |
| 14 | MYL4 | −2.2 | 3.6 × 10−4 | myosin light chain 4 [Source:HGNC Symbol;Acc:HGNC:7585] |
| 15 | NEAT1_3 | −2.7 | 4.4 × 10−2 | Nuclear enriched abundant transcript 1 conserved region 3 [Source:RFAM;Acc:RF01957] |
| 16 | OSBP2 | −2.2 | 7.1 × 10−4 | oxysterol binding protein 2 [Source:HGNC Symbol;Acc:HGNC:8504] |
| 17 | PDZK1IP1 | −2.6 | 2.8 × 10−6 | PDZK1 interacting protein 1 [Source:HGNC Symbol;Acc:HGNC:16887] |
| 18 | PLVAP | −2.6 | 7.3 × 10−4 | plasmalemma vesicle associated protein [Source:HGNC Symbol;Acc:HGNC:13635] |
| 19 | RUNDC3A | −2.2 | 2.1 × 10−4 | RUN domain containing 3A [Source:HGNC Symbol;Acc:HGNC:16984] |
| 20 | SELENBP1 | −2.2 | 2.3 × 10−3 | selenium binding protein 1 [Source:HGNC Symbol;Acc:HGNC:10719] |
| 21 | SHISA4 | −2.2 | 1.9 × 10−2 | shisa family member 4 [Source:HGNC Symbol;Acc:HGNC:27139] |
| 22 | SLC4A1 | −2.1 | 1.8 × 10−3 | solute carrier family 4 member 1 (Diego blood group) [Source:HGNC Symbol;Acc:HGNC:11027] |
| 23 | SNCA | −2.2 | 4.7 × 10−3 | synuclein alpha [Source:HGNC Symbol;Acc:HGNC:11138] |
| 24 | TNS1 | −2.1 | 1.4 × 10−3 | tensin 1 [Source:HGNC Symbol;Acc:HGNC:11973] |
| 25 | TRIM58 | −2.0 | 6.9 × 10−3 | tripartite motif containing 58 [Source:HGNC Symbol;Acc:HGNC:24150] |
| 26 | VWCE | −2.4 | 8.1 × 10−4 | von Willebrand factor C and EGF domains [Source:HGNC Symbol;Acc:HGNC:26487] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merzah, M.; Póliska, S.; Balogh, L.; Sándor, J.; Szász, I.; Natae, S.; Fiatal, S. A Transcriptomic Analysis of Smoking-Induced Gene Expression Alterations in Coronary Artery Disease Patients. Int. J. Mol. Sci. 2023, 24, 13920. https://doi.org/10.3390/ijms241813920
Merzah M, Póliska S, Balogh L, Sándor J, Szász I, Natae S, Fiatal S. A Transcriptomic Analysis of Smoking-Induced Gene Expression Alterations in Coronary Artery Disease Patients. International Journal of Molecular Sciences. 2023; 24(18):13920. https://doi.org/10.3390/ijms241813920
Chicago/Turabian StyleMerzah, Mohammed, Szilárd Póliska, László Balogh, János Sándor, István Szász, Shewaye Natae, and Szilvia Fiatal. 2023. "A Transcriptomic Analysis of Smoking-Induced Gene Expression Alterations in Coronary Artery Disease Patients" International Journal of Molecular Sciences 24, no. 18: 13920. https://doi.org/10.3390/ijms241813920
APA StyleMerzah, M., Póliska, S., Balogh, L., Sándor, J., Szász, I., Natae, S., & Fiatal, S. (2023). A Transcriptomic Analysis of Smoking-Induced Gene Expression Alterations in Coronary Artery Disease Patients. International Journal of Molecular Sciences, 24(18), 13920. https://doi.org/10.3390/ijms241813920