
Droplet Digital PCR Is a Novel Screening Method Identifying Potential Cardiac G-Protein-Coupled Receptors as Candidate Pharmacological Targets in a Rat Model of Pressure-Overload-Induced Cardiac Dysfunction
 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Echocardiographic Characterization of Pressure-Overload-Induced Cardiac Dysfunction in Rats
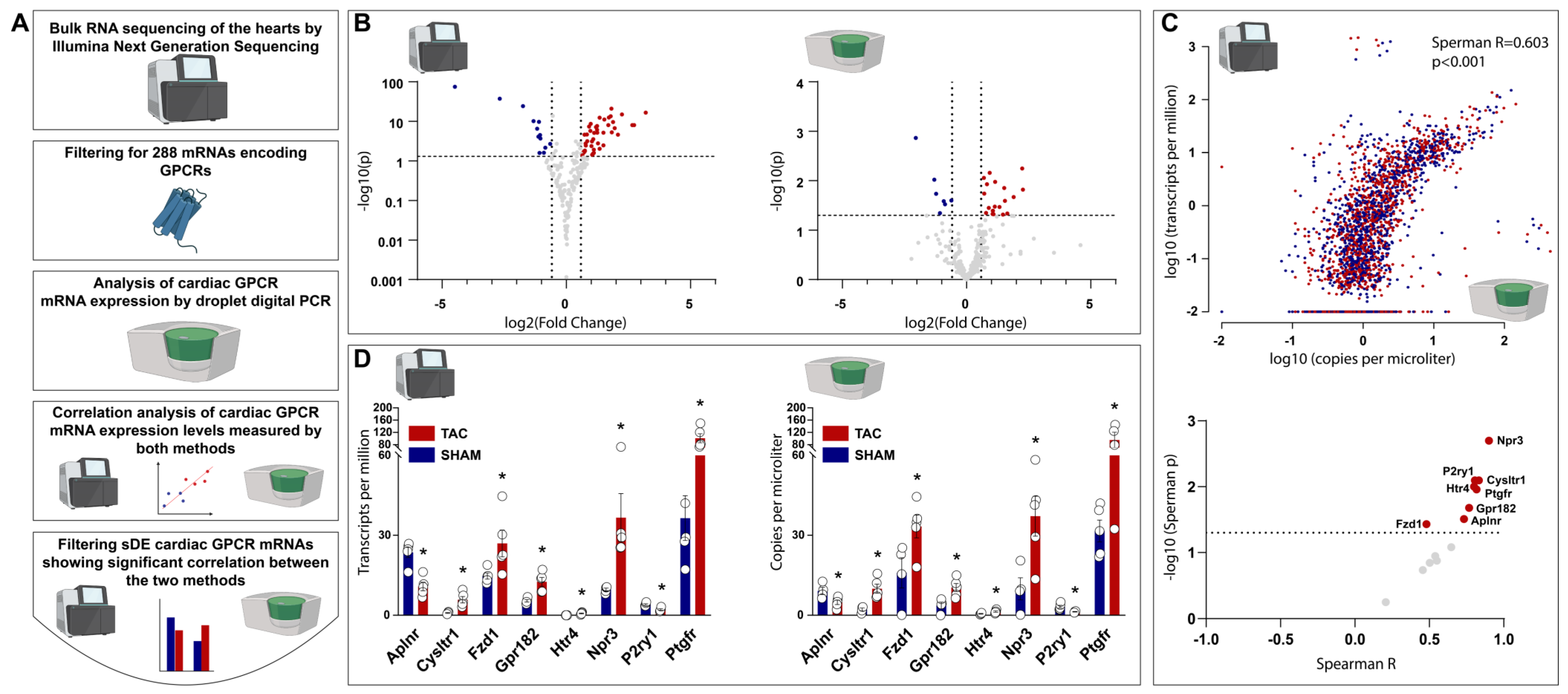
2.2. Bulk RNA Sequencing Identified 69, and ddPCR Identified 27 Cardiac GPCR Genes to Be Differentially Expressed in TAC vs. SHAM Rat Hearts
2.3. Comparative Analysis of Cardiac GPCR Gene Expression Profiles Measured via Bulk RNA Sequencing and ddPCR Shows Significant Correlation
2.4. Filtering for Novel GPCR Targets Identifies Prostaglandin F2α Receptor to Be a Potential GPCR Target with Relevant Clinical Translatability in Heart Failure
2.5. Ptgfr Is Expressed by Cardiac Fibroblasts and Cardiomyocytes
2.6. Ptgfr Inhibition by AL-8810 Reverts Angiotensin-II Induced Hypertrophy of Neonatal Rat Cardiomyocytes
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
4.2. Rat Model of Transverse Aortic Constriction Induced Cardiac Dysfunction and Hypertrophy
4.3. Echocardiography
4.4. RNA Isolation
4.5. RNA Sequencing and Bioinformatic Analysis
4.6. Droplet Digital PCR
4.7. Neonatal Rat Cardiomyocyte Model of Hypertrophy
4.8. RNA Scope® In Situ Hybridization Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, S.P.; Ibrahim, N.E.; Januzzi, J.L. Heart Failure with Reduced Ejection Fraction. JAMA 2020, 324, 488–504. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.S.; Xu, H.; Matsouaka, R.A.; Bhatt, D.L.; Heidenreich, P.A.; Hernandez, A.F.; Devore, A.D.; Yancy, C.W.; Fonarow, G.C. Heart Failure with Preserved, Borderline, and Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 2476–2486. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Ghionzoli, N.; Innocenti, L.; Taddei, C.; Giannoni, A.; Valleggi, A.; Borrelli, C.; Senni, M.; Passino, C.; Emdin, M. Noncardiac Versus Cardiac Mortality in Heart Failure with Preserved, Midrange, and Reduced Ejection Fraction. J. Am. Heart Assoc. 2019, 8, e013441. [Google Scholar] [CrossRef]
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A.J.S. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2022, 118, 3272–3287. [Google Scholar] [CrossRef] [PubMed]
- Sertkaya, A.; Birkenbach, A.; Berlind, A.; Eyraud, J. Examination of Clinical Trial Costs and Barriers for Drug Development. U.S. Department of Health and Human Services. 2014. Available online: https://aspe.hhs.gov/sites/default/files/migrated_legacy_files/44516/rpt_erg.pdf (accessed on 4 September 2023).
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein–Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y. Transmembrane protein western blotting: Impact of sample preparation on detection of SLC11A2 (DMT1) and SLC40A1 (ferroportin). PLoS ONE 2020, 15, e0235563. [Google Scholar] [CrossRef] [PubMed]
- Corney, D.C. RNA-seq Using Next Generation Sequencing. Mater. Methods 2013, 3, 1–26. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef] [PubMed]
- Onódi, Z.; Ruppert, M.; Kucsera, D.; Sayour, A.A.; Tóth, V.E.; Koncsos, G.; Novák, J.; Brenner, G.B.; Makkos, A.; Baranyai, T.; et al. AIM2-driven inflammasome activation in heart failure. Cardiovasc. Res. 2021, 117, 2639–2651. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, M.; Lakatos, B.K.; Braun, S.; Tokodi, M.; Karime, C.; Oláh, A.; Sayour, A.A.; Hizoh, I.; Barta, B.A.; Merkely, B.; et al. Longitudinal Strain Reflects Ventriculoarterial Coupling Rather Than Mere Contractility in Rat Models of Hemodynamic Overload–Induced Heart Failure. J. Am. Soc. Echocardiogr. 2020, 33, 1264–1275. [Google Scholar] [CrossRef]
- Miyazaki, T.; Otani, K.; Chiba, A.; Nishimura, H.; Tokudome, T.; Takano-Watanabe, H.; Matsuo, A.; Ishikawa, H.; Shimamoto, K.; Fukui, H.; et al. A New Secretory Peptide of Natriuretic Peptide Family, Osteocrin, Suppresses the Progression of Congestive Heart Failure after Myocardial Infarction. Circ. Res. 2018, 122, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Brattelid, T.; Qvigstad, E.; Moltzau, L.R.; Bekkevold, S.V.S.; Sandnes, D.L.; Birkeland, J.A.K.; Skomedal, T.; Osnes, J.-B.; Sjaastad, I.; Levy, F.O. The Cardiac Ventricular 5-HT4 Receptor Is Functional in Late Foetal Development and Is Reactivated in Heart Failure. PLoS ONE 2012, 7, e45489. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lucitt, M.B.; Stubbe, J.; Cheng, Y.; Friis, U.G.; Hansen, P.B.; Jensen, B.L.; Smyth, E.M.; FitzGerald, G.A. Prostaglandin F 2α elevates blood pressure and promotes atherosclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7985–7990. [Google Scholar] [CrossRef]
- Wong, S.L.; Leung, F.P.; Lau, C.W.; Au, C.L.; Yung, L.M.; Yao, X.; Chen, Z.-Y.; Vanhoutte, P.M.; Gollasch, M.; Huang, Y.; et al. Cyclooxygenase-2–Derived Prostaglandin F 2α Mediates Endothelium-Dependent Contractions in the Aortae of Hamsters with Increased Impact during Aging. Circ. Res. 2009, 104, 228–235. [Google Scholar] [CrossRef]
- Oga, T.; Matsuoka, T.; Yao, C.; Nonomura, K.; Kitaoka, S.; Sakata, D.; Kita, Y.; Tanizawa, K.; Taguchi, Y.; Chin, K.; et al. Prostaglandin F2α receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-β. Nat. Med. 2009, 15, 1426–1430. [Google Scholar] [CrossRef]
- Ding, W.-Y.; Ti, Y.; Wang, J.; Wang, Z.-H.; Xie, G.-L.; Shang, Y.-Y.; Tang, M.-X.; Zhang, Y.; Zhang, W.; Zhong, M. Prostaglandin F2α facilitates collagen synthesis in cardiac fibroblasts via an F-prostanoid receptor/protein kinase C/Rho kinase pathway independent of transforming growth factor β1. Int. J. Biochem. Cell Biol. 2012, 44, 1031–1039. [Google Scholar] [CrossRef]
- Wu, Y.; Cui, C.; Bi, F.-F.; Wu, C.-Y.; Li, J.-R.; Hou, Y.-M.; Jing, Z.-H.; Pan, Q.-M.; Cao, M.; Lv, L.-F.; et al. Montelukast, cysteinyl leukotriene receptor 1 antagonist, inhibits cardiac fibrosis by activating APJ. Eur. J. Pharmacol. 2022, 923, 174892. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Principal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Schoger, E.; Bleckwedel, F.; Germena, G.; Rocha, C.; Tucholla, P.; Sobitov, I.; Möbius, W.; Sitte, M.; Lenz, C.; Samak, M.; et al. Single-cell transcriptomics reveal extracellular vesicles secretion with a cardiomyocyte proteostasis signature during pathological remodeling. Commun. Biol. 2023, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Onódi, Z.; Visnovitz, T.; Kiss, B.; Hambalkó, S.; Koncz, A.; Ágg, B.; Váradi, B.; Tóth, V.; Nagy, R.N.; Gergely, T.G.; et al. Systematic transcriptomic and phenotypic characterization of human and murine cardiac myocyte cell lines and primary cardiomyocytes reveals serious limitations and low resemblances to adult cardiac phenotype. J. Mol. Cell. Cardiol. 2021, 165, 19–30. [Google Scholar] [CrossRef]
- Fujimori, K.; Ueno, T.; Nagata, N.; Kashiwagi, K.; Aritake, K.; Amano, F.; Urade, Y. Suppression of Adipocyte Differentiation by Aldo-keto Reductase 1B3 Acting as Prostaglandin F2α Synthase. J. Biol. Chem. 2010, 285, 8880–8886. [Google Scholar] [CrossRef]
- Goupil, E.; Wisehart, V.; Khoury, E.; Zimmerman, B.; Jaffal, S.; Hébert, T.E.; Laporte, S.A. Biasing the Prostaglandin F2α Receptor Responses toward EGFR-Dependent Transactivation of MAPK. Mol. Endocrinol. 2012, 26, 1189–1202. [Google Scholar] [CrossRef]
- Harks, E.G.A.; Peters, P.H.J.; van Dongen, J.L.J.; van Zoelen, E.J.J.; Theuvenet, A.P.R. Autocrine production of prostaglandin F2αenhances phenotypic transformation of normal rat kidney fibroblasts. Am. J. Physiol. Cell Physiol. 2005, 289, C130–C137. [Google Scholar] [CrossRef]
- Jelemenský, M.; Kovácsházi, C.; Ferenczyová, K.; Hofbauerová, M.; Kiss, B.; Pállinger, É.; Kittel, Á.; Sayour, V.N.; Görbe, A.; Pelyhe, C.; et al. Helium Conditioning Increases Cardiac Fibroblast Migration Which Effect Is Not Propagated via Soluble Factors or Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 10504. [Google Scholar] [CrossRef]
- Weber, B.Y.; Brenner, G.B.; Kiss, B.; Gergely, T.G.; Sayour, N.V.; Tian, H.; Makkos, A.; Görbe, A.; Ferdinandy, P.; Giricz, Z. Rosiglitazone Does Not Show Major Hidden Cardiotoxicity in Models of Ischemia/Reperfusion but Abolishes Ischemic Preconditioning-Induced Antiarrhythmic Effects in Rats In Vivo. Pharmaceuticals 2022, 15, 1055. [Google Scholar] [CrossRef]
- Brenner, G.B.; Makkos, A.; Nagy, C.T.; Onódi, Z.; Sayour, N.V.; Gergely, T.G.; Kiss, B.; Görbe, A.; Sághy, É.; Zádori, Z.S.; et al. Hidden Cardiotoxicity of Rofecoxib Can be Revealed in Experimental Models of Ischemia/Reperfusion. Cells 2020, 9, 551. [Google Scholar] [CrossRef]
- Sayour, N.V.; Brenner, G.B.; Makkos, A.; Kiss, B.; Kovácsházi, C.; Gergely, T.G.; Aukrust, S.G.; Tian, H.; Zenkl, V.; Gömöri, K.; et al. Cardioprotective efficacy of limb remote ischaemic preconditioning in rats: Discrepancy between a meta-analysis and a three-centre in vivo study. Cardiovasc. Res. 2023, 119, 1336–1351. [Google Scholar] [CrossRef] [PubMed]
- Brenner, G.B.; Giricz, Z.; Garamvölgyi, R.; Makkos, A.; Onódi, Z.; Sayour, N.V.; Gergely, T.G.; Baranyai, T.; Petneházy, Ö.; Kőrösi, D.; et al. Post-Myocardial Infarction Heart Failure in Closed-chest Coronary Occlusion/Reperfusion Model in Göttingen Minipigs and Landrace Pigs. J. Vis. Exp. 2021, e61901. [Google Scholar] [CrossRef]
- Chaffin, M.; Papangeli, I.; Simonson, B.; Akkad, A.-D.; Hill, M.C.; Arduini, A.; Fleming, S.J.; Melanson, M.; Hayat, S.; Kost-Alimova, M.; et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature 2022, 608, 174–180. [Google Scholar] [CrossRef]
- Zhang, J.; Gong, Y.; Yu, Y. PG F2α Receptor: A Promising Therapeutic Target for Cardiovascular Disease. Front. Pharmacol. 2010, 1, 116. [Google Scholar] [CrossRef] [PubMed]
- Beccacece, L.; Abondio, P.; Bini, C.; Pelotti, S.; Luiselli, D. The Link between Prostanoids and Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 4193. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, B.; Arad, M.; Elazar, E.; Klein, R.; Zahav, Y.H. Epicardial versus endocardial "in mirror" changes in prostaglandin synthesis after short periods of ischemia and reperfusion. Eicosanoids 1992, 5, 163–167. [Google Scholar] [PubMed]
- Yoshida, M.; Sagawa, N.; Itoh, H.; Yura, S.; Takemura, M.; Wada, Y.; Sato, T.; Ito, A.; Fujii, S. Prostaglandin F2alpha, cytokines and cyclic mechanical stretch augment matrix metalloproteinase-1 secretion from cultured human uterine cervical fibroblast cells. Mol. Hum. Reprod. 2002, 8, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Almirza, W.; Dernison, M.; Peters, P.; van Zoelen, E.; Theuvenet, A. Role of the prostanoid FP receptor in action potential generation and phenotypic transformation of NRK fibroblasts. Cell. Signal. 2008, 20, 2022–2029. [Google Scholar] [CrossRef]
- Lai, J.; Jin, H.; Yang, R.; Winer, J.; Li, W.; Yen, R.; King, K.L.; Zeigler, F.; Ko, A.; Cheng, J.; et al. Prostaglandin F2 alpha induces cardiac myocyte hypertrophy in vitro and cardiac growth in vivo. Am. J. Physiol. Circ. Physiol. 1996, 271, H2197–H2208. [Google Scholar] [CrossRef]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated Levels of 8-iso-Prostaglandin F 2α in Pericardial Fluid of Patients with Heart Failure. Circulation 1998, 97, 1536–1539. [Google Scholar] [CrossRef]
- Cohen, D.; Koh, G.Y.; Nikonova, L.N.; Porter, J.G.; Maack, T. Molecular Determinants of the Clearance Function of Type C Receptors of Natriuretic Peptides. J. Biol. Chem. 1996, 271, 9863–9869. [Google Scholar] [CrossRef] [PubMed]
- Nussenzveig, D.R.; Lewicki, J.A.; Maack, T. Cellular mechanisms of the clearance function of type C receptors of atrial natriuretic factor. Perspect. Surg. 1990, 265, 20952–20958. [Google Scholar] [CrossRef]
- Venkatesan, B.; Tumala, A.; Subramanian, V.; Vellaichamy, E. Transient silencing of Npr3 gene expression improved the circulatory levels of atrial natriuretic peptides and attenuated β-adrenoceptor activation- induced cardiac hypertrophic growth in experimental rats. Eur. J. Pharmacol. 2016, 782, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Wee, A.S.; Lim, J.Y.; Ng, J.Y.; Chong, J.P.; Liew, O.W.; Lilyanna, S.; Martinez, E.C.; Ackers-Johnson, M.A.; Vardy, L.A.; et al. Natriuretic peptide receptor 3 (NPR3) is regulated by microRNA-100. J. Mol. Cell. Cardiol. 2015, 82, 13–21. [Google Scholar] [CrossRef]
- Wang, J.; Tong, K.S.; Wong, L.L.; Liew, O.-W.; Raghuram, D.; Richards, A.M.; Chen, Y.-T. MicroRNA-143 modulates the expression of Natriuretic Peptide Receptor 3 in cardiac cells. Sci. Rep. 2018, 8, 7055. [Google Scholar] [CrossRef]
- Sato, T.; Sato, C.; Kadowaki, A.; Watanabe, H.; Ho, L.; Ishida, J.; Yamaguchi, T.; Kimura, A.; Fukamizu, A.; Penninger, J.M.; et al. ELABELA-APJ axis protects from pressure overload heart failure and angiotensin II-induced cardiac damage. Cardiovasc. Res. 2017, 113, 760–769. [Google Scholar] [CrossRef]
- Azizi, Y.; Faghihi, M.; Imani, A.; Roghani, M.; Nazari, A. Post-infarct treatment with [Pyr1]-apelin-13 reduces myocardial damage through reduction of oxidative injury and nitric oxide enhancement in the rat model of myocardial infarction. Peptides 2013, 46, 76–82. [Google Scholar] [CrossRef]
- Wang, W.; McKinnie, S.M.; Patel, V.B.; Haddad, G.; Wang, Z.; Zhabyeyev, P.; Das, S.K.; Basu, R.; McLean, B.; Kandalam, V.; et al. Loss of Apelin Exacerbates Myocardial Infarction Adverse Remodeling and Ischemia-reperfusion Injury: Therapeutic Potential of Synthetic Apelin Analogues. J. Am. Heart Assoc. 2013, 2, e000249. [Google Scholar] [CrossRef]
- Hoxha, M.; Tedesco, C.C.; Quaglin, S.; Malaj, V.; Pustina, L.; Capra, V.; Evans, J.F.; Sala, A.; Rovati, G.E. Montelukast Use Decreases Cardiovascular Events in Asthmatics. Front. Pharmacol. 2021, 11, 611561. [Google Scholar] [CrossRef]
- Ingelsson, E.; Yin, L.; Bäck, M. Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J. Allergy Clin. Immunol. 2012, 129, 702–707. [Google Scholar] [CrossRef]
- Sarszegi, Z.; Szabo, D.; Gaszner, B.; Konyi, A.; Reglodi, D.; Nemeth, J.; Lelesz, B.; Polgar, B.; Jungling, A.; Tamas, A. Examination of Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) as a Potential Biomarker in Heart Failure Patients. J. Mol. Neurosci. 2018, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Szabó, D.; Sárszegi, Z.; Polgár, B.; Sághy, É.; Reglődi, D.; Tóth, T.; Onódi, Z.; Leszek, P.; Varga, Z.V.; Helyes, Z.; et al. PACAP-38 and PAC1 Receptor Alterations in Plasma and Cardiac Tissue Samples of Heart Failure Patients. Int. J. Mol. Sci. 2022, 23, 3715. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; McMurray, J.J.; Krum, H.; Kiowski, W.; Massie, B.M.; Caspi, A.; Pratt, C.M.; Petrie, M.C.; DeMets, D.; Kobrin, I.; et al. Long-Term Effect of Endothelin Receptor Antagonism with Bosentan on the Morbidity and Mortality of Patients with Severe Chronic Heart Failure. JACC Heart Fail. 2017, 5, 317–326. [Google Scholar] [CrossRef]
- Gao, C.; Ren, S.V.; Yu, J.; Baal, U.; Thai, D.; Lu, J.; Zeng, C.; Yan, H.; Wang, Y. Glucagon Receptor Antagonism Ameliorates Progression of Heart Failure. JACC Basic Transl. Sci. 2019, 4, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Kolur, V.; Vastrad, B.; Vastrad, C.; Kotturshetti, S.; Tengli, A. Identification of candidate biomarkers and therapeutic agents for heart failure by bioinformatics analysis. BMC Cardiovasc. Disord. 2021, 21, 329. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Williams, C.R.; Baccarella, A.; Parrish, J.Z.; Kim, C.C. Trimming of sequence reads alters RNA-Seq gene expression estimates. BMC Bioinform. 2016, 17, 103. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gorbe, A.; Giricz, Z.; Szunyog, A.; Csont, T.; Burley, D.S.; Baxter, G.F.; Ferdinandy, P. Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res. Cardiol. 2010, 105, 643–650. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef]
- Feng, D.; Nagy, J.A.; Pyne, K.; Dvorak, H.F.; Dvorak, A.M. Ultrastructural Localization of Platelet Endothelial Cell Adhesion Molecule (PECAM-1, CD31) in Vascular Endothelium. J. Histochem. Cytochem. 2004, 52, 87–101. [Google Scholar] [CrossRef]
- Tamiolakis, D.; Papadopoulos, N.; Sivridis, E.; Anastasiadis, P.; Karamanidis, D.; Romanidis, C.; Kotini, A.; Bounovas, A.; Simopoulos, C. Expression of the intermediate filament vimentin and fibrillar proteins of the extracellular matrix related to embryonal heart development. Clin. Exp. Obstet. Gynecol. 2001, 28, 193–195. [Google Scholar]
- Lawson, J.S.; Syme, H.M.; Wheeler-Jones, C.P.D.; Elliott, J. Characterisation of feline renal cortical fibroblast cultures and their transcriptional response to transforming growth factor β1. BMC Vet. Res. 2018, 14, 76. [Google Scholar] [CrossRef]
- Lees-Miller, J.P.; Heeley, D.H.; Smillie, L.B. An abundant and novel protein of 22 kDa (SM22) is widely distributed in smooth muscles. Purification from bovine aorta. Biochem. J. 1987, 244, 705–709. [Google Scholar] [CrossRef]
- Greaves, D.R.; Gordon, S. Macrophage-Specific Gene Expression: Current Paradigms and Future Challenges. Int. J. Hematol. 2002, 76, 6–15. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayour, N.V.; Tóth, V.É.; Nagy, R.N.; Vörös, I.; Gergely, T.G.; Onódi, Z.; Nagy, N.; Bödör, C.; Váradi, B.; Ruppert, M.; et al. Droplet Digital PCR Is a Novel Screening Method Identifying Potential Cardiac G-Protein-Coupled Receptors as Candidate Pharmacological Targets in a Rat Model of Pressure-Overload-Induced Cardiac Dysfunction. Int. J. Mol. Sci. 2023, 24, 13826. https://doi.org/10.3390/ijms241813826
Sayour NV, Tóth VÉ, Nagy RN, Vörös I, Gergely TG, Onódi Z, Nagy N, Bödör C, Váradi B, Ruppert M, et al. Droplet Digital PCR Is a Novel Screening Method Identifying Potential Cardiac G-Protein-Coupled Receptors as Candidate Pharmacological Targets in a Rat Model of Pressure-Overload-Induced Cardiac Dysfunction. International Journal of Molecular Sciences. 2023; 24(18):13826. https://doi.org/10.3390/ijms241813826
Chicago/Turabian StyleSayour, Nabil V., Viktória É. Tóth, Regina N. Nagy, Imre Vörös, Tamás G. Gergely, Zsófia Onódi, Noémi Nagy, Csaba Bödör, Barnabás Váradi, Mihály Ruppert, and et al. 2023. "Droplet Digital PCR Is a Novel Screening Method Identifying Potential Cardiac G-Protein-Coupled Receptors as Candidate Pharmacological Targets in a Rat Model of Pressure-Overload-Induced Cardiac Dysfunction" International Journal of Molecular Sciences 24, no. 18: 13826. https://doi.org/10.3390/ijms241813826
APA StyleSayour, N. V., Tóth, V. É., Nagy, R. N., Vörös, I., Gergely, T. G., Onódi, Z., Nagy, N., Bödör, C., Váradi, B., Ruppert, M., Radovits, T., Bleckwedel, F., Zelarayán, L. C., Pacher, P., Ágg, B., Görbe, A., Ferdinandy, P., & Varga, Z. V. (2023). Droplet Digital PCR Is a Novel Screening Method Identifying Potential Cardiac G-Protein-Coupled Receptors as Candidate Pharmacological Targets in a Rat Model of Pressure-Overload-Induced Cardiac Dysfunction. International Journal of Molecular Sciences, 24(18), 13826. https://doi.org/10.3390/ijms241813826