
Multivariate Genomic Hybrid Prediction with Kernels and Parental Information

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Trait DTF
2.2. Trait DTH
2.3. Trait YIELD
2.4. Across Traits
3. Discussion
4. Materials and Methods
4.1. Phenotypic Data
4.2. Genotypic Data
4.3. Multivariate Statistical Model
4.4. Evaluation of Prediction Performance
4.5. Kernel Methods
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
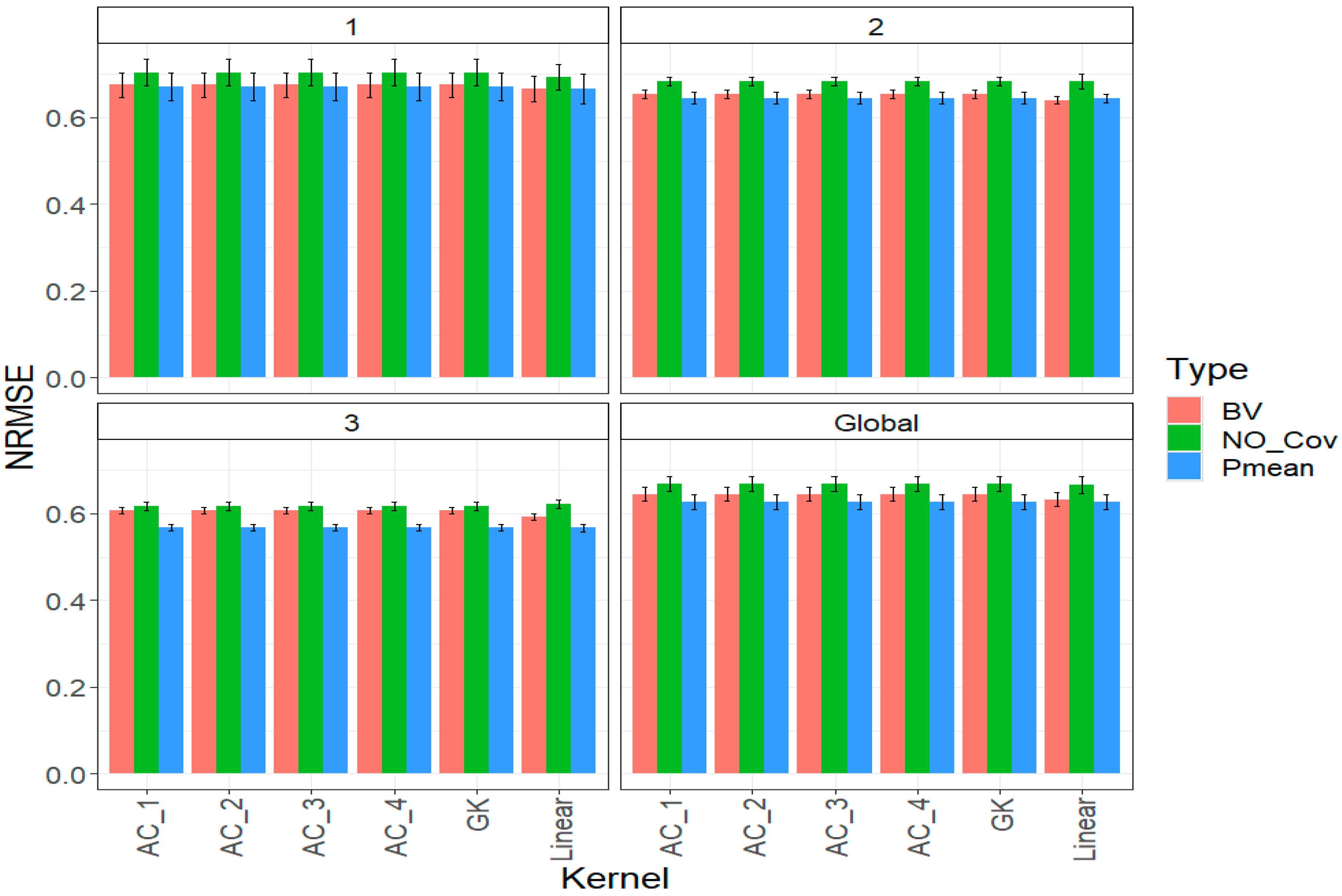
| Type | Kernel | Year | NRMSE | RE__Ker | RE_Env |
|---|---|---|---|---|---|
| BV | AC_1 | 1 | 0.675 | 0.000 | 0.000 |
| BV | AC_2 | 1 | 0.675 | 0.000 | 0.000 |
| BV | AC_3 | 1 | 0.675 | 0.000 | 0.000 |
| BV | AC_4 | 1 | 0.675 | 0.000 | 0.000 |
| BV | GK | 1 | 0.675 | 0.000 | 0.000 |
| BV | Linear | 1 | 0.665 | 1.534 | 0.000 |
| NO_Cov | AC_1 | 1 | 0.703 | 0.000 | 0.000 |
| NO_Cov | AC_2 | 1 | 0.703 | 0.000 | 0.000 |
| NO_Cov | AC_3 | 1 | 0.703 | 0.000 | 0.000 |
| NO_Cov | AC_4 | 1 | 0.703 | 0.000 | 0.000 |
| NO_Cov | GK | 1 | 0.703 | 0.000 | 0.000 |
| NO_Cov | Linear | 1 | 0.693 | 1.428 | 0.000 |
| Pmean | AC_1 | 1 | 0.670 | 0.000 | 0.000 |
| Pmean | AC_2 | 1 | 0.670 | 0.000 | 0.000 |
| Pmean | AC_3 | 1 | 0.670 | 0.000 | 0.000 |
| Pmean | AC_4 | 1 | 0.670 | 0.000 | 0.000 |
| Pmean | GK | 1 | 0.670 | 0.000 | 0.000 |
| Pmean | Linear | 1 | 0.666 | 0.706 | 0.000 |
| BV | AC_1 | 2 | 0.654 | 0.000 | 3.321 |
| BV | AC_2 | 2 | 0.654 | 0.000 | 3.321 |
| BV | AC_3 | 2 | 0.654 | 0.000 | 3.321 |
| BV | AC_4 | 2 | 0.654 | 0.000 | 3.321 |
| BV | GK | 2 | 0.654 | 0.000 | 3.321 |
| BV | Linear | 2 | 0.640 | 2.125 | 3.922 |
| NO_Cov | AC_1 | 2 | 0.683 | 0.234 | 3.048 |
| NO_Cov | AC_2 | 2 | 0.683 | 0.234 | 3.048 |
| NO_Cov | AC_3 | 2 | 0.683 | 0.234 | 3.048 |
| NO_Cov | AC_4 | 2 | 0.683 | 0.234 | 3.048 |
| NO_Cov | GK | 2 | 0.683 | 0.234 | 3.048 |
| NO_Cov | Linear | 2 | 0.684 | 0.000 | 1.359 |
| Pmean | AC_1 | 2 | 0.645 | 0.000 | 3.955 |
| Pmean | AC_2 | 2 | 0.645 | 0.000 | 3.955 |
| Pmean | AC_3 | 2 | 0.645 | 0.000 | 3.955 |
| Pmean | AC_4 | 2 | 0.645 | 0.000 | 3.955 |
| Pmean | GK | 2 | 0.645 | 0.000 | 3.955 |
| Pmean | Linear | 2 | 0.644 | 0.062 | 3.290 |
| BV | AC_1 | 3 | 0.606 | 0.000 | 11.382 |
| BV | AC_2 | 3 | 0.606 | 0.000 | 11.382 |
| BV | AC_3 | 3 | 0.606 | 0.000 | 11.382 |
| BV | AC_4 | 3 | 0.606 | 0.000 | 11.382 |
| BV | GK | 3 | 0.606 | 0.000 | 11.382 |
| BV | Linear | 3 | 0.592 | 2.330 | 12.255 |
| NO_Cov | AC_1 | 3 | 0.617 | 0.616 | 13.950 |
| NO_Cov | AC_2 | 3 | 0.617 | 0.616 | 13.950 |
| NO_Cov | AC_3 | 3 | 0.617 | 0.616 | 13.950 |
| NO_Cov | AC_4 | 3 | 0.617 | 0.616 | 13.950 |
| NO_Cov | GK | 3 | 0.617 | 0.616 | 13.950 |
| NO_Cov | Linear | 3 | 0.621 | 0.000 | 11.659 |
| Pmean | AC_1 | 3 | 0.567 | 0.000 | 18.139 |
| Pmean | AC_2 | 3 | 0.567 | 0.000 | 18.139 |
| Pmean | AC_3 | 3 | 0.567 | 0.000 | 18.139 |
| Pmean | AC_4 | 3 | 0.567 | 0.000 | 18.139 |
| Pmean | GK | 3 | 0.567 | 0.000 | 18.139 |
| Pmean | Linear | 3 | 0.567 | 0.106 | 17.434 |
| BV | AC_1 | Global | 0.645 | 0.000 | 4.688 |
| BV | AC_2 | Global | 0.645 | 0.000 | 4.688 |
| BV | AC_3 | Global | 0.645 | 0.000 | 4.688 |
| BV | AC_4 | Global | 0.645 | 0.000 | 4.688 |
| BV | GK | Global | 0.645 | 0.000 | 4.688 |
| BV | Linear | Global | 0.632 | 1.982 | 5.149 |
| NO_Cov | AC_1 | Global | 0.668 | 0.000 | 5.337 |
| NO_Cov | AC_2 | Global | 0.668 | 0.000 | 5.337 |
| NO_Cov | AC_3 | Global | 0.668 | 0.000 | 5.337 |
| NO_Cov | AC_4 | Global | 0.668 | 0.000 | 5.337 |
| NO_Cov | GK | Global | 0.668 | 0.000 | 5.337 |
| NO_Cov | Linear | Global | 0.666 | 0.225 | 4.088 |
| Pmean | AC_1 | Global | 0.627 | 0.000 | 6.822 |
| Pmean | AC_2 | Global | 0.627 | 0.000 | 6.822 |
| Pmean | AC_3 | Global | 0.627 | 0.000 | 6.822 |
| Pmean | AC_4 | Global | 0.627 | 0.000 | 6.822 |
| Pmean | GK | Global | 0.627 | 0.000 | 6.822 |
| Pmean | Linear | Global | 0.626 | 0.304 | 6.395 |
| Type | Kernel | Env | NRMSE | RE_Ker | RE_Env |
|---|---|---|---|---|---|
| BV | AC_1 | 1 | 0.651 | 0.000 | 0.000 |
| BV | AC_2 | 1 | 0.651 | 0.000 | 0.000 |
| BV | AC_3 | 1 | 0.651 | 0.000 | 0.000 |
| BV | AC_4 | 1 | 0.651 | 0.000 | 0.000 |
| BV | GK | 1 | 0.651 | 0.000 | 0.000 |
| BV | Linear | 1 | 0.641 | 1.577 | 0.000 |
| NO_Cov | AC_1 | 1 | 0.678 | 0.000 | 0.000 |
| NO_Cov | AC_2 | 1 | 0.678 | 0.000 | 0.000 |
| NO_Cov | AC_3 | 1 | 0.678 | 0.000 | 0.000 |
| NO_Cov | AC_4 | 1 | 0.678 | 0.000 | 0.000 |
| NO_Cov | GK | 1 | 0.678 | 0.000 | 0.000 |
| NO_Cov | Linear | 1 | 0.670 | 1.224 | 0.000 |
| Pmean | AC_1 | 1 | 0.645 | 0.000 | 0.000 |
| Pmean | AC_2 | 1 | 0.645 | 0.000 | 0.000 |
| Pmean | AC_3 | 1 | 0.645 | 0.000 | 0.000 |
| Pmean | AC_4 | 1 | 0.645 | 0.000 | 0.000 |
| Pmean | GK | 1 | 0.645 | 0.000 | 0.000 |
| Pmean | Linear | 1 | 0.642 | 0.468 | 0.000 |
| BV | AC_1 | 2 | 0.630 | 0.000 | 3.204 |
| BV | AC_2 | 2 | 0.630 | 0.000 | 3.204 |
| BV | AC_3 | 2 | 0.630 | 0.000 | 3.204 |
| BV | AC_4 | 2 | 0.630 | 0.000 | 3.204 |
| BV | GK | 2 | 0.630 | 0.000 | 3.204 |
| BV | Linear | 2 | 0.619 | 1.875 | 3.507 |
| NO_Cov | AC_1 | 2 | 0.659 | 0.410 | 2.991 |
| NO_Cov | AC_2 | 2 | 0.659 | 0.410 | 2.991 |
| NO_Cov | AC_3 | 2 | 0.659 | 0.410 | 2.991 |
| NO_Cov | AC_4 | 2 | 0.659 | 0.410 | 2.991 |
| NO_Cov | GK | 2 | 0.659 | 0.410 | 2.991 |
| NO_Cov | Linear | 2 | 0.661 | 0.000 | 1.331 |
| Pmean | AC_1 | 2 | 0.625 | 0.000 | 3.103 |
| Pmean | AC_2 | 2 | 0.625 | 0.000 | 3.103 |
| Pmean | AC_3 | 2 | 0.625 | 0.000 | 3.103 |
| Pmean | AC_4 | 2 | 0.625 | 0.000 | 3.103 |
| Pmean | GK | 2 | 0.625 | 0.000 | 3.103 |
| Pmean | Linear | 2 | 0.625 | 0.000 | 2.607 |
| BV | AC_1 | 3 | 0.607 | 0.000 | 7.201 |
| BV | AC_2 | 3 | 0.607 | 0.000 | 7.201 |
| BV | AC_3 | 3 | 0.607 | 0.000 | 7.201 |
| BV | AC_4 | 3 | 0.607 | 0.000 | 7.201 |
| BV | GK | 3 | 0.607 | 0.000 | 7.201 |
| BV | Linear | 3 | 0.592 | 2.448 | 8.120 |
| NO_Cov | AC_1 | 3 | 0.619 | 0.679 | 9.667 |
| NO_Cov | AC_2 | 3 | 0.619 | 0.679 | 9.667 |
| NO_Cov | AC_3 | 3 | 0.619 | 0.679 | 9.667 |
| NO_Cov | AC_4 | 3 | 0.619 | 0.679 | 9.667 |
| NO_Cov | GK | 3 | 0.619 | 0.679 | 9.667 |
| NO_Cov | Linear | 3 | 0.623 | 0.000 | 7.611 |
| Pmean | AC_1 | 3 | 0.565 | 0.000 | 14.048 |
| Pmean | AC_2 | 3 | 0.565 | 0.000 | 14.048 |
| Pmean | AC_3 | 3 | 0.565 | 0.000 | 14.048 |
| Pmean | AC_4 | 3 | 0.565 | 0.000 | 14.048 |
| Pmean | GK | 3 | 0.565 | 0.000 | 14.048 |
| Pmean | Linear | 3 | 0.564 | 0.142 | 13.678 |
| BV | AC_1 | Global | 0.629 | 0.000 | 3.385 |
| BV | AC_2 | Global | 0.629 | 0.000 | 3.385 |
| BV | AC_3 | Global | 0.629 | 0.000 | 3.385 |
| BV | AC_4 | Global | 0.629 | 0.000 | 3.385 |
| BV | GK | Global | 0.629 | 0.000 | 3.385 |
| BV | Linear | Global | 0.617 | 1.955 | 3.770 |
| NO_Cov | AC_1 | Global | 0.652 | 0.000 | 4.065 |
| NO_Cov | AC_2 | Global | 0.652 | 0.000 | 4.065 |
| NO_Cov | AC_3 | Global | 0.652 | 0.000 | 4.065 |
| NO_Cov | AC_4 | Global | 0.652 | 0.000 | 4.065 |
| NO_Cov | GK | Global | 0.652 | 0.000 | 4.065 |
| NO_Cov | Linear | Global | 0.651 | 0.067 | 2.876 |
| Pmean | AC_1 | Global | 0.612 | 0.000 | 5.384 |
| Pmean | AC_2 | Global | 0.612 | 0.000 | 5.384 |
| Pmean | AC_3 | Global | 0.612 | 0.000 | 5.384 |
| Pmean | AC_4 | Global | 0.612 | 0.000 | 5.384 |
| Pmean | GK | Global | 0.612 | 0.000 | 5.384 |
| Pmean | Linear | Global | 0.610 | 0.202 | 5.106 |
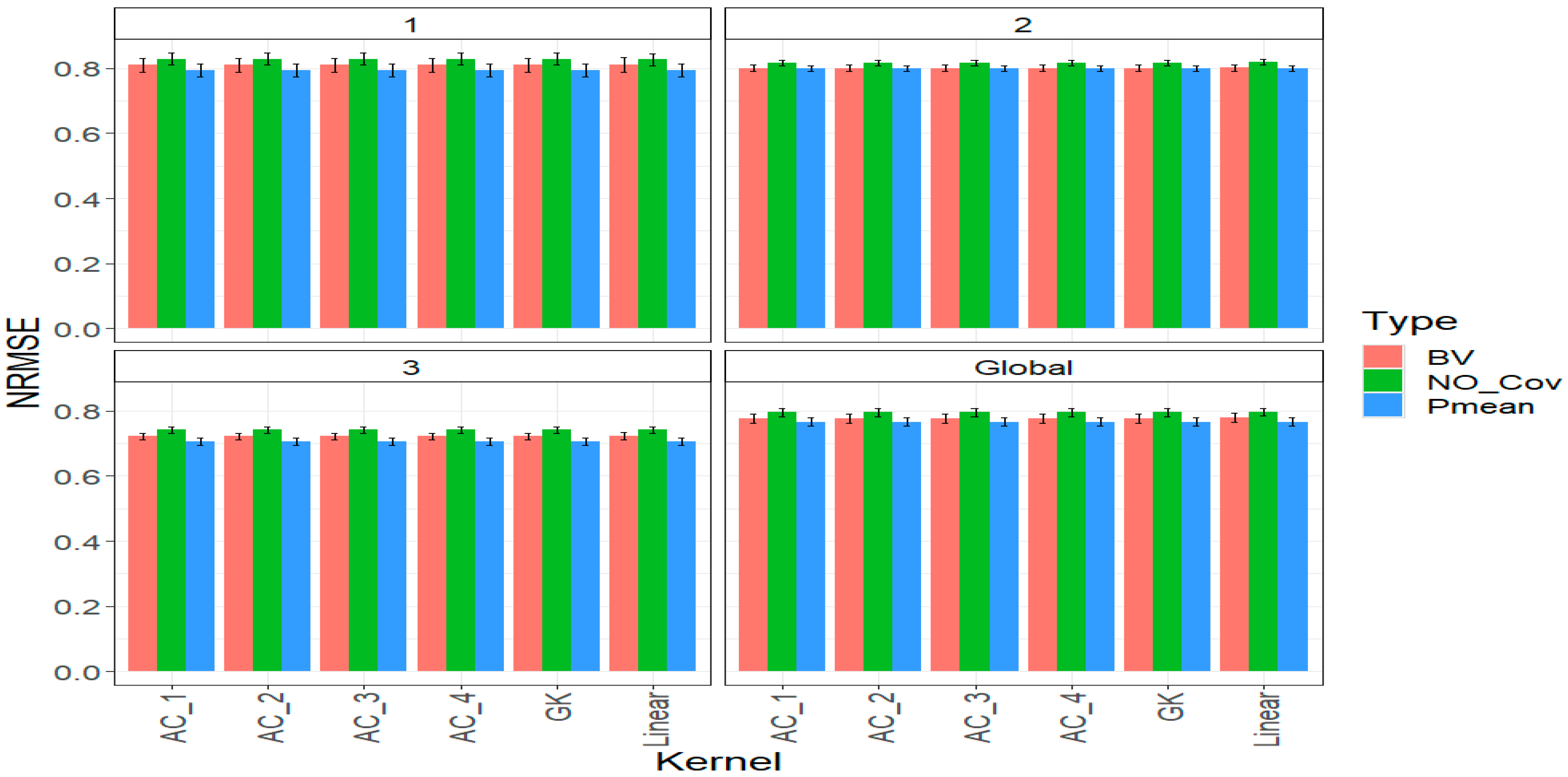
| Type | Kernel | Env | NRMSE | GRE_Entre_Ker | GRE_Entre_Env |
|---|---|---|---|---|---|
| BV | AC_1 | 1 | 0.810 | 0.099 | 0.000 |
| BV | AC_2 | 1 | 0.810 | 0.099 | 0.000 |
| BV | AC_3 | 1 | 0.810 | 0.099 | 0.000 |
| BV | AC_4 | 1 | 0.810 | 0.099 | 0.000 |
| BV | GK | 1 | 0.810 | 0.099 | 0.000 |
| BV | Linear | 1 | 0.811 | 0.000 | 0.000 |
| NO_Cov | AC_1 | 1 | 0.828 | 0.000 | 0.000 |
| NO_Cov | AC_2 | 1 | 0.828 | 0.000 | 0.000 |
| NO_Cov | AC_3 | 1 | 0.828 | 0.000 | 0.000 |
| NO_Cov | AC_4 | 1 | 0.828 | 0.000 | 0.000 |
| NO_Cov | GK | 1 | 0.828 | 0.000 | 0.000 |
| NO_Cov | Linear | 1 | 0.827 | 0.169 | 0.000 |
| Pmean | AC_1 | 1 | 0.793 | 0.000 | 0.757 |
| Pmean | AC_2 | 1 | 0.793 | 0.000 | 0.757 |
| Pmean | AC_3 | 1 | 0.793 | 0.000 | 0.757 |
| Pmean | AC_4 | 1 | 0.793 | 0.000 | 0.757 |
| Pmean | GK | 1 | 0.793 | 0.000 | 0.757 |
| Pmean | Linear | 1 | 0.793 | 0.000 | 0.769 |
| BV | AC_1 | 2 | 0.800 | 0.075 | 1.162 |
| BV | AC_2 | 2 | 0.800 | 0.075 | 1.162 |
| BV | AC_3 | 2 | 0.800 | 0.075 | 1.162 |
| BV | AC_4 | 2 | 0.800 | 0.075 | 1.162 |
| BV | GK | 2 | 0.800 | 0.075 | 1.162 |
| BV | Linear | 2 | 0.801 | 0.000 | 1.186 |
| NO_Cov | AC_1 | 2 | 0.818 | 0.281 | 1.333 |
| NO_Cov | AC_2 | 2 | 0.818 | 0.281 | 1.333 |
| NO_Cov | AC_3 | 2 | 0.818 | 0.281 | 1.333 |
| NO_Cov | AC_4 | 2 | 0.818 | 0.281 | 1.333 |
| NO_Cov | GK | 2 | 0.818 | 0.281 | 1.333 |
| NO_Cov | Linear | 2 | 0.820 | 0.000 | 0.878 |
| Pmean | AC_1 | 2 | 0.799 | 0.013 | 0.000 |
| Pmean | AC_2 | 2 | 0.799 | 0.013 | 0.000 |
| Pmean | AC_3 | 2 | 0.799 | 0.013 | 0.000 |
| Pmean | AC_4 | 2 | 0.799 | 0.013 | 0.000 |
| Pmean | GK | 2 | 0.799 | 0.013 | 0.000 |
| Pmean | Linear | 2 | 0.799 | 0.000 | 0.000 |
| BV | AC_1 | 3 | 0.722 | 0.194 | 12.162 |
| BV | AC_2 | 3 | 0.722 | 0.194 | 12.162 |
| BV | AC_3 | 3 | 0.722 | 0.194 | 12.162 |
| BV | AC_4 | 3 | 0.722 | 0.194 | 12.162 |
| BV | GK | 3 | 0.722 | 0.194 | 12.162 |
| BV | Linear | 3 | 0.723 | 0.000 | 12.056 |
| NO_Cov | AC_1 | 3 | 0.742 | 0.054 | 11.659 |
| NO_Cov | AC_2 | 3 | 0.742 | 0.054 | 11.659 |
| NO_Cov | AC_3 | 3 | 0.742 | 0.054 | 11.659 |
| NO_Cov | AC_4 | 3 | 0.742 | 0.054 | 11.659 |
| NO_Cov | GK | 3 | 0.742 | 0.054 | 11.659 |
| NO_Cov | Linear | 3 | 0.742 | 0.000 | 11.410 |
| Pmean | AC_1 | 3 | 0.706 | 0.071 | 13.128 |
| Pmean | AC_2 | 3 | 0.706 | 0.071 | 13.128 |
| Pmean | AC_3 | 3 | 0.706 | 0.071 | 13.128 |
| Pmean | AC_4 | 3 | 0.706 | 0.071 | 13.128 |
| Pmean | GK | 3 | 0.706 | 0.071 | 13.128 |
| Pmean | Linear | 3 | 0.707 | 0.000 | 13.063 |
| BV | AC_1 | Global | 0.777 | 0.120 | 4.164 |
| BV | AC_2 | Global | 0.777 | 0.120 | 4.164 |
| BV | AC_3 | Global | 0.777 | 0.120 | 4.164 |
| BV | AC_4 | Global | 0.777 | 0.120 | 4.164 |
| BV | GK | Global | 0.777 | 0.120 | 4.164 |
| BV | Linear | Global | 0.778 | 0.000 | 4.142 |
| NO_Cov | AC_1 | Global | 0.796 | 0.054 | 4.079 |
| NO_Cov | AC_2 | Global | 0.796 | 0.054 | 4.079 |
| NO_Cov | AC_3 | Global | 0.796 | 0.054 | 4.079 |
| NO_Cov | AC_4 | Global | 0.796 | 0.054 | 4.079 |
| NO_Cov | GK | Global | 0.796 | 0.054 | 4.079 |
| NO_Cov | Linear | Global | 0.796 | 0.000 | 3.847 |
| Pmean | AC_1 | Global | 0.766 | 0.026 | 4.296 |
| Pmean | AC_2 | Global | 0.766 | 0.026 | 4.296 |
| Pmean | AC_3 | Global | 0.766 | 0.026 | 4.296 |
| Pmean | AC_4 | Global | 0.766 | 0.026 | 4.296 |
| Pmean | GK | Global | 0.766 | 0.026 | 4.296 |
| Pmean | Linear | Global | 0.766 | 0.000 | 4.281 |
| Phenotypic covariance | |||
| yield_blue | DTH_blue | DTF_blue | |
| yield_blue | 0.943 | 3.318 | 2.981 |
| DTH_blue | 3.318 | 35.301 | 33.398 |
| DTF_blue | 2.981 | 33.398 | 31.872 |
| Phenotypic correlation | |||
| yield_blue | DTH_blue | DTF_blue | |
| yield_blue | 1.000 | 0.575 | 0.544 |
| DTH_blue | 0.575 | 1.000 | 0.996 |
| DTF_blue | 0.544 | 0.996 | 1.000 |
| Genetic covariance | |||
| yield_blue | DTH_blue | DTF_blue | |
| yield_blue | 2.787 × 10−10 | 1.726 × 10−10 | 1.142 × 10−10 |
| DTH_blue | 1.726 × 10−10 | 5.383 × 10−9 | 5.147 × 10−9 |
| DTF_blue | 1.142 × 10−10 | 5.147 × 10−9 | 5.037 × 10−9 |
| Genetic correlation | |||
| yield_blue | DTH_blue | DTF_blue | |
| yield_blue | 1.000 | 0.141 | 0.096 |
| DTH_blue | 0.141 | 1.000 | 0.988 |
| DTF_blue | 0.096 | 0.988 | 1.000 |
| Residual covariance | |||
| yield_blue | DTH_blue | DTF_blue | |
| yield_blue | 0.144 | −0.017 | −0.009 |
| DTH_blue | −0.017 | 1.575 | 1.531 |
| DTF_blue | −0.009 | 1.531 | 1.619 |
| Residual correlation | |||
| yield_blue | DTH_blue | DTF_blue | |
| yield_blue | 1.000 | −0.035 | −0.019 |
| DTH_blue | −0.035 | 1.000 | 0.958 |
| DTF_blue | −0.019 | 0.958 | 1.000 |
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.-L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Hickey, J.M.; Gorjanc, G. Simulated data for genomic selection and cross-validation trials in plant and animal breeding. G3 Genes Genomes Genet. 2020, 10, 1925–1931. [Google Scholar]
- Spindel, J.E.; Begum, H.; Akdemir, D.; Collard, B.; Redoña, E.; Jannink, J.-L.; McCouch, S. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 2016, 116, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Van Raden, P.M. Genomic measures of relationship and inbreeding. Interbull Bull. 2007, 52, 11–16. [Google Scholar]
- Jannink, J.L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigacker, S. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome 2012, 5, 103–113. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez, P.; Hickey, J.; Burgueño, J.; Ornella, L. Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Riedelsheimer, C.; Czedik-Eysenberg, A.; Grieder, C.; Lisec, J.; Technow, F.; Sulpice, R.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 2012, 44, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.V.; Crossa, J.; Singh, P.K.; Bains, N.S.; Singh, K.; Sharma, I. Multi-trait and multi-environment QTL analyses for resistance to maize lethal necrosis disease and grain yield. PLoS ONE 2012, 7, e38008. [Google Scholar] [CrossRef]
- Montesinos-López, O.A.; Montesinos-López, J.C.; Montesinos-López, A.; Ramírez-Alcaraz, J.M.; Poland, J.; Singh, R.; Dreisigacker, S.; Crespo, L.; Mondal, S.; Govidan, V.; et al. Bayesian multitrait kernel methods improve multi-environment genome-based prediction. G3 Genes Genomes Genet. 2022, 12, jkab406. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J. Multivariate Statistical Machine Learning Methods for Genomic Prediction; Springer International Publishing: Cham, Switzerland, 2022; ISBN 978-3-030-89010-0. [Google Scholar]
- Gianola, D.; van Kaam, J.B. Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics 2008, 178, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Fernando, R.L.; Dekkers, J.C. The impact of genetic relationship information on genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, Y.; Wang, X.; Ma, Y.; Li, P.; Yang, Z.; Zhang, X.; Xu, C.; Xu, S. Incorporation of parental phenotypic data into multi-omic models improves prediction of yield-related traits in hybrid rice. Plant Biotechnol. J. 2021, 19, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shi, J.; Zhu, J.; Wu, R. The use of parental information to improve genomic prediction in plant breeding. Crop Sci. 2012, 52, 1476–1487. [Google Scholar]
- Pérez-Rodríguez, P.; de Los Campos, G. Multitrait Bayesian shrinkage and variable selection models with the BGLR-R package. Genetics 2022, 222, iyac112. [Google Scholar] [CrossRef] [PubMed]
- Technow, F.; Riedelsheimer, C.; Schrag, T.A. Genomic prediction of hybrid performance in maize with models incorporating dominance and population specific marker effects. Theor. Appl. Genet. 2012, 125, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los Campos, G. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Saul, L. Kernel Methods for Deep Learning. In Proceedings of the NIPS’09 the 22nd International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 7–10 December 2009; pp. 342–350. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montesinos-López, O.A.; Crossa, J.; Saint Pierre, C.; Gerard, G.; Valenzo-Jiménez, M.A.; Vitale, P.; Valladares-Cellis, P.E.; Buenrostro-Mariscal, R.; Montesinos-López, A.; Crespo-Herrera, L. Multivariate Genomic Hybrid Prediction with Kernels and Parental Information. Int. J. Mol. Sci. 2023, 24, 13799. https://doi.org/10.3390/ijms241813799
Montesinos-López OA, Crossa J, Saint Pierre C, Gerard G, Valenzo-Jiménez MA, Vitale P, Valladares-Cellis PE, Buenrostro-Mariscal R, Montesinos-López A, Crespo-Herrera L. Multivariate Genomic Hybrid Prediction with Kernels and Parental Information. International Journal of Molecular Sciences. 2023; 24(18):13799. https://doi.org/10.3390/ijms241813799
Chicago/Turabian StyleMontesinos-López, Osval A., José Crossa, Carolina Saint Pierre, Guillermo Gerard, Marco Alberto Valenzo-Jiménez, Paolo Vitale, Patricia Edwigis Valladares-Cellis, Raymundo Buenrostro-Mariscal, Abelardo Montesinos-López, and Leonardo Crespo-Herrera. 2023. "Multivariate Genomic Hybrid Prediction with Kernels and Parental Information" International Journal of Molecular Sciences 24, no. 18: 13799. https://doi.org/10.3390/ijms241813799
APA StyleMontesinos-López, O. A., Crossa, J., Saint Pierre, C., Gerard, G., Valenzo-Jiménez, M. A., Vitale, P., Valladares-Cellis, P. E., Buenrostro-Mariscal, R., Montesinos-López, A., & Crespo-Herrera, L. (2023). Multivariate Genomic Hybrid Prediction with Kernels and Parental Information. International Journal of Molecular Sciences, 24(18), 13799. https://doi.org/10.3390/ijms241813799