
Integrated Metabolome and Transcriptome Analyses Reveal Amino Acid Biosynthesis Mechanisms during the Physiological Maturity of Grains in Yunnan Hulled Wheat (Triticum aestivum ssp. yunnanense King)

Abstract
:1. Introduction
2. Results
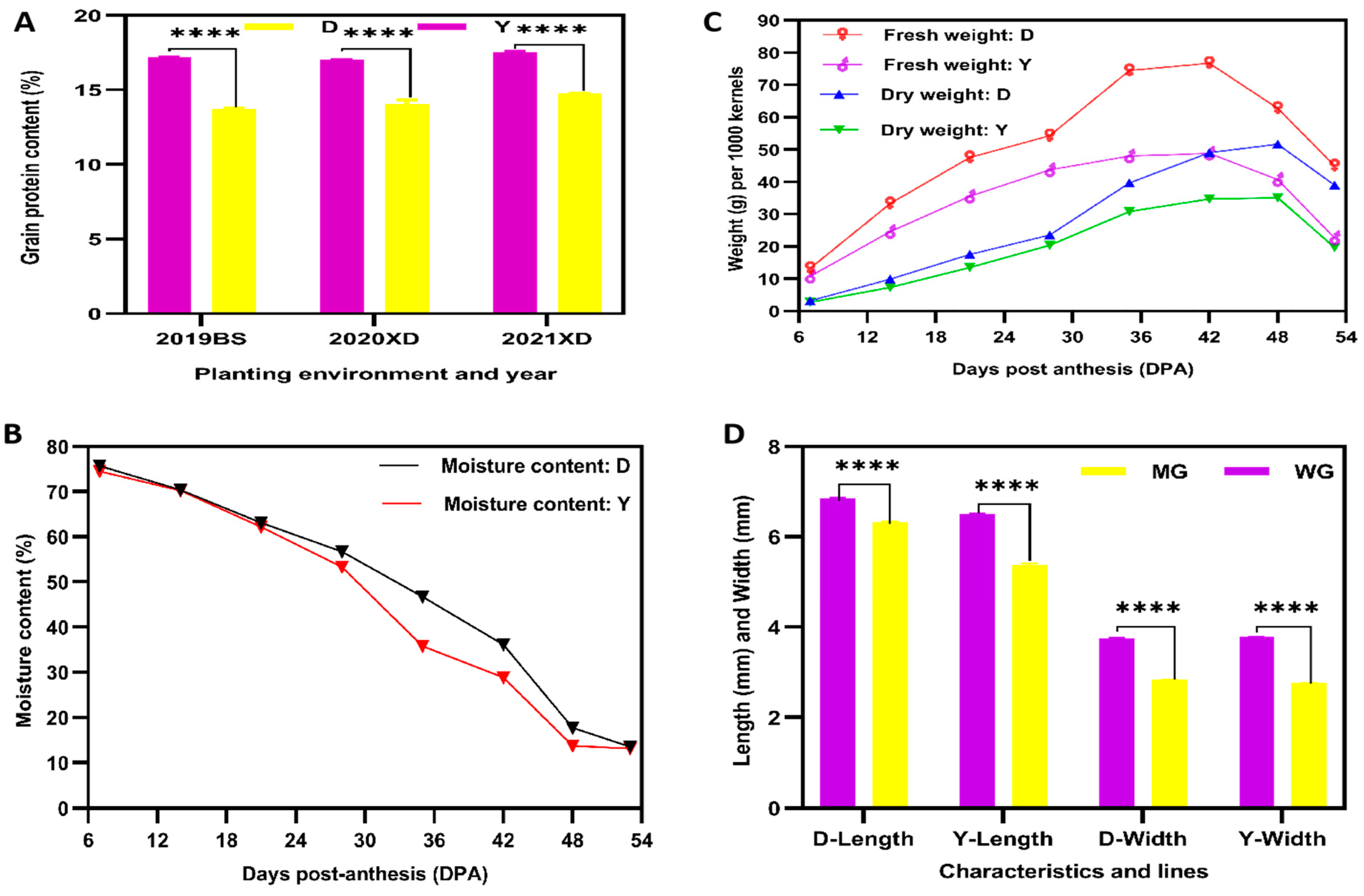
2.1. Plant Material and Physiological Maturity Selection of Two YHW Lines
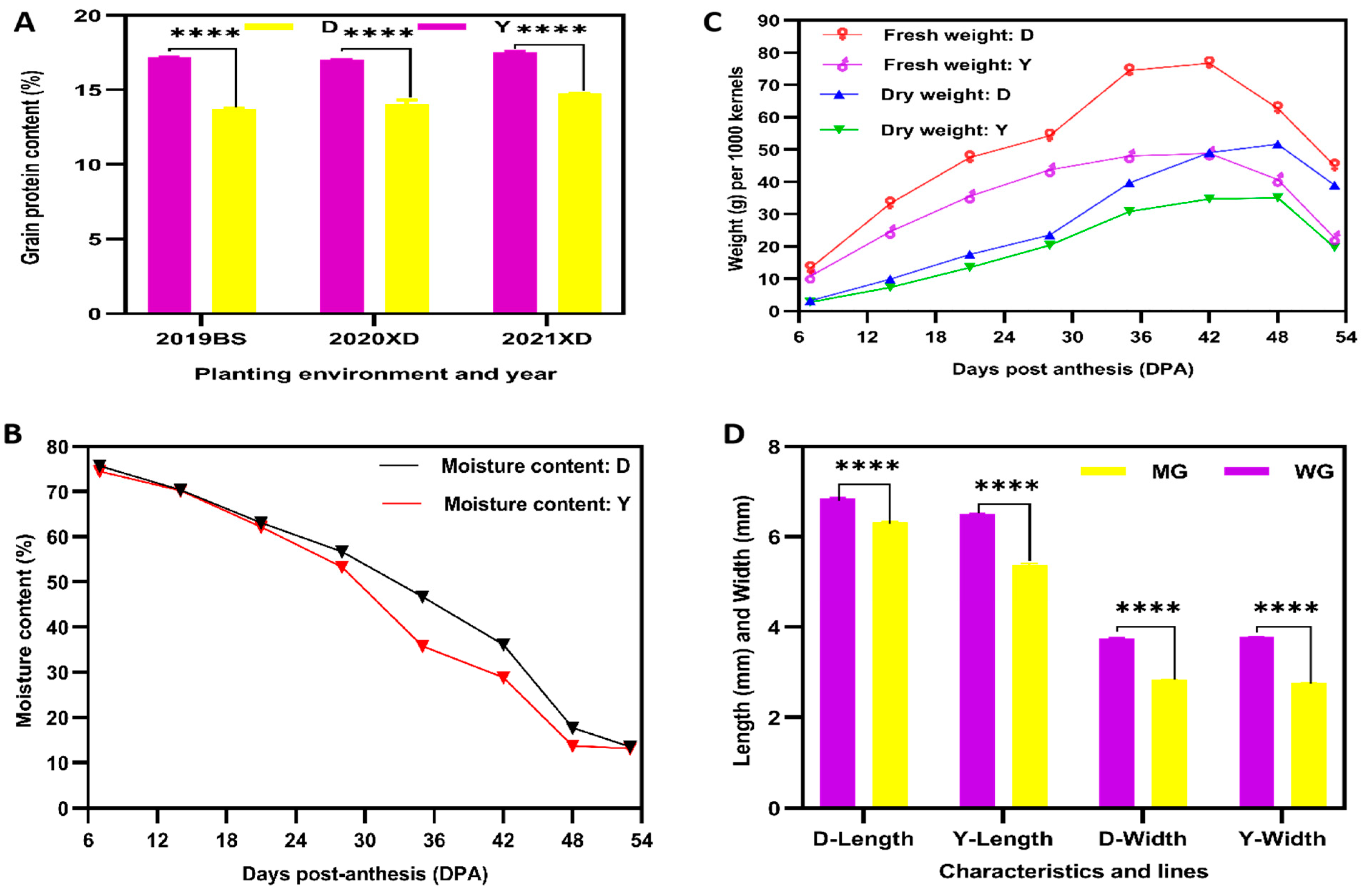
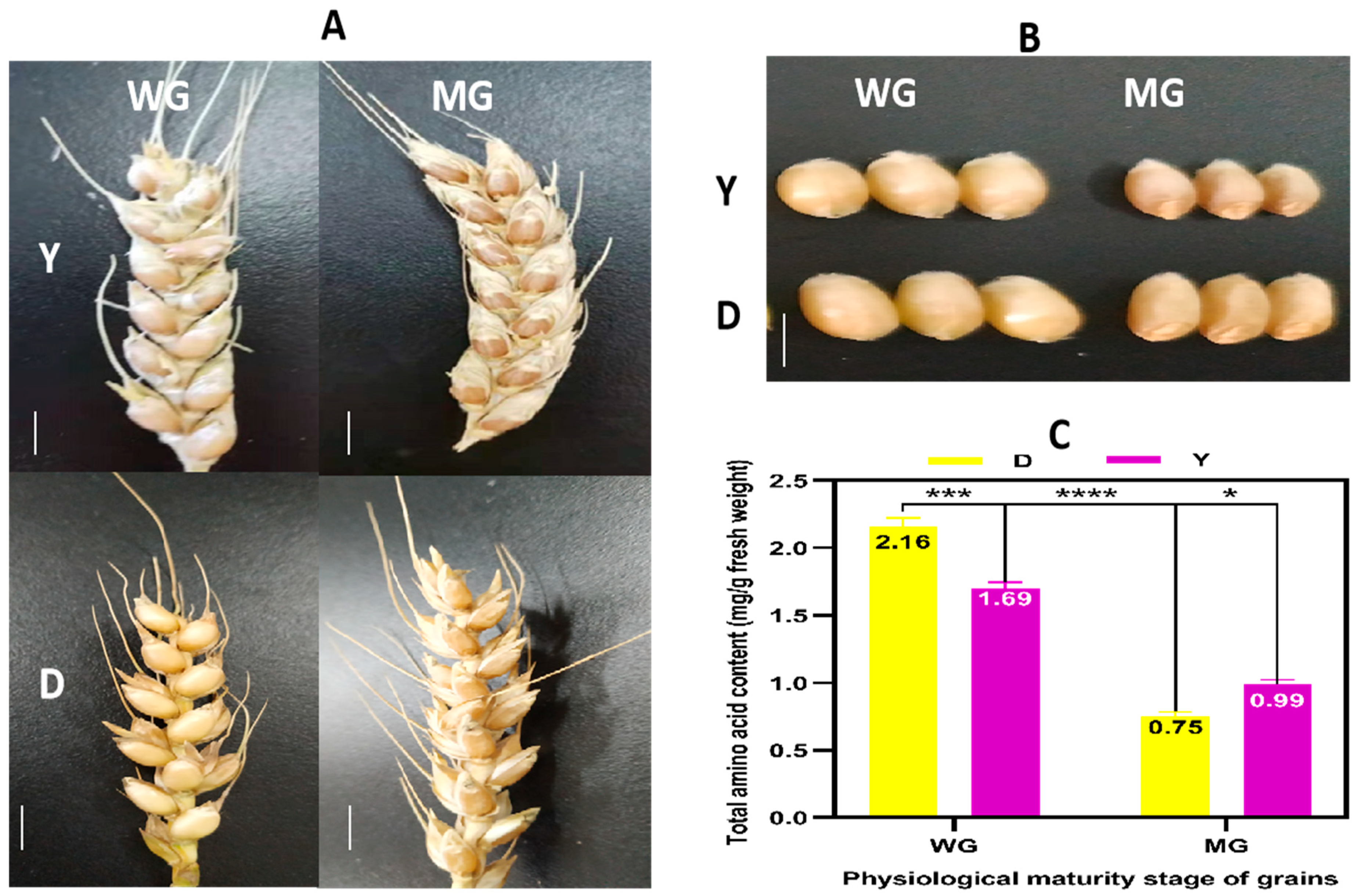
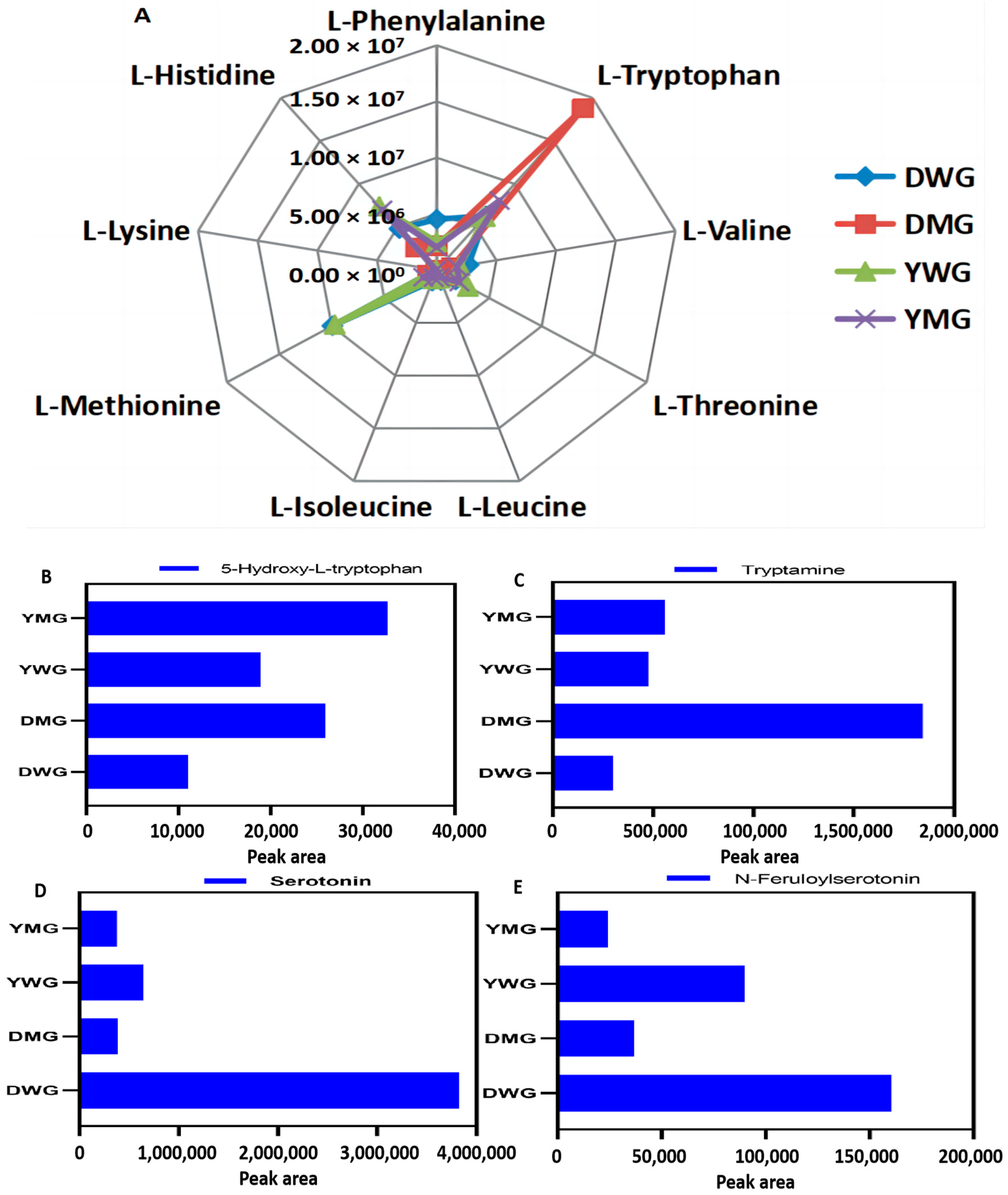
2.2. Phenotype and Total Amino Acid Contents of D and Y
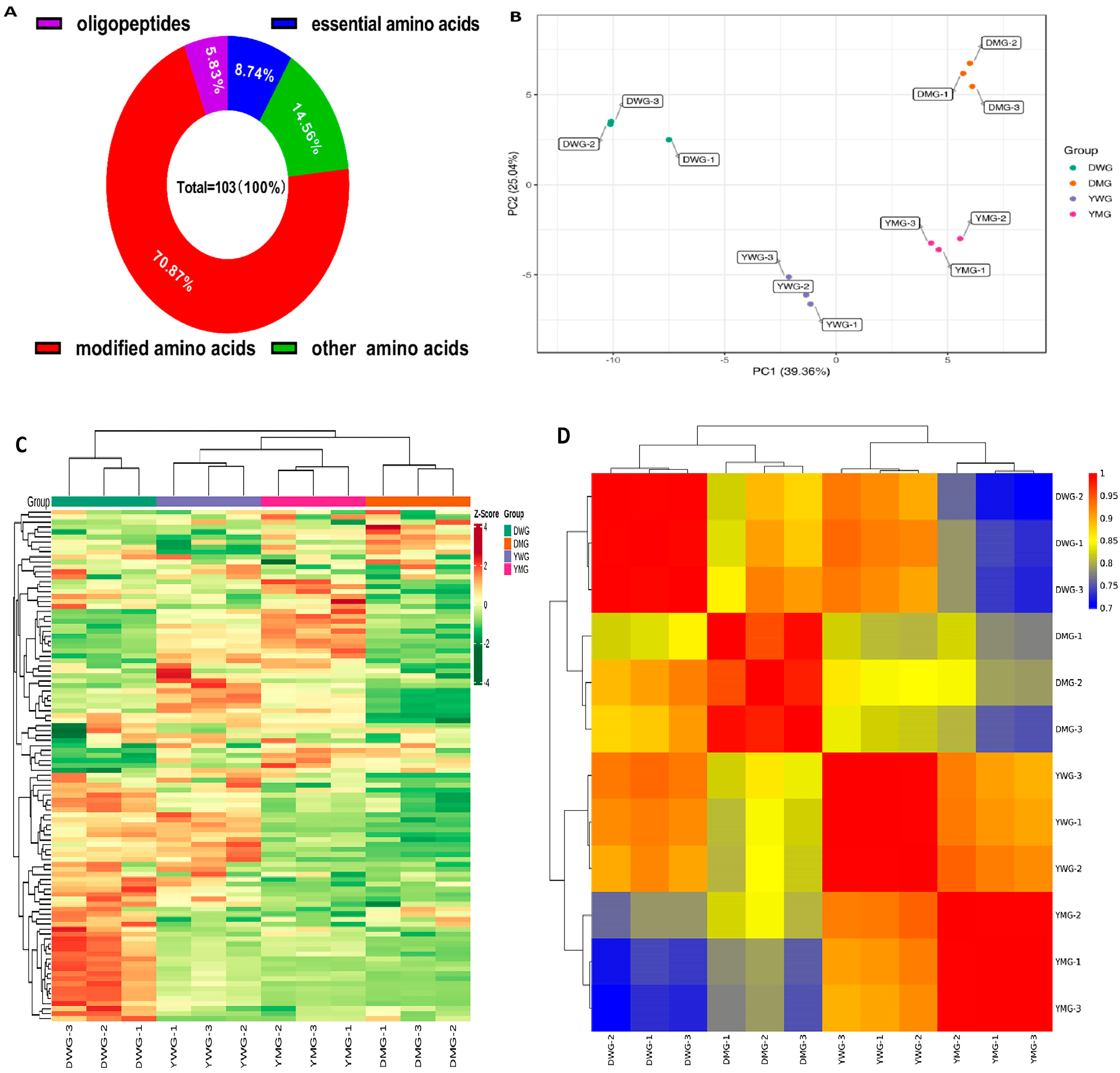
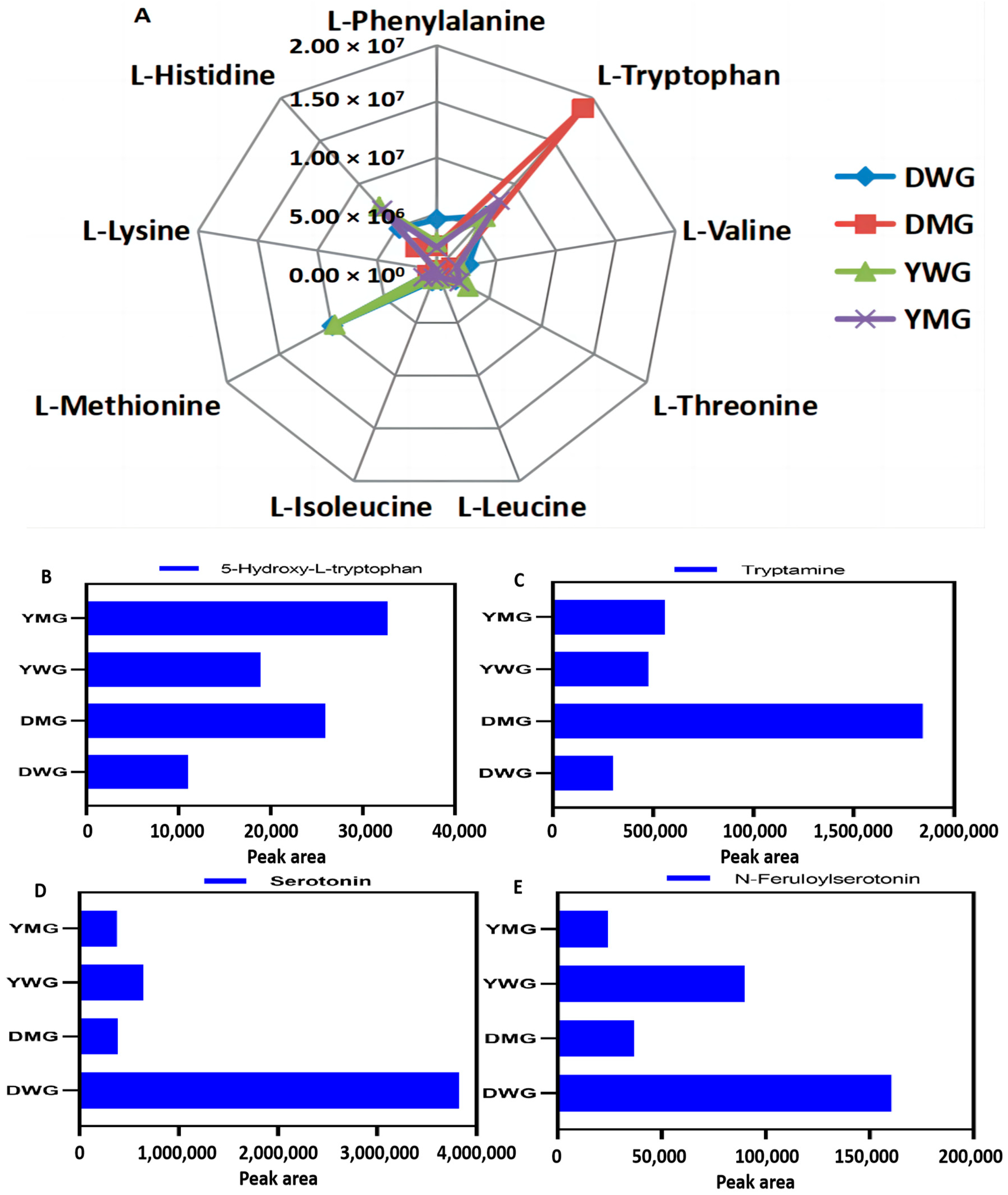
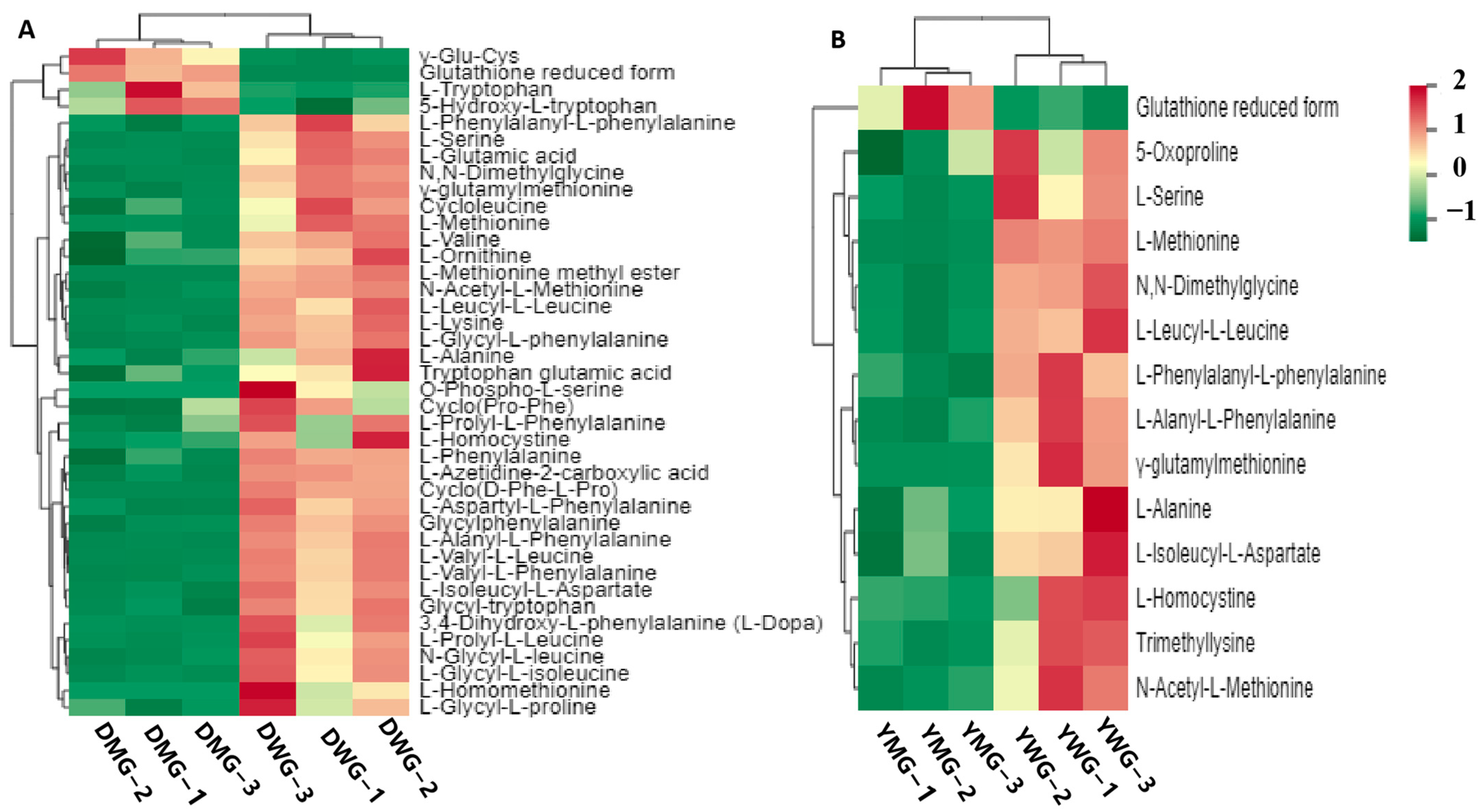
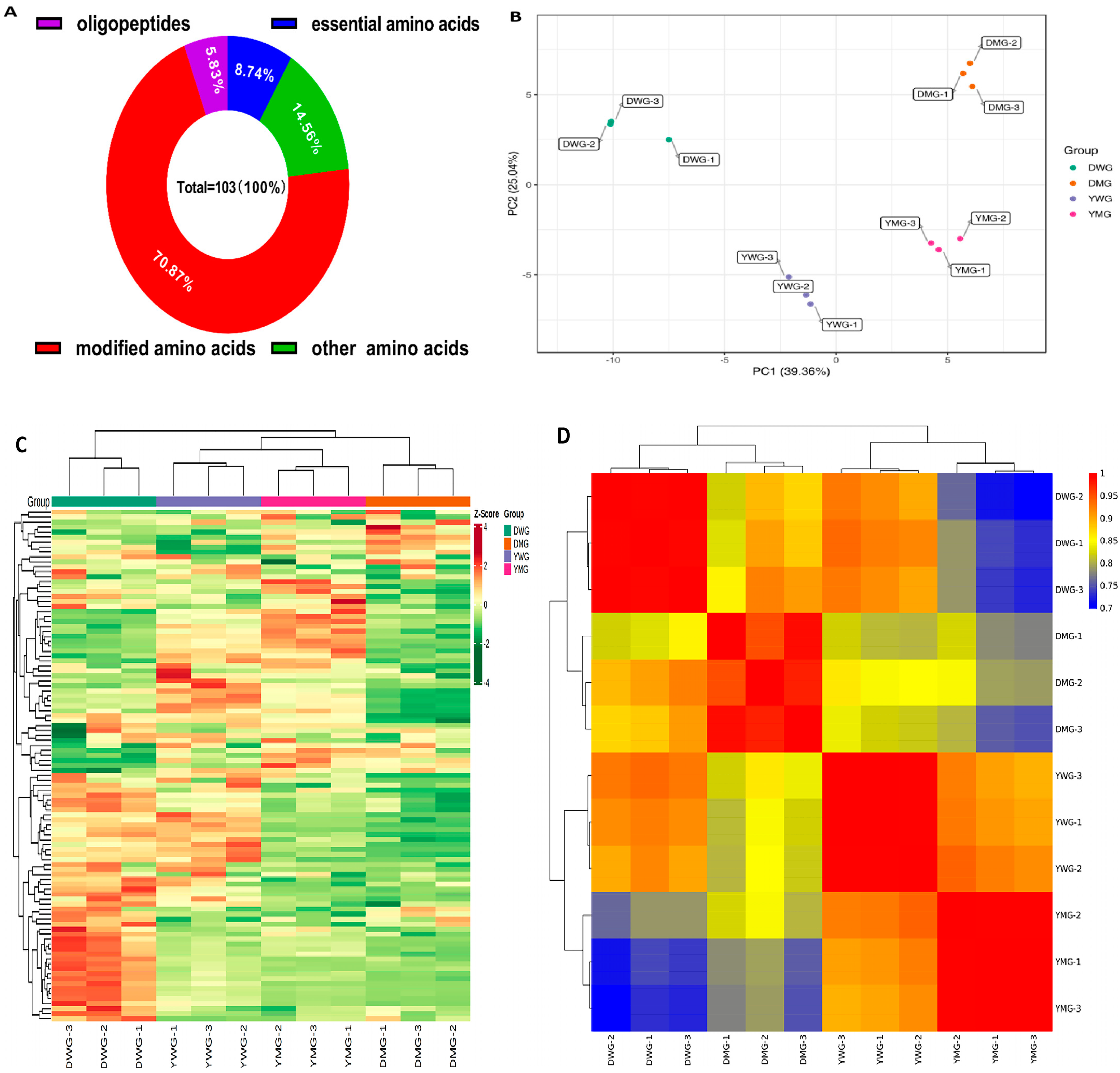
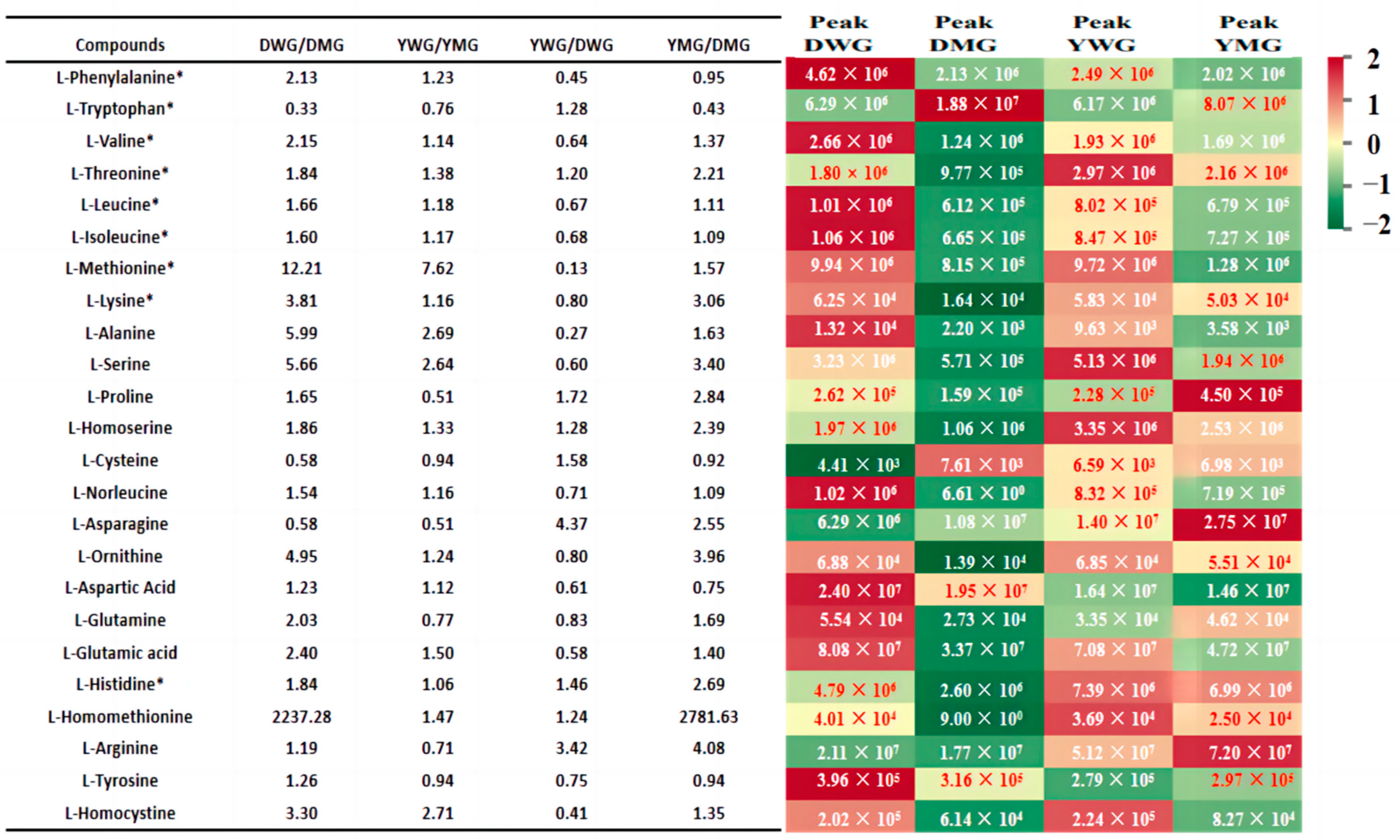
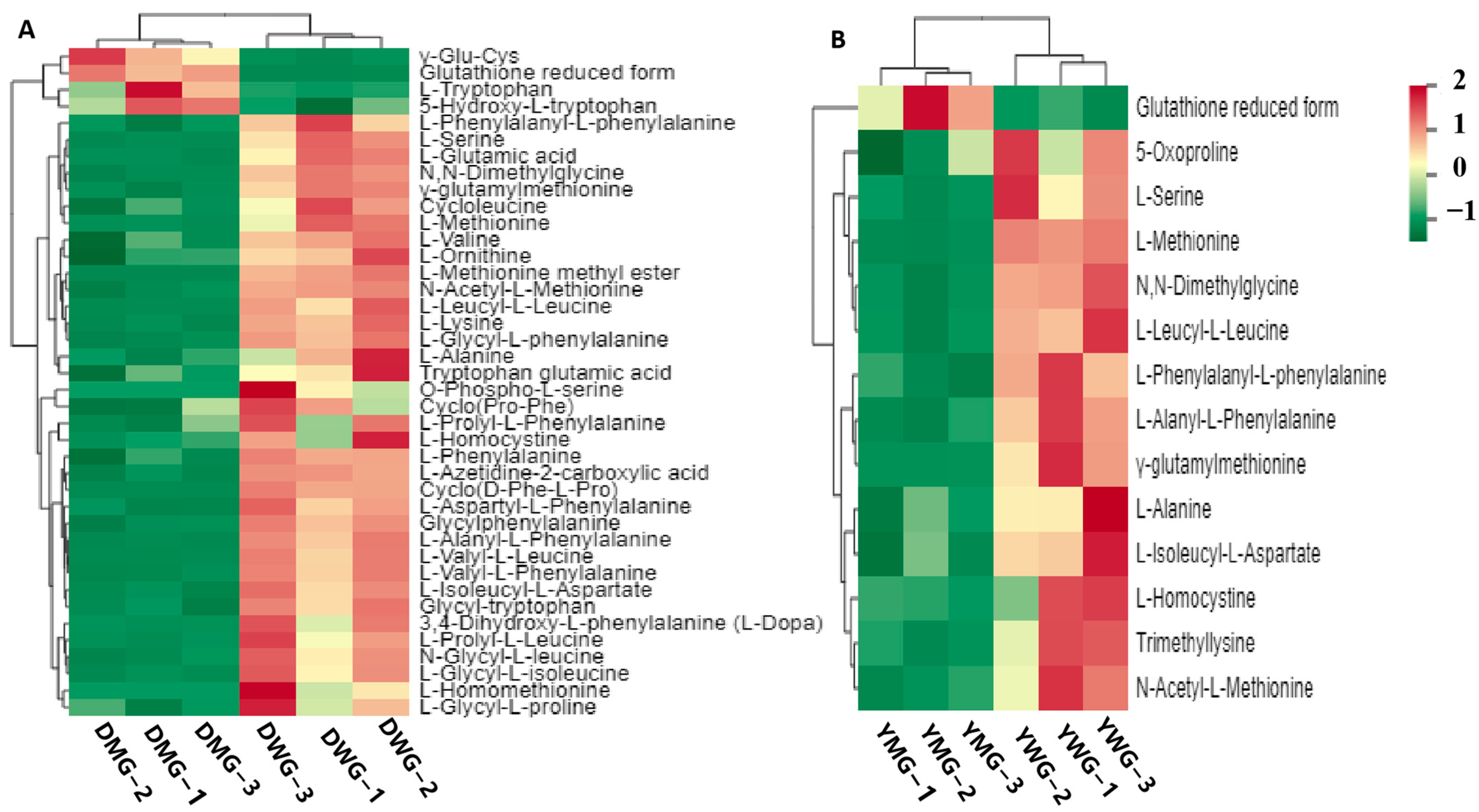
2.3. Metabolome Analyses of AAs and Derivatives in WG and MG of Y and D
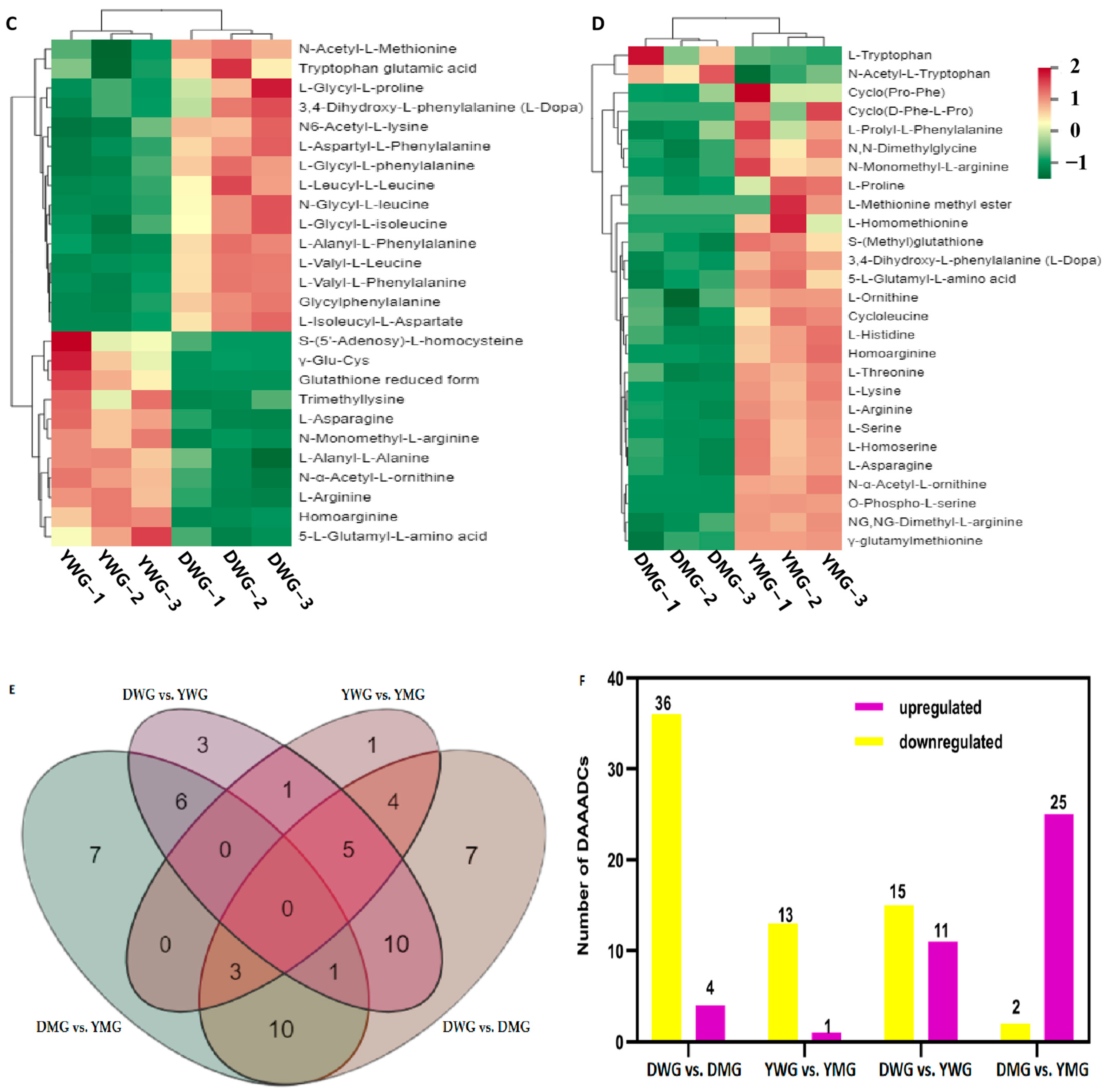
2.4. Changes in AADCs during Developmental Stages Differed between the Two Wheat Lines
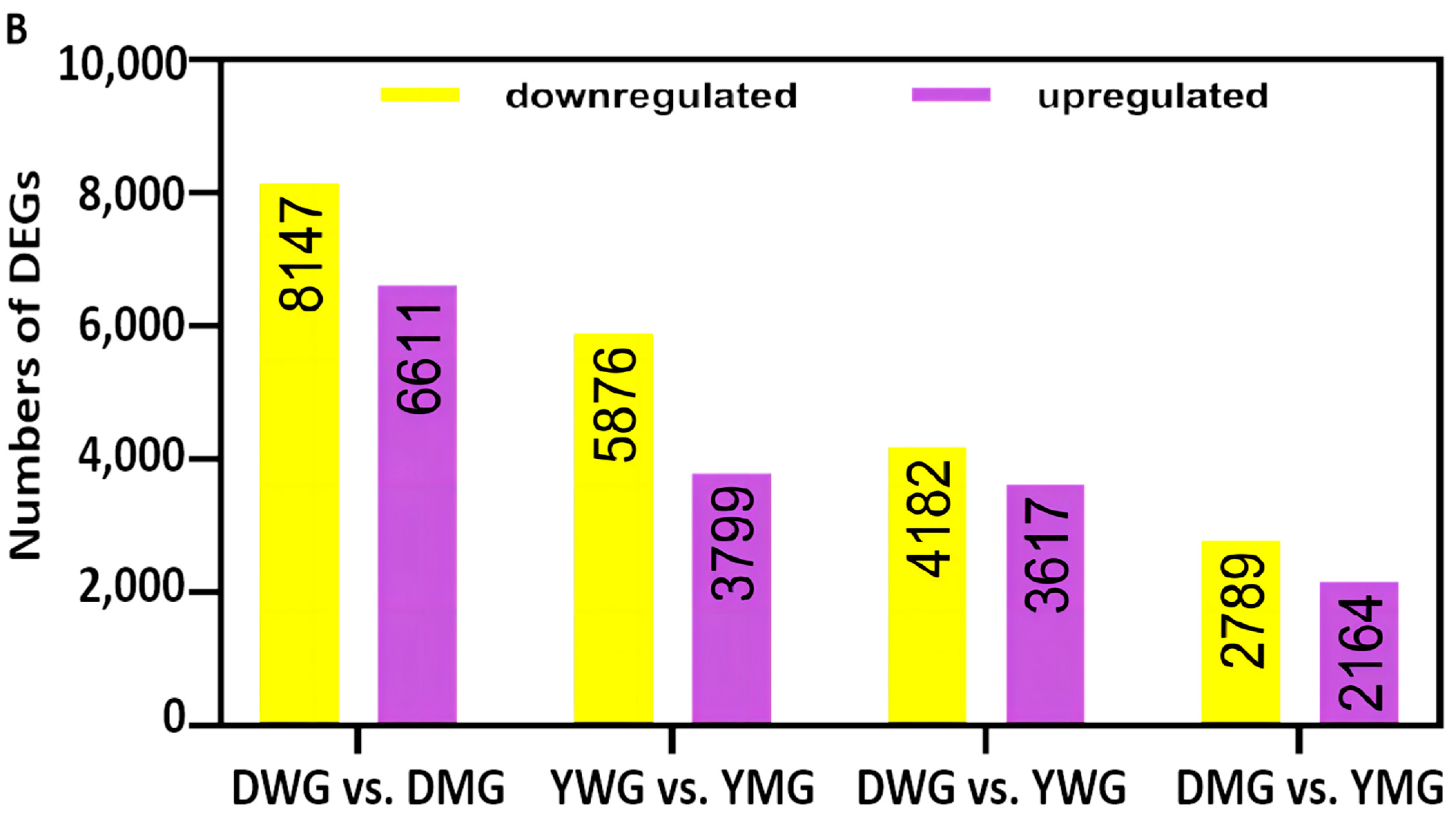
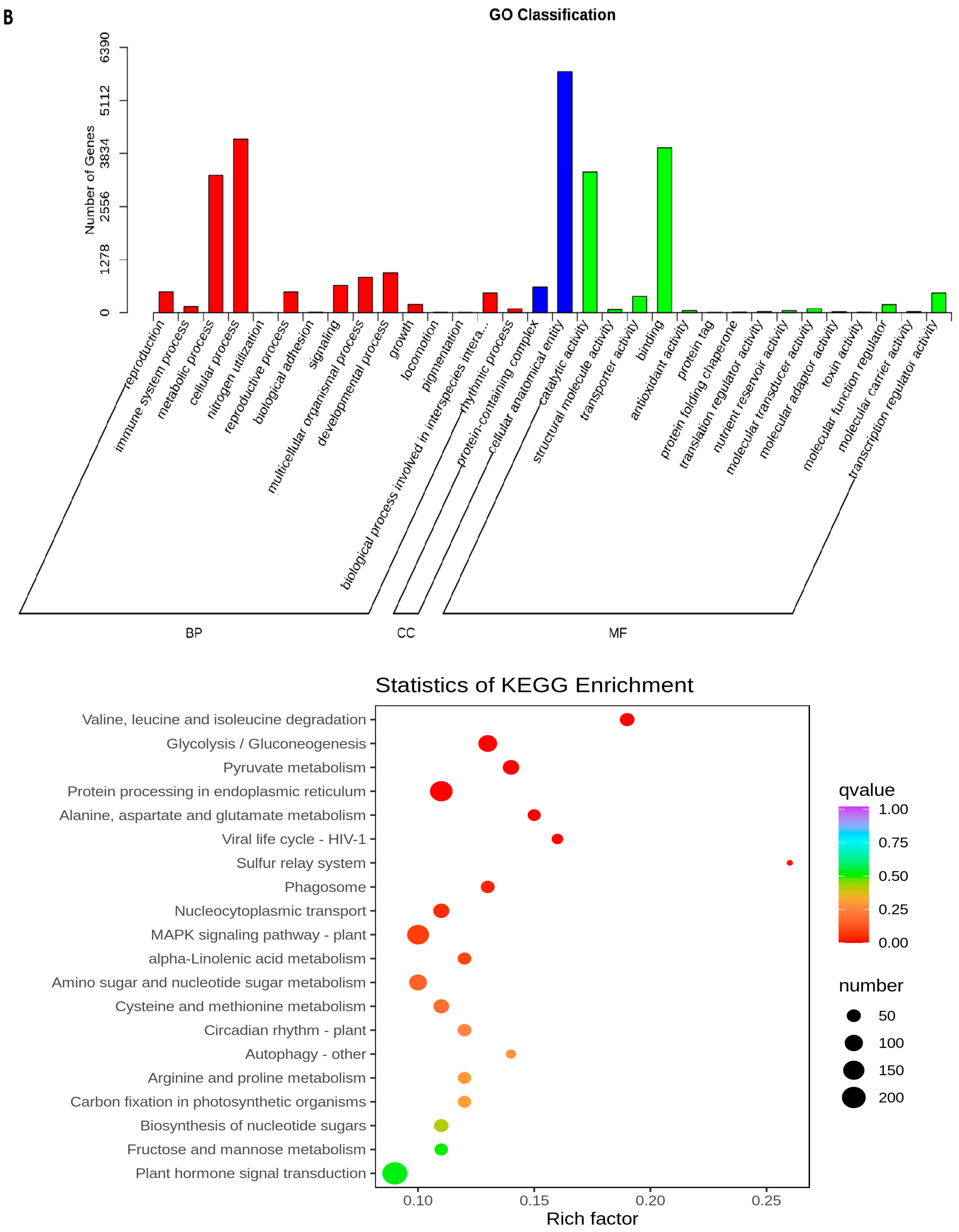
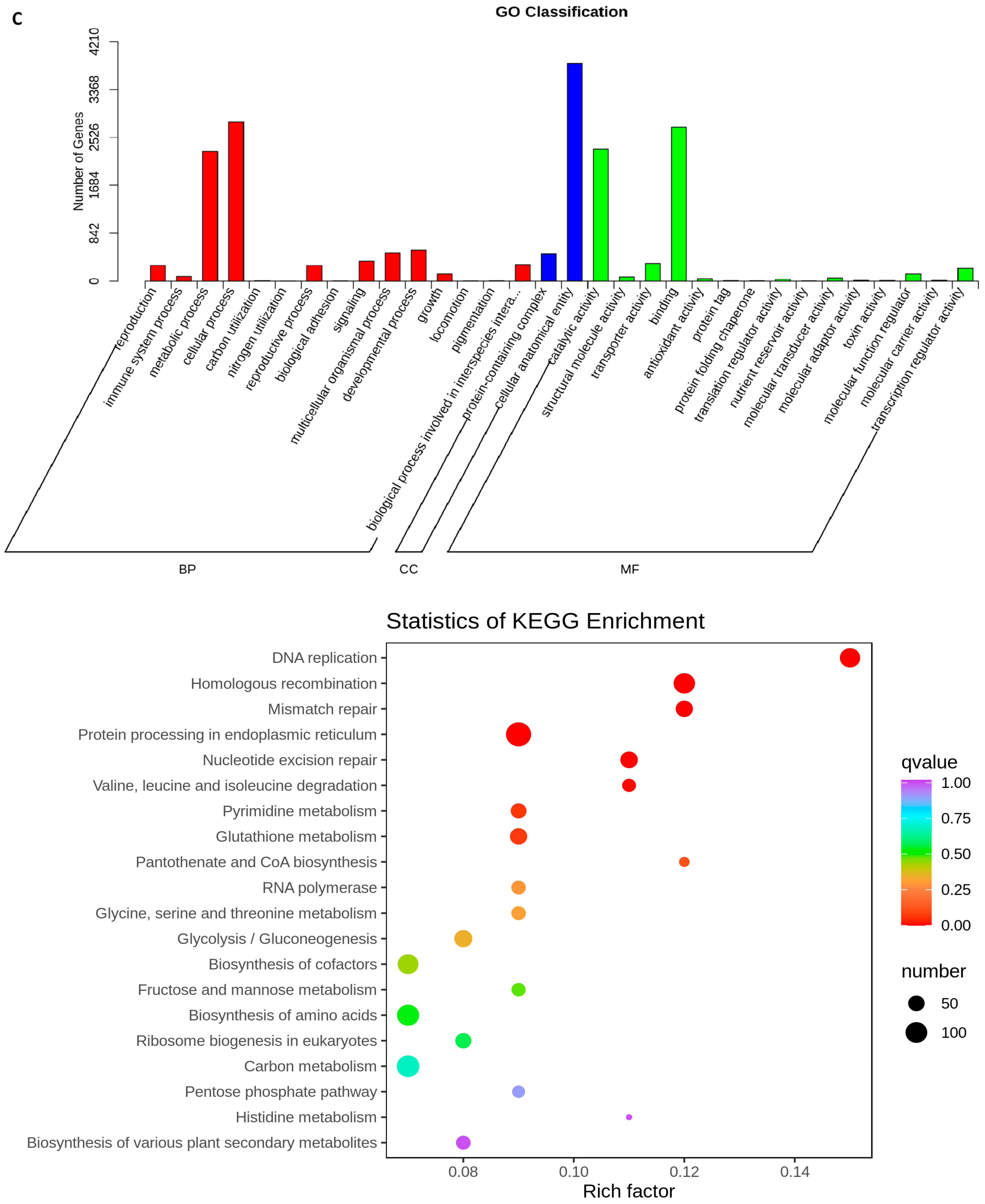
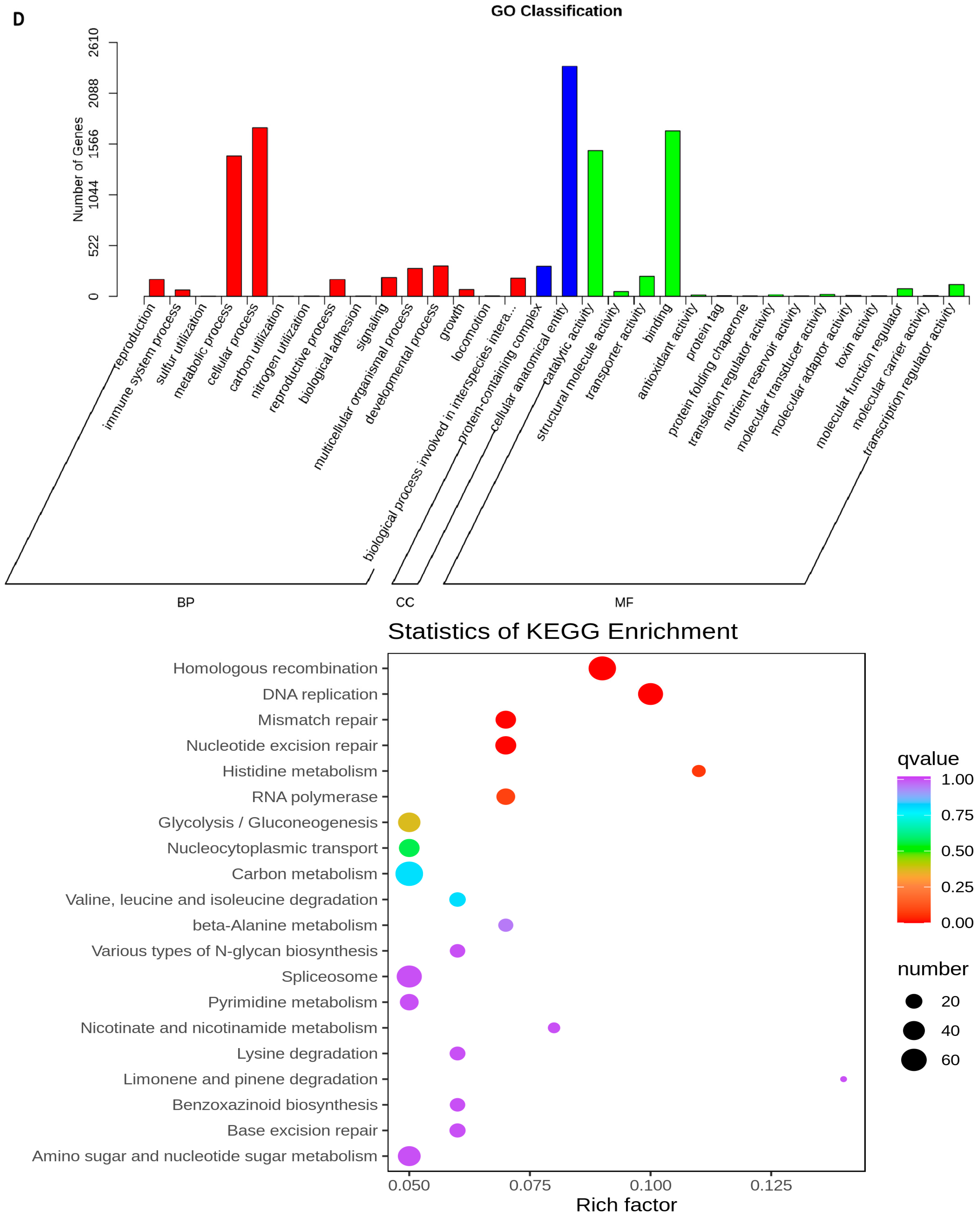
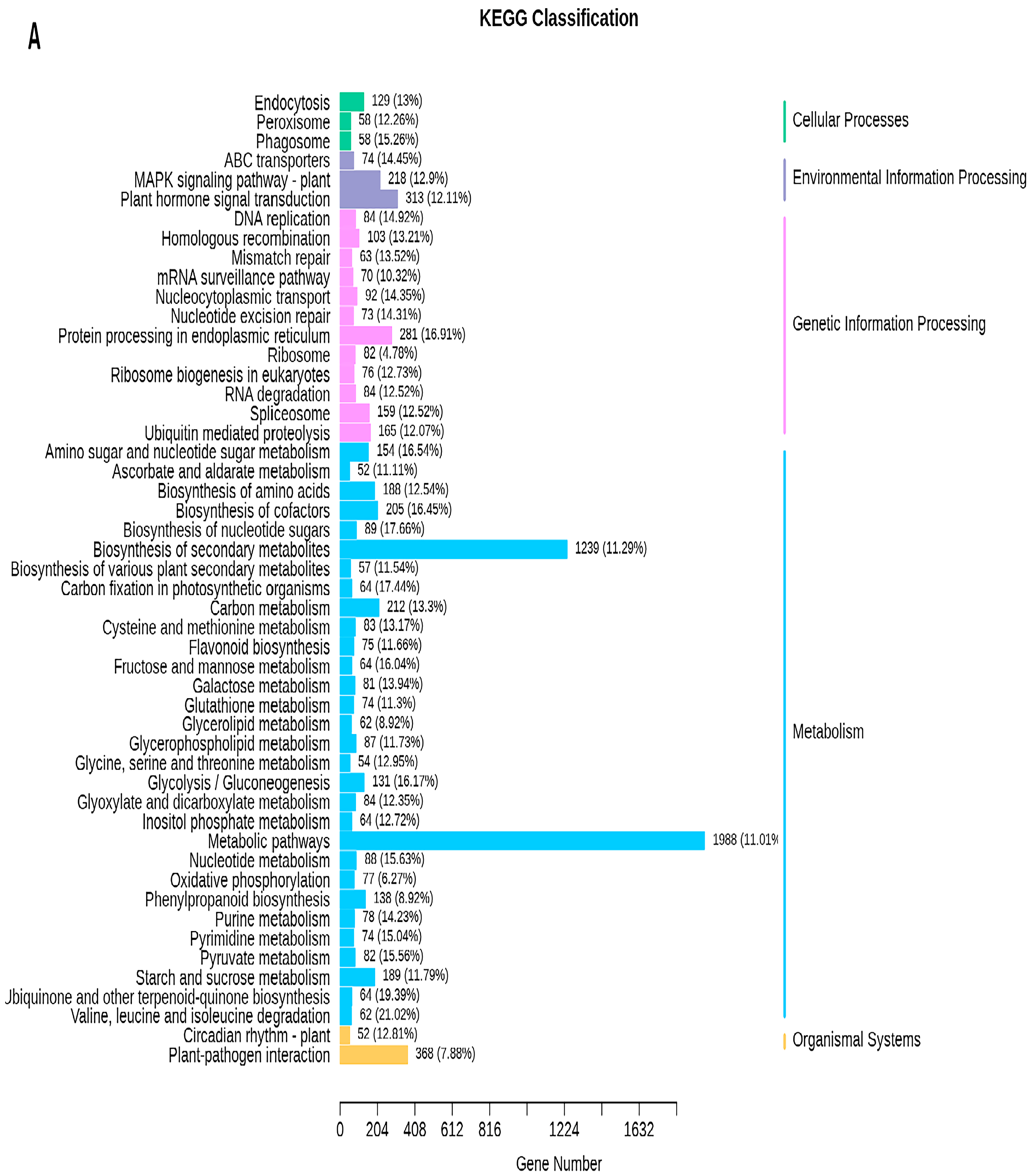
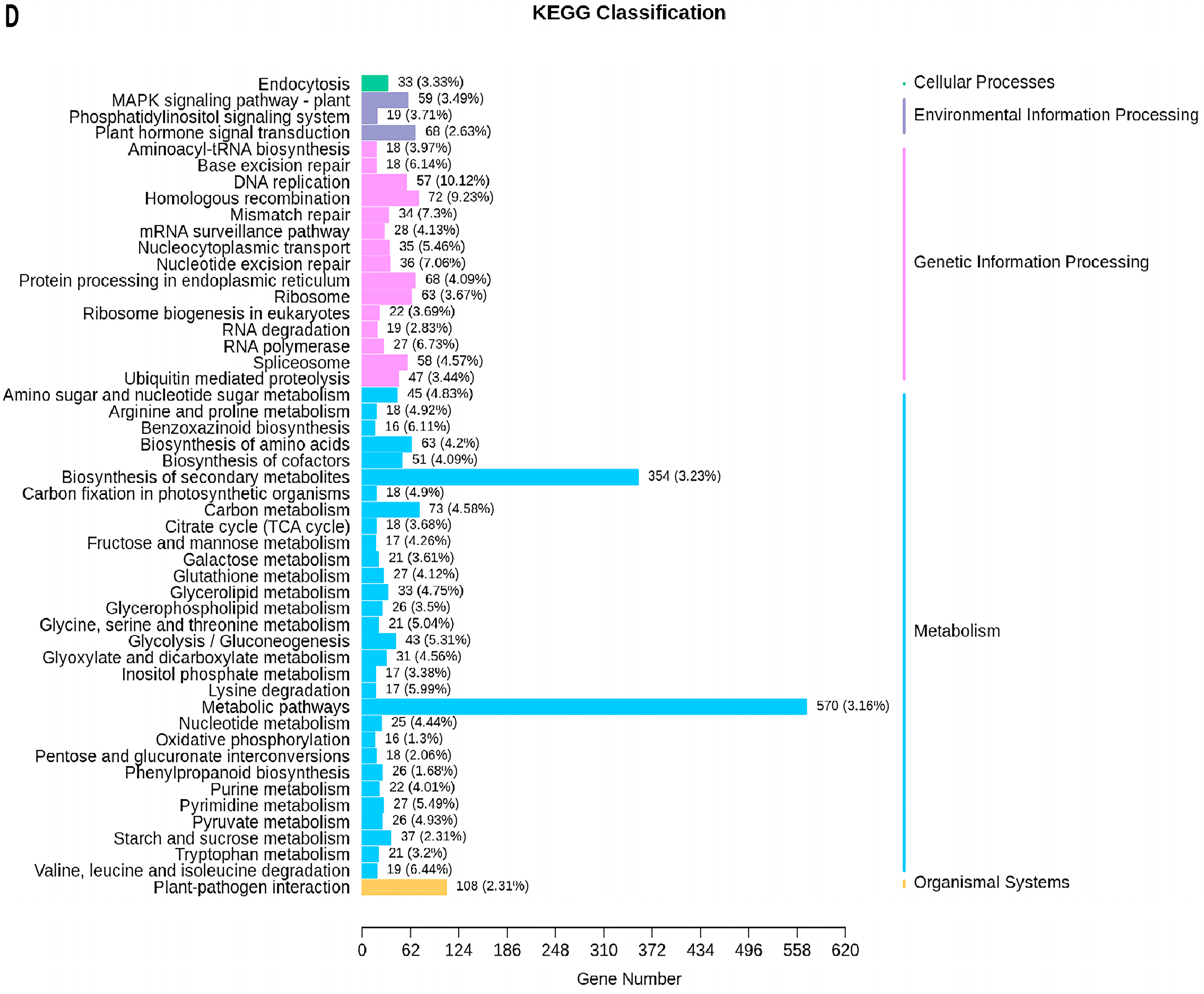
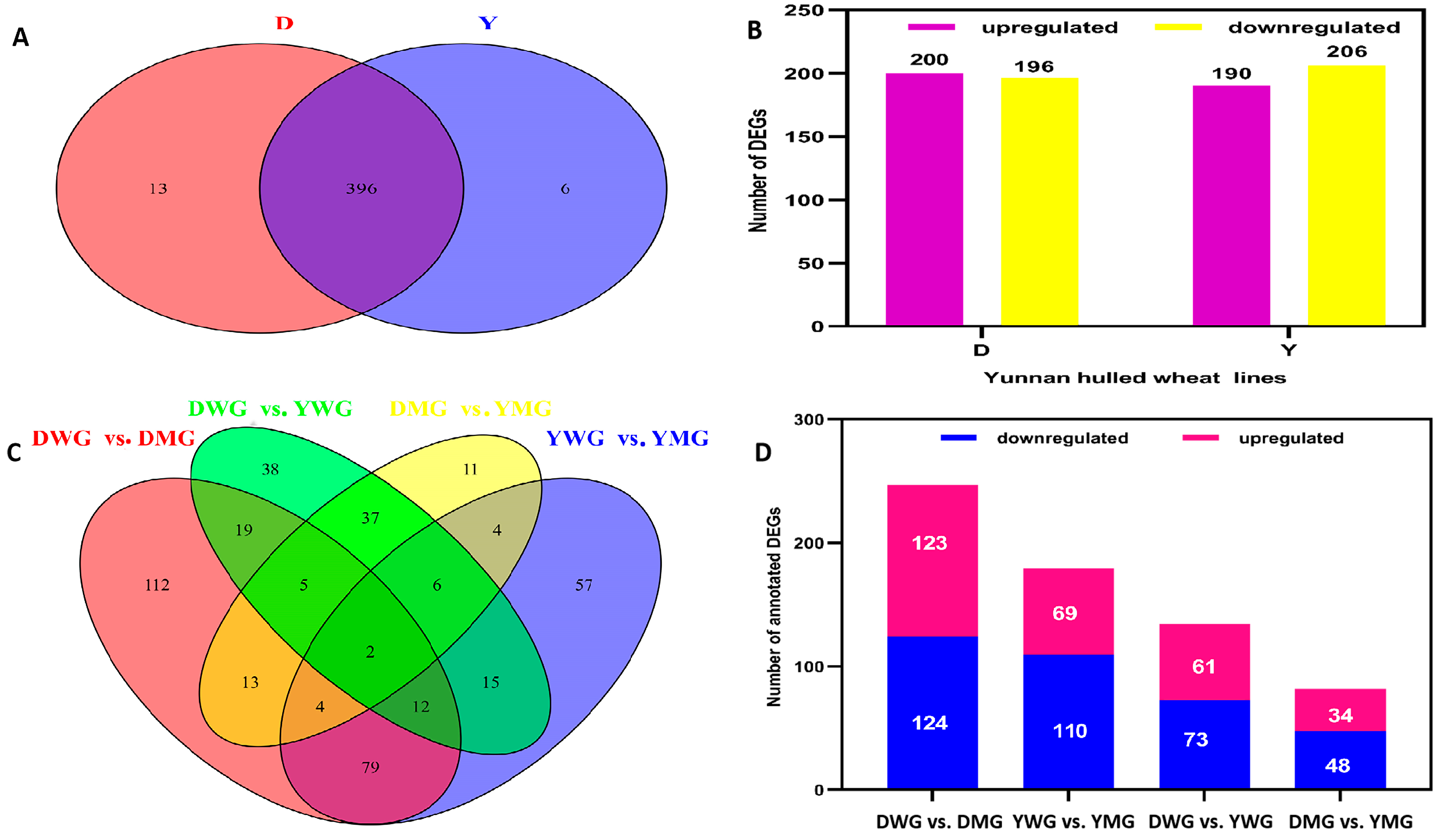
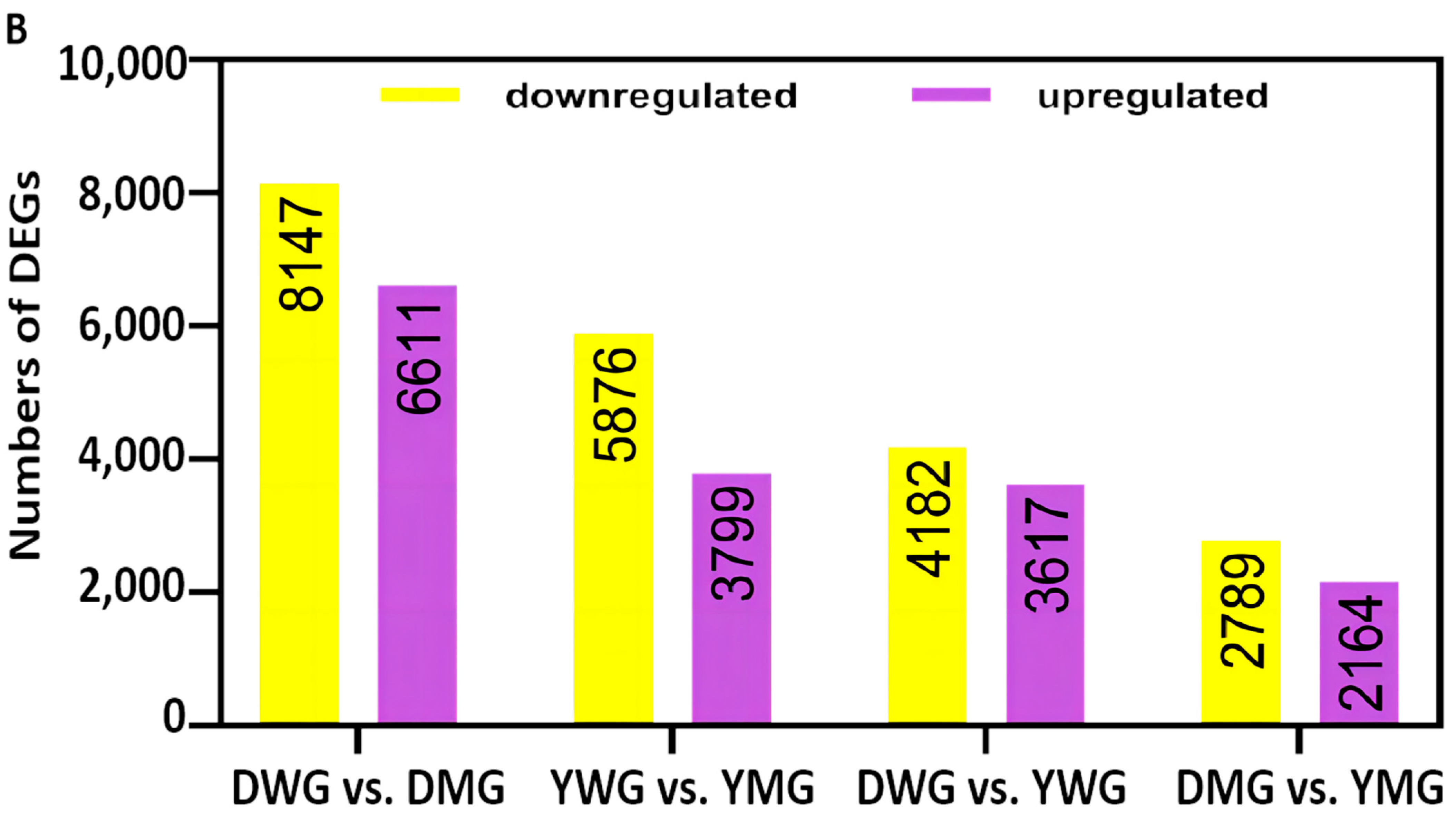
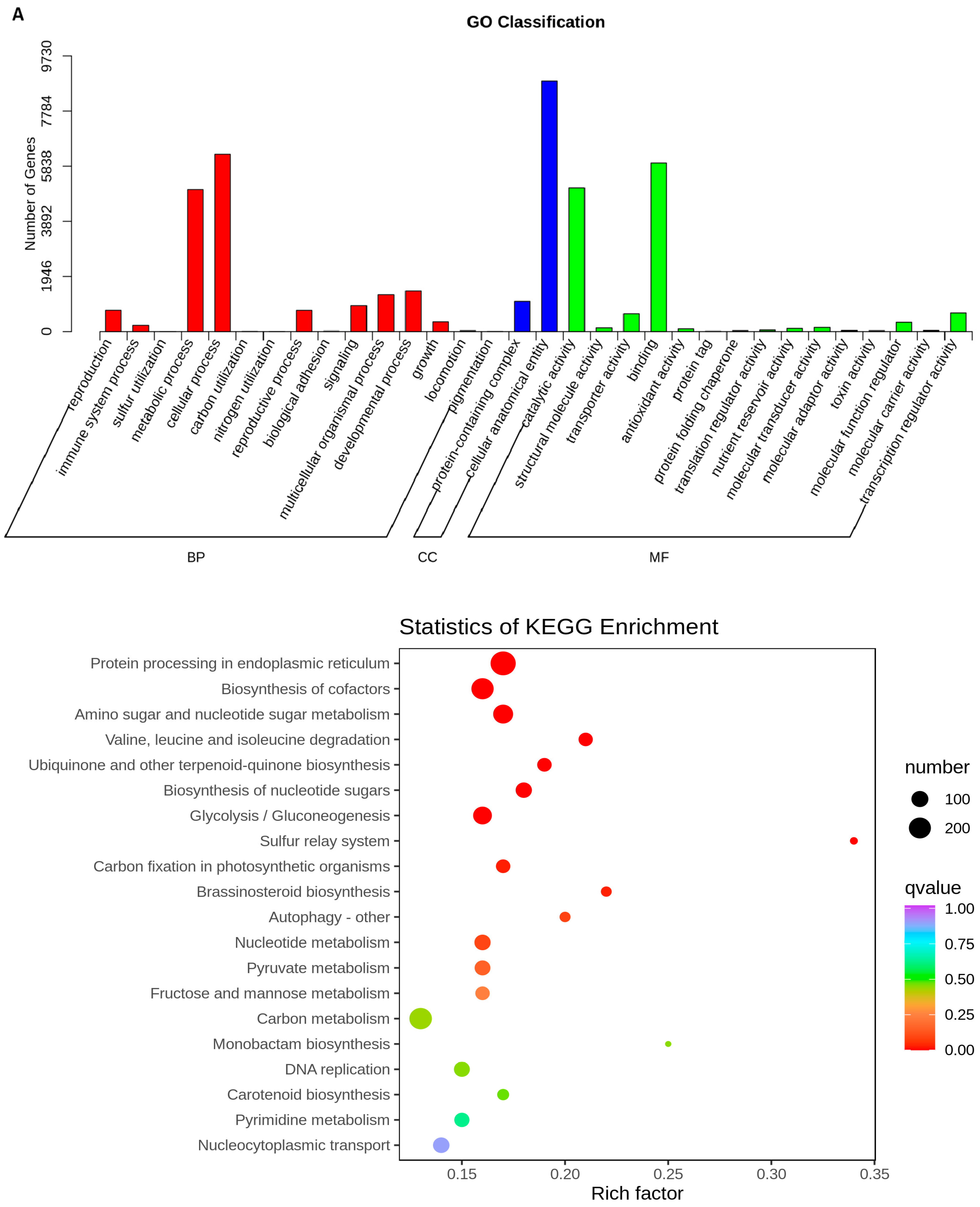
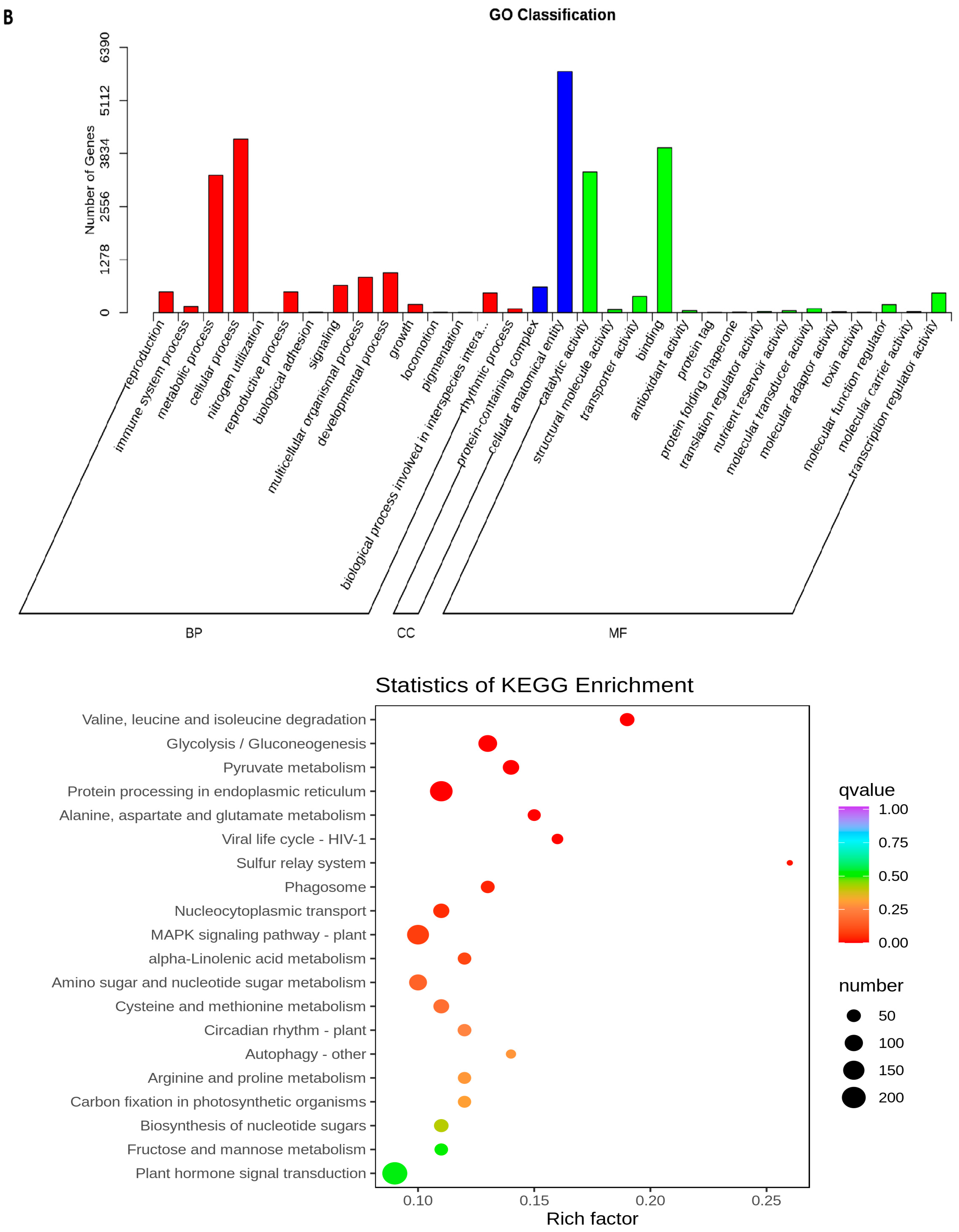
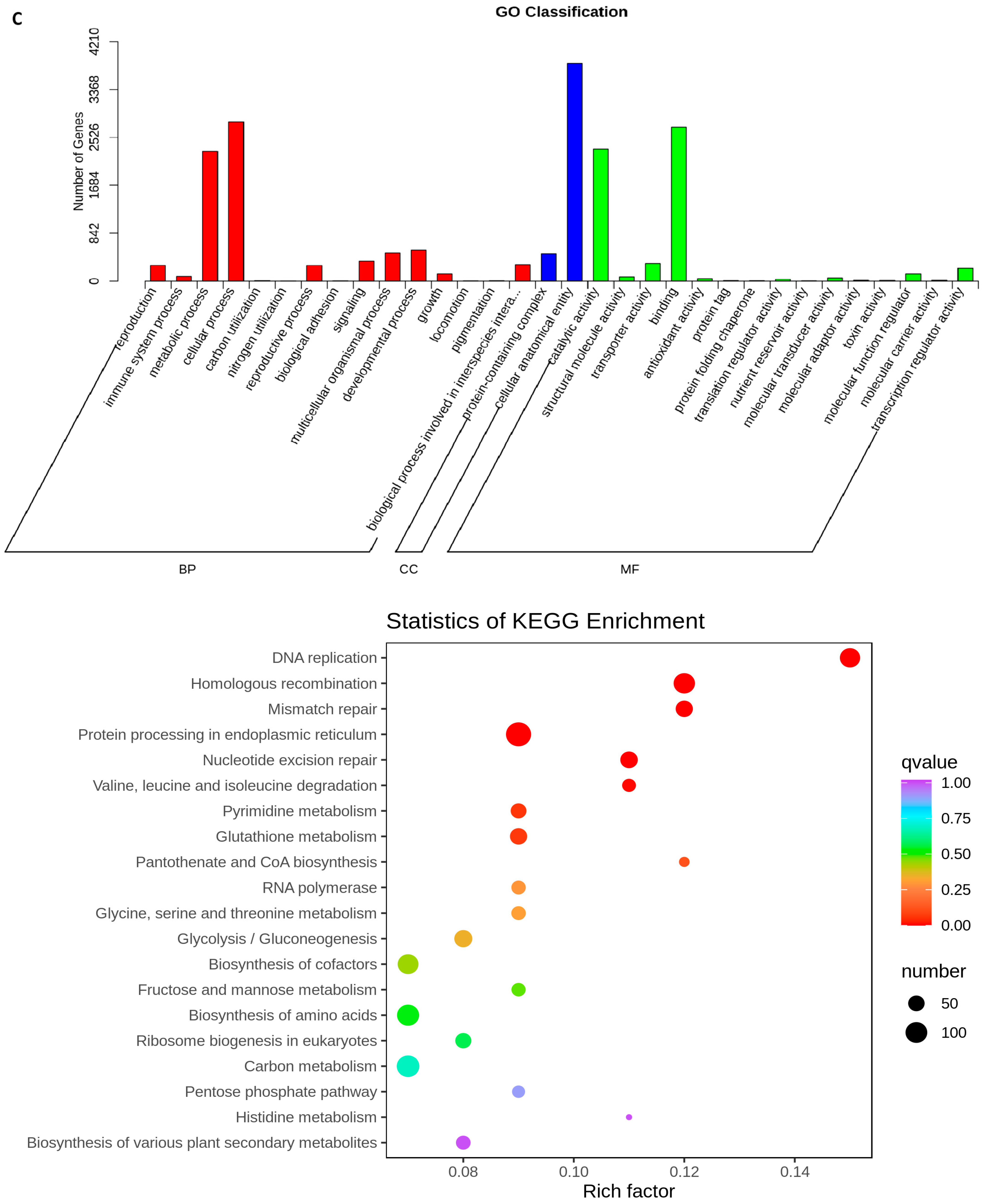
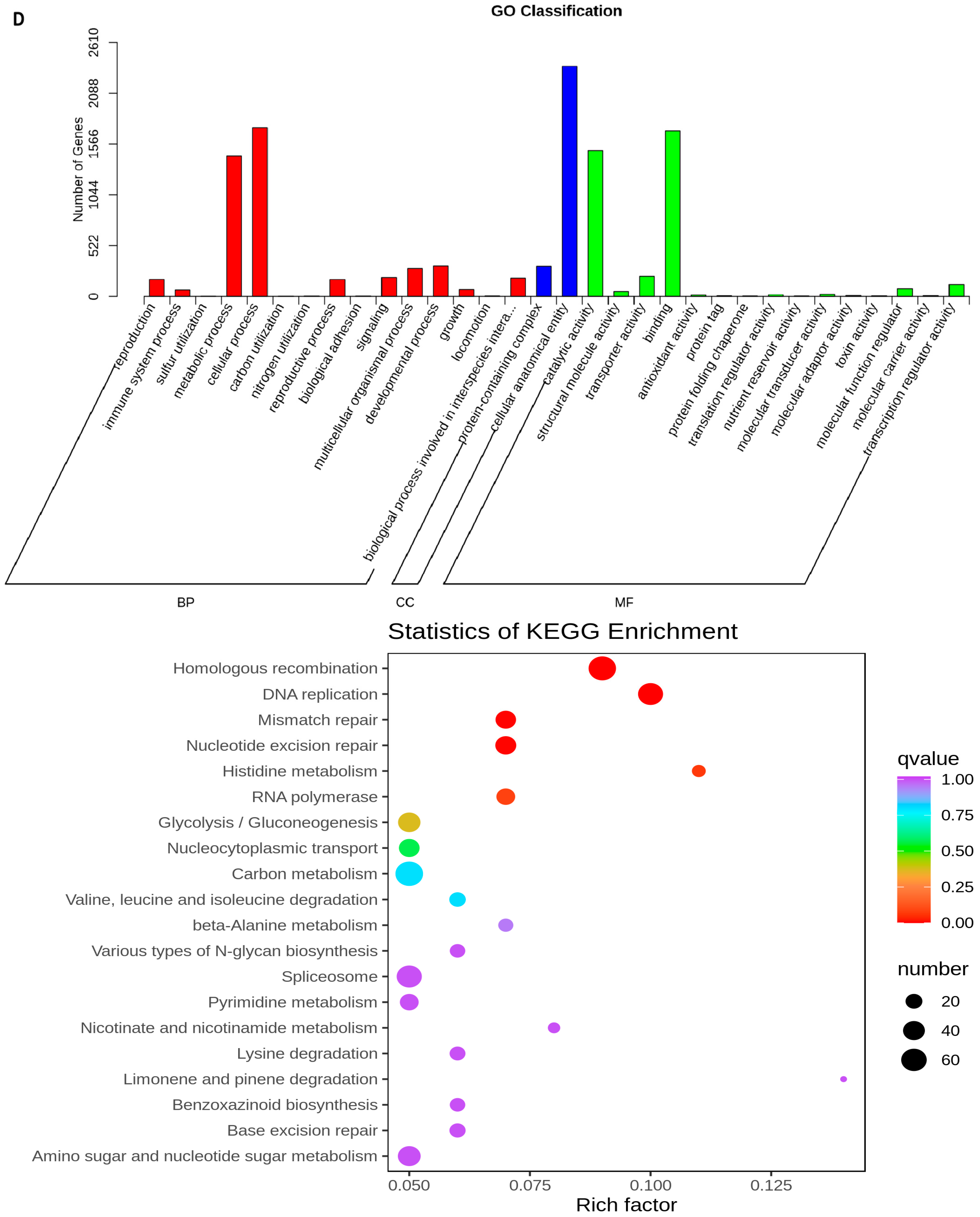
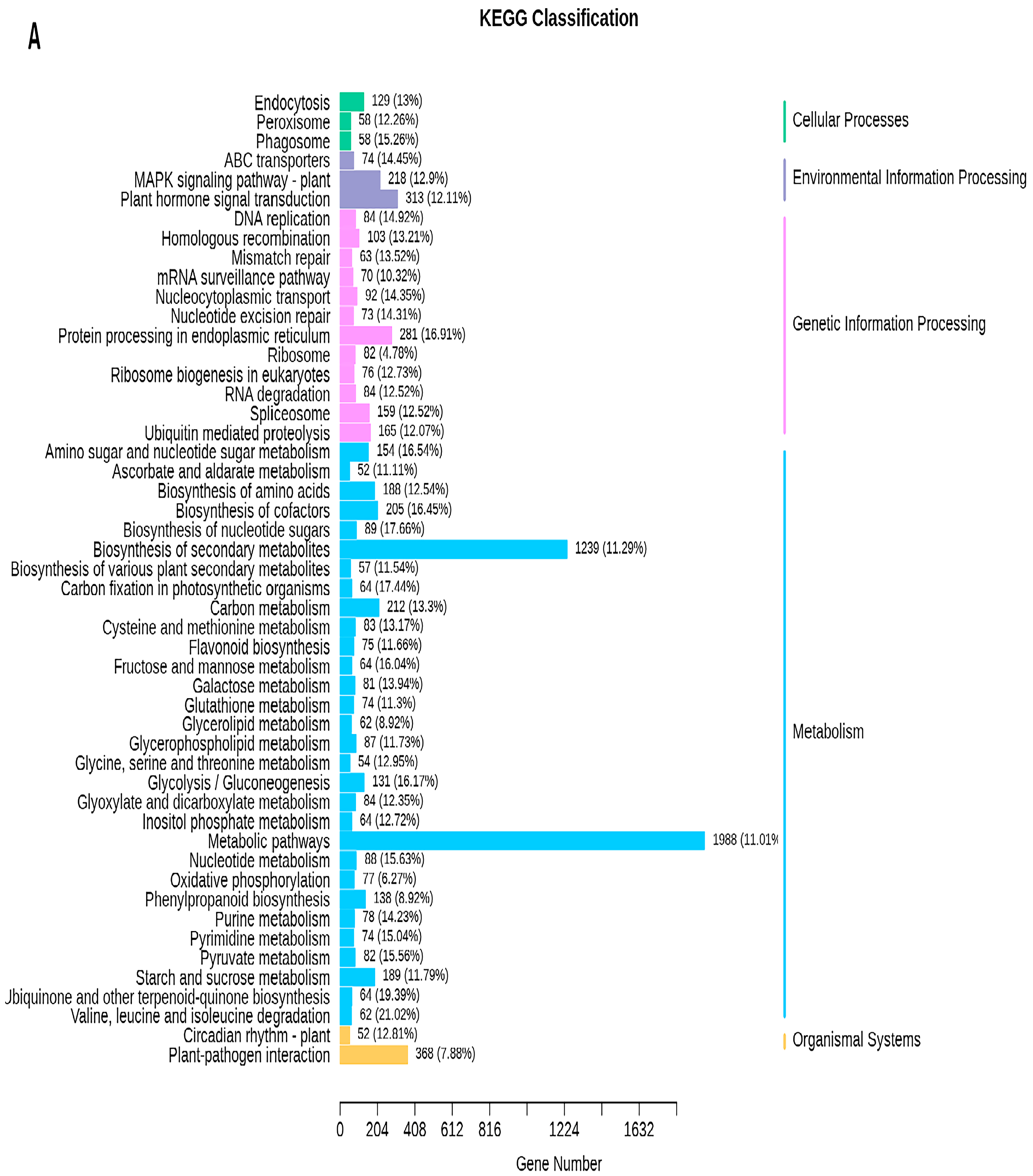
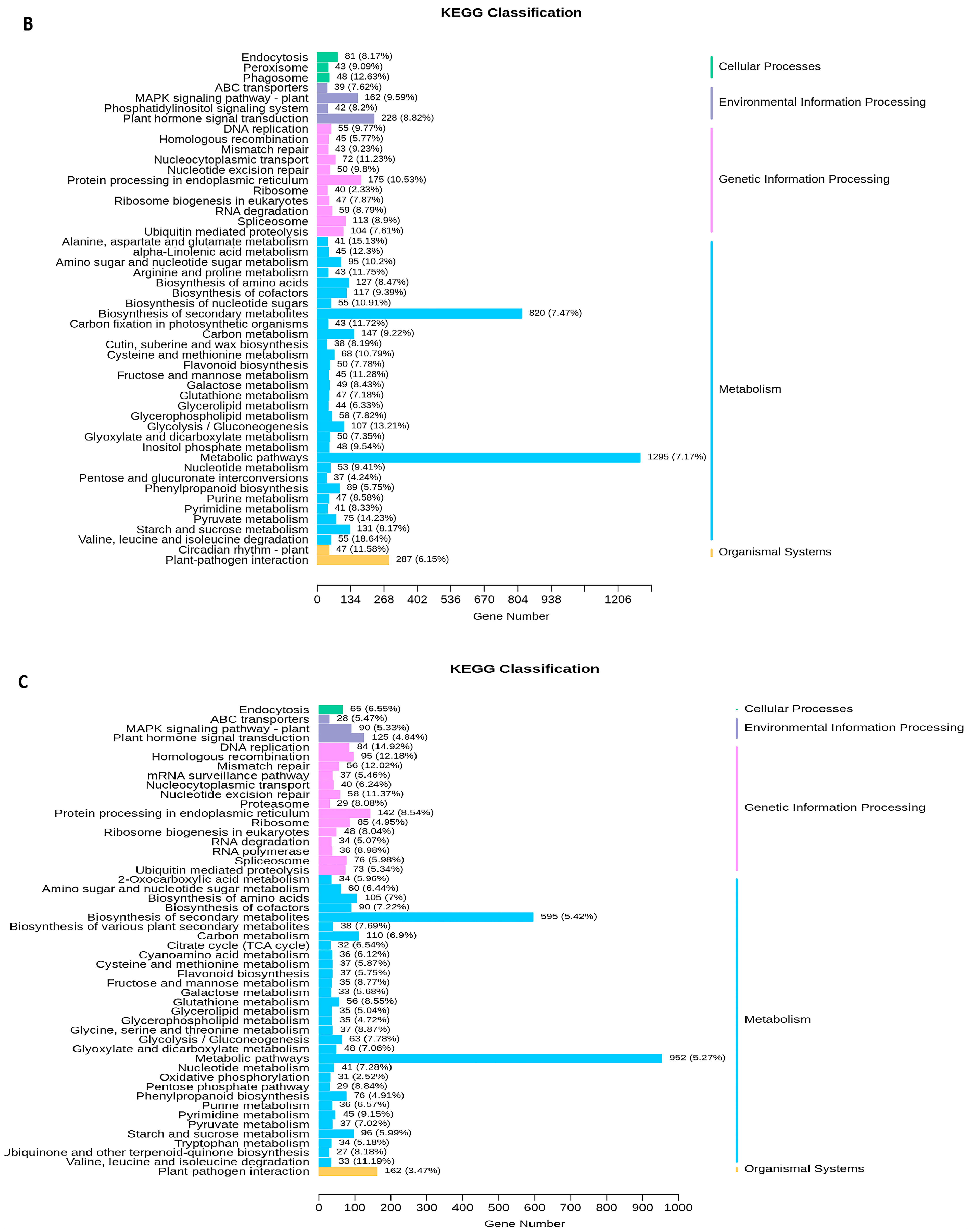
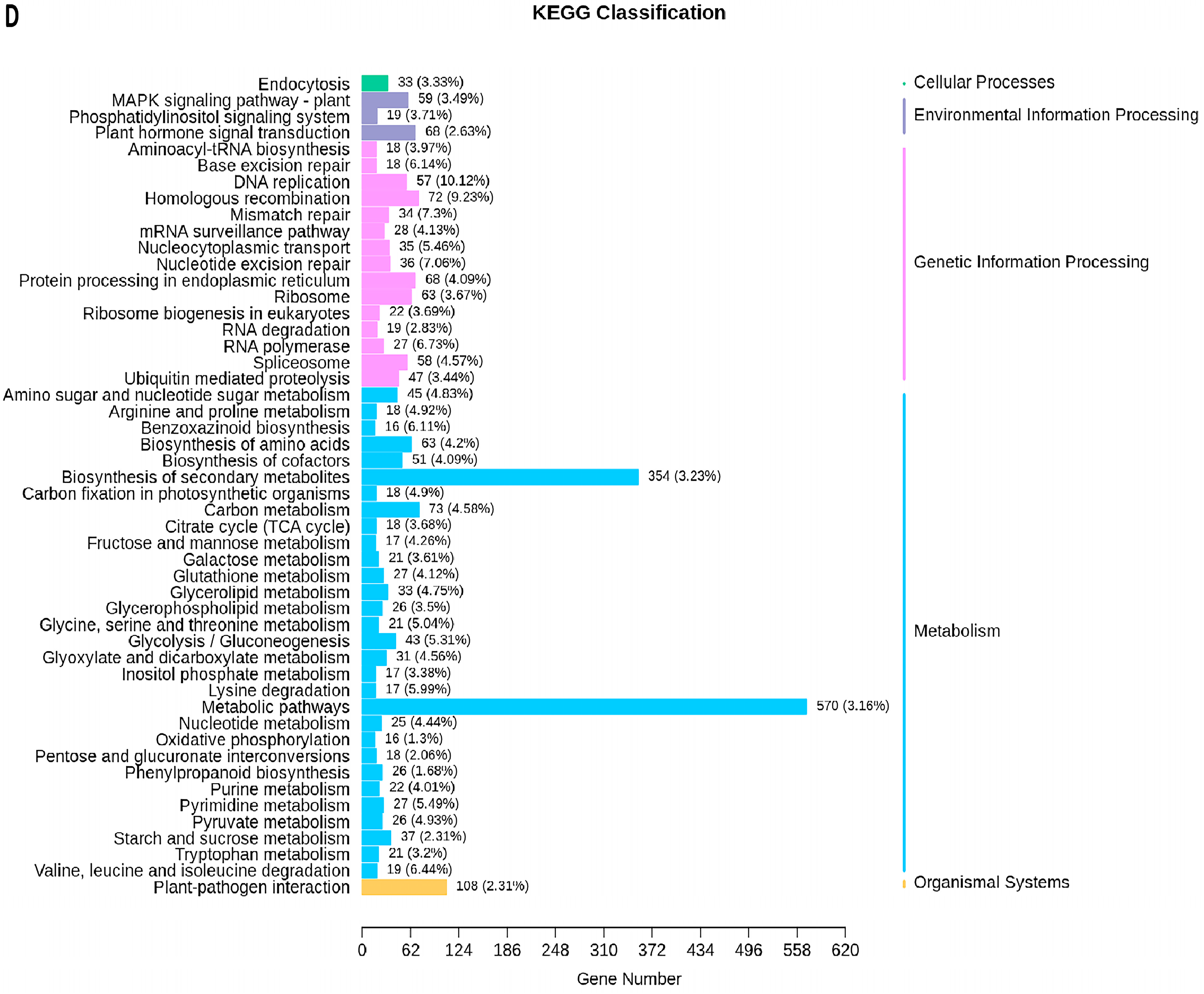
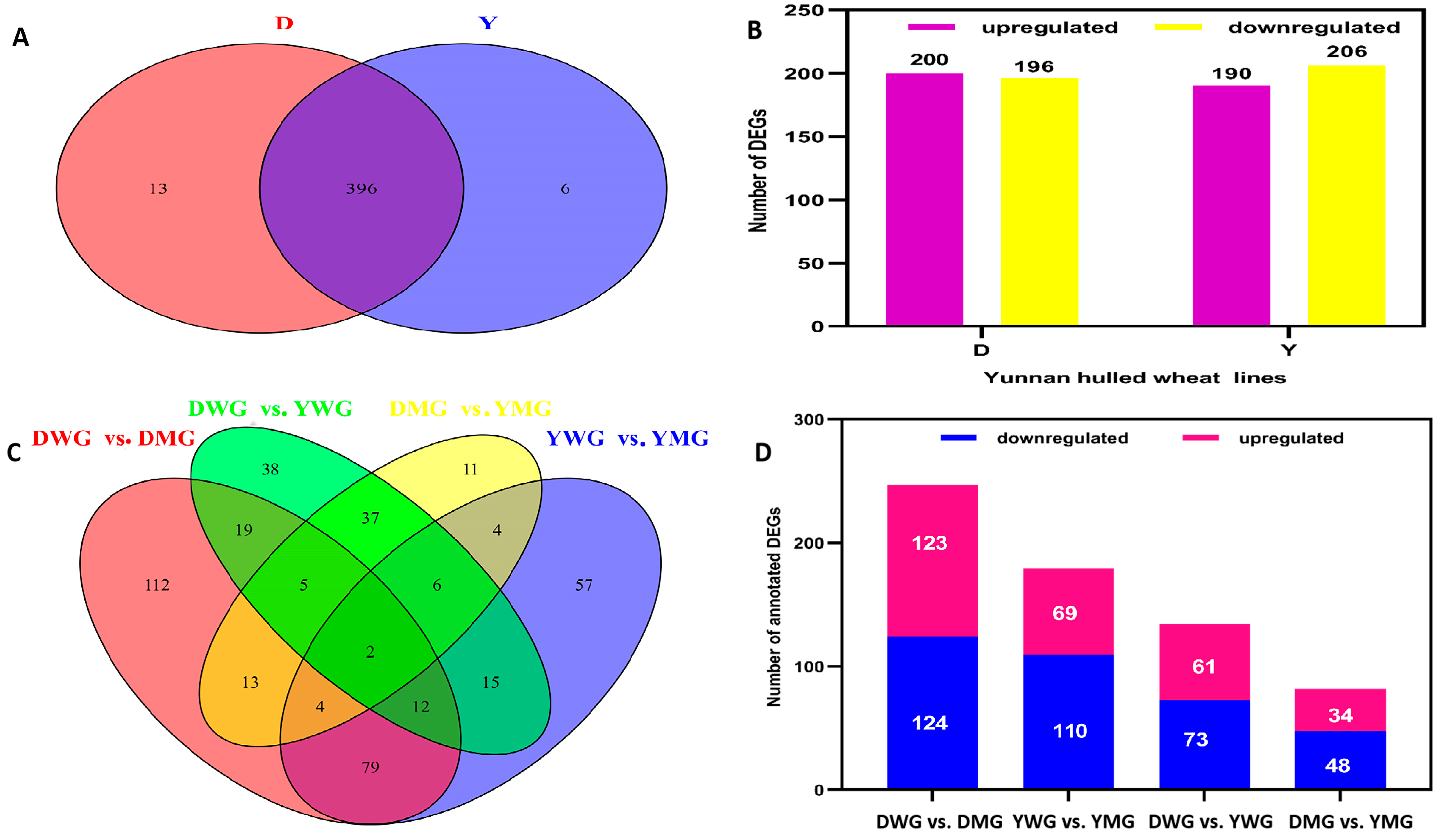
2.5. Transcriptome Analyses of the Two Wheat Lines at Different Developmental Stages
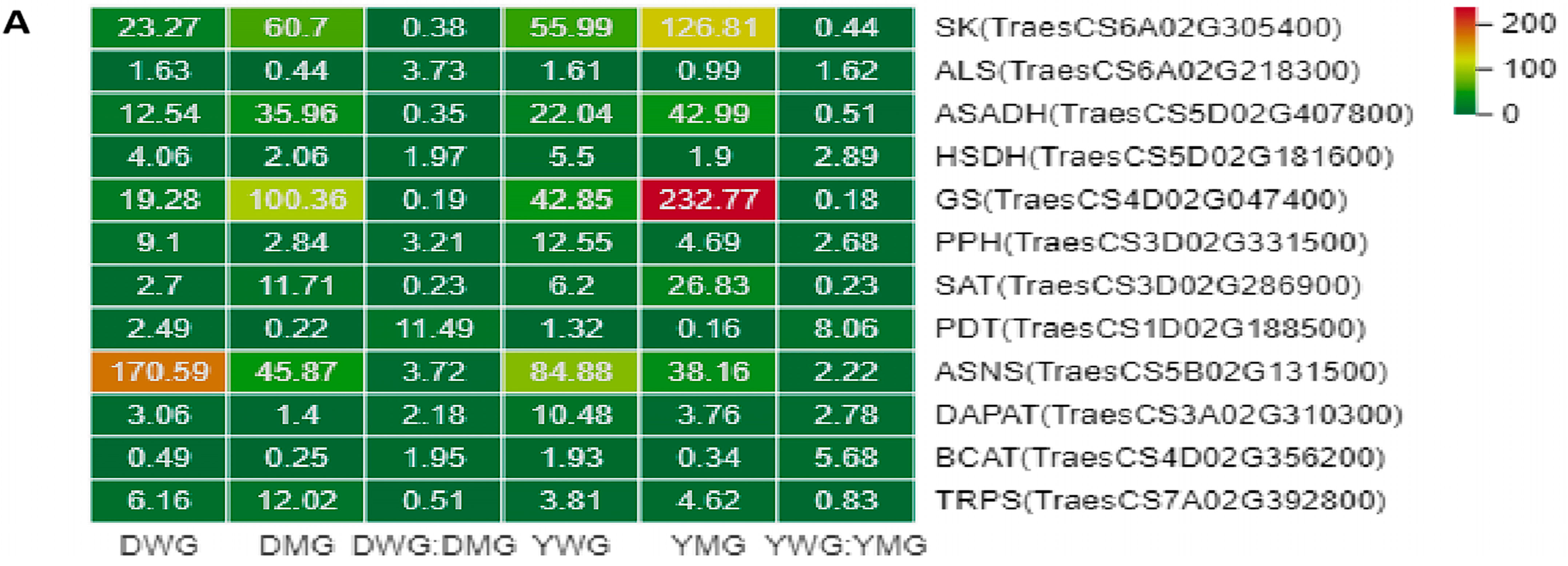
2.6. Differential Expression of AA Biosynthesis and Regulatory Genes between WG and MG
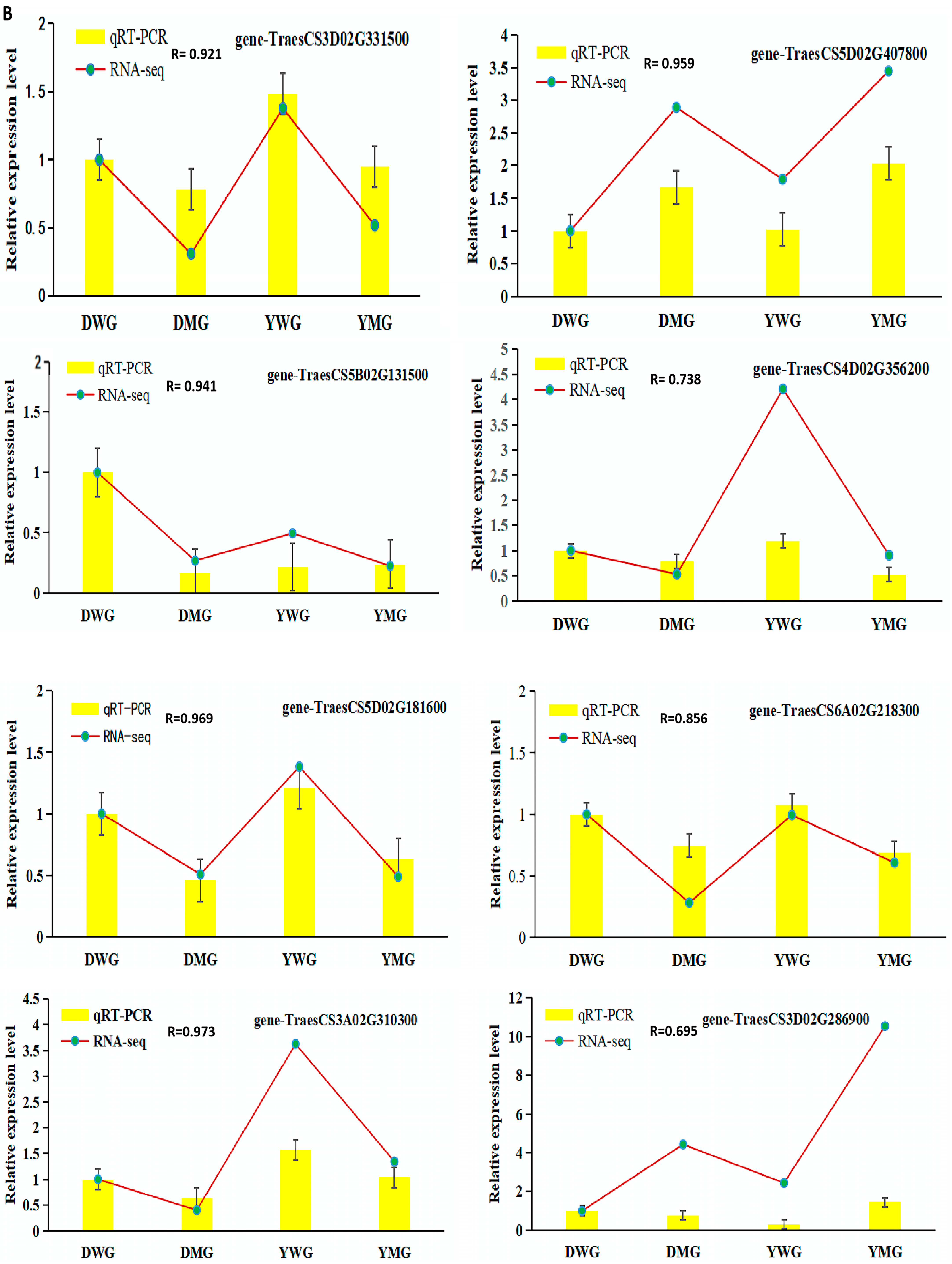
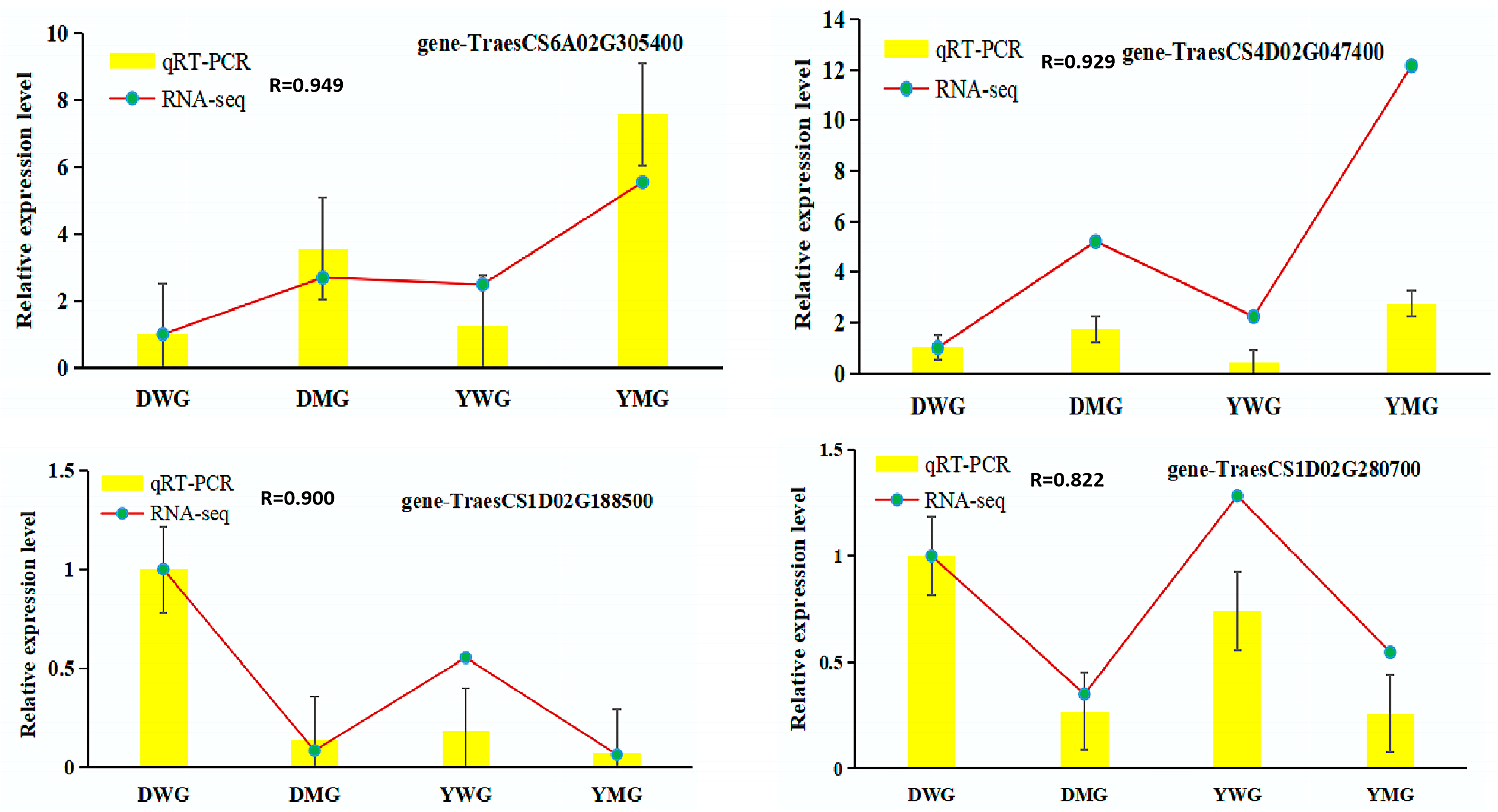
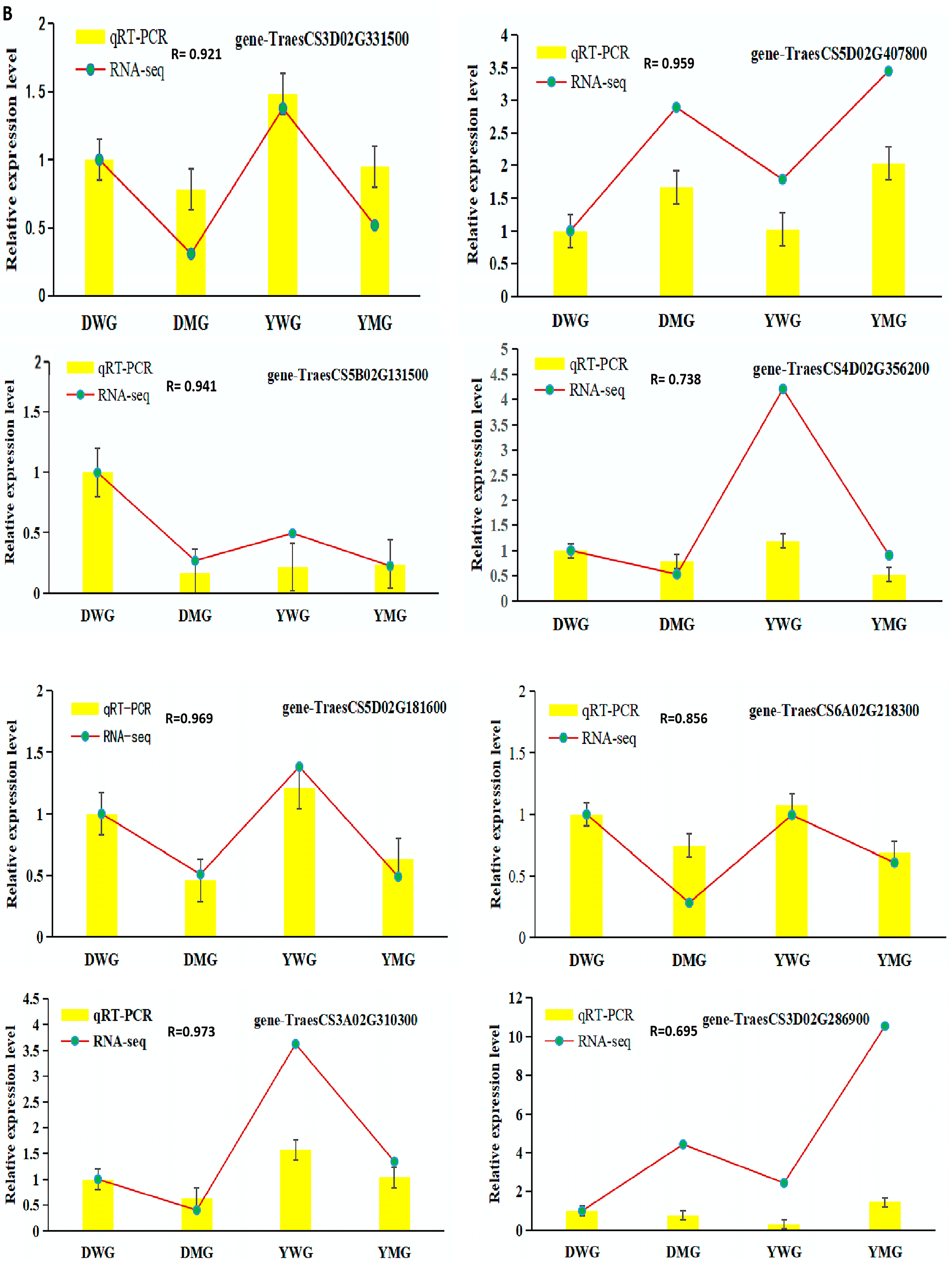
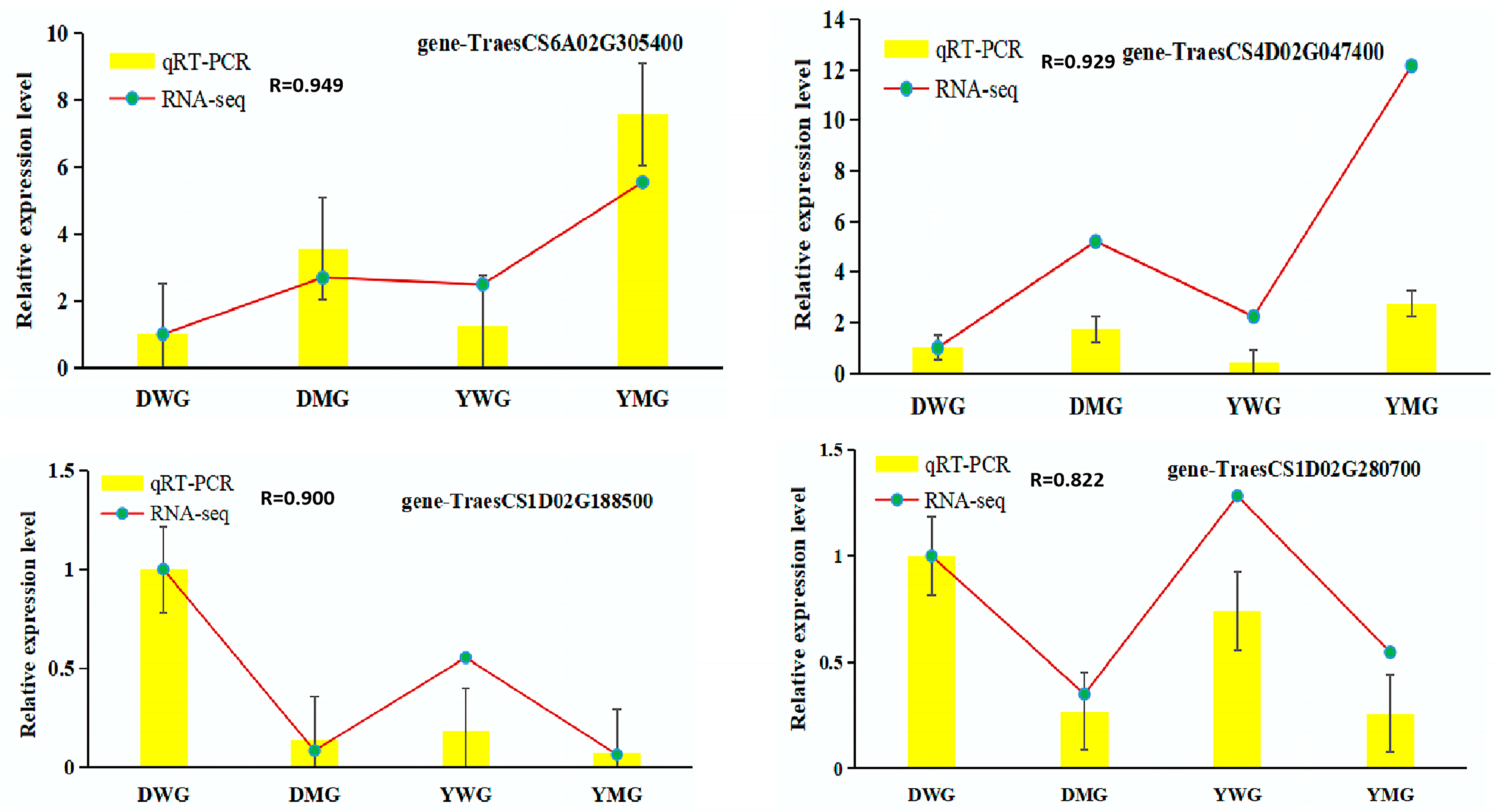
2.7. Confirmation of RNA-Seq Data Using Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
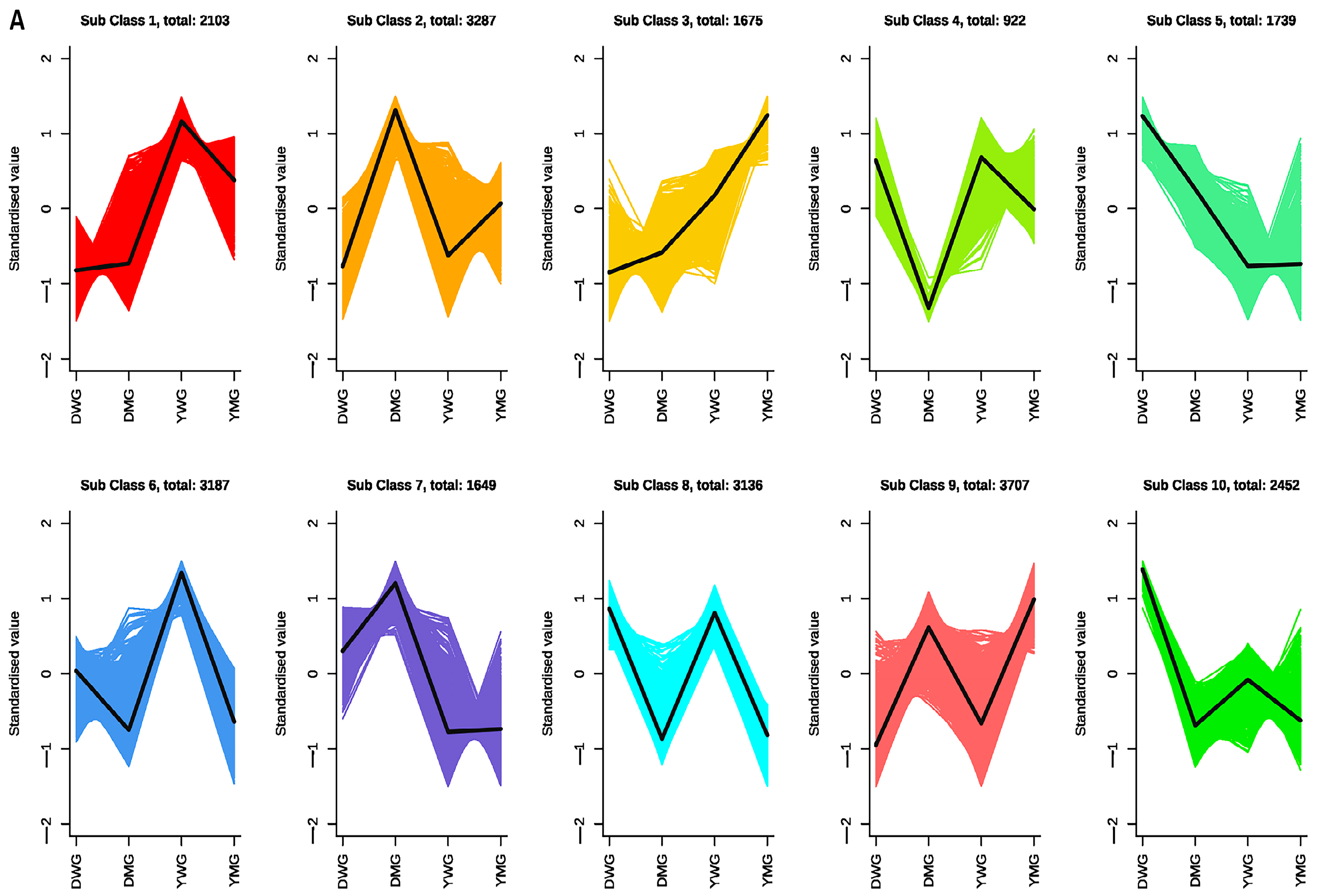
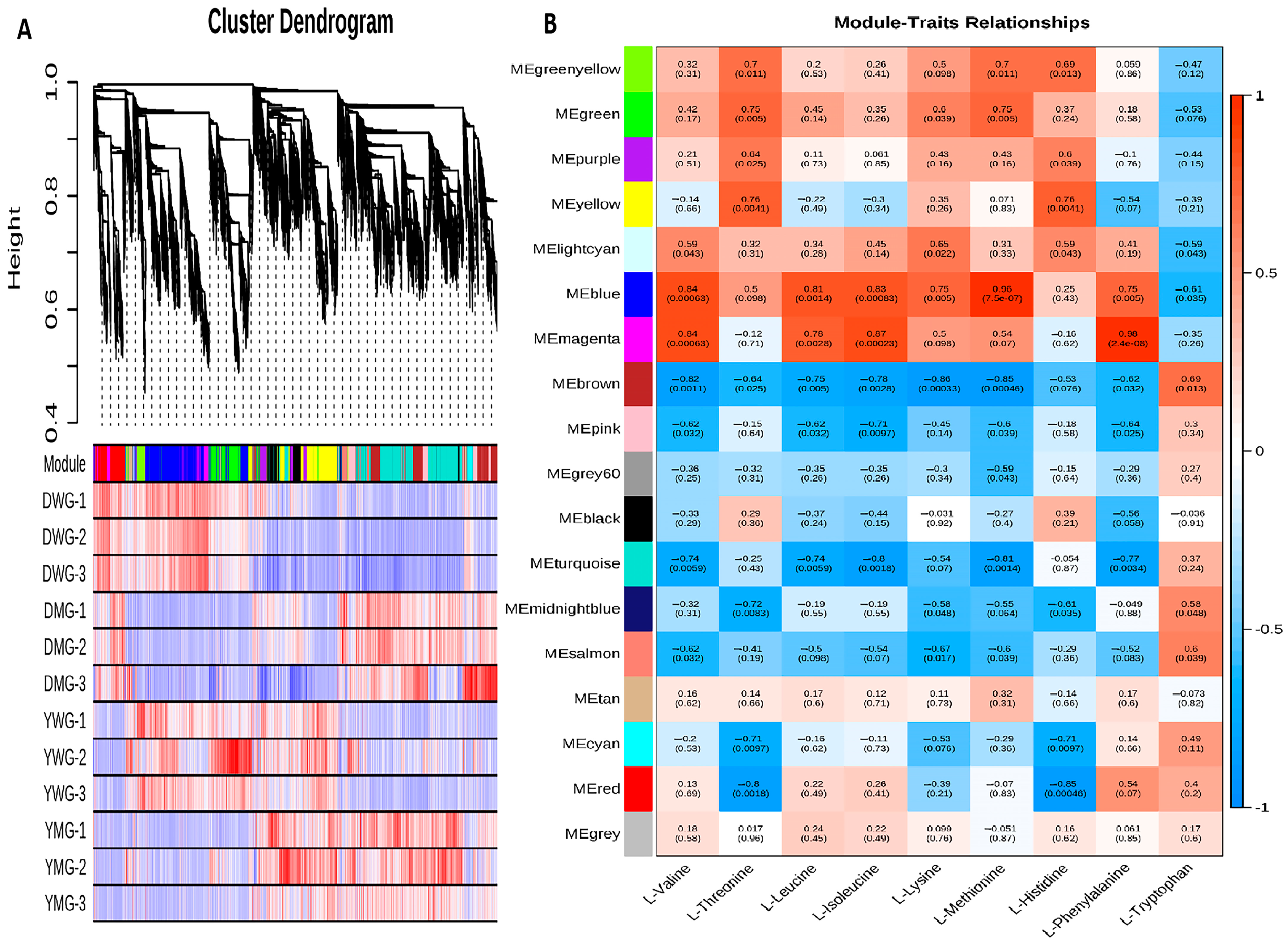
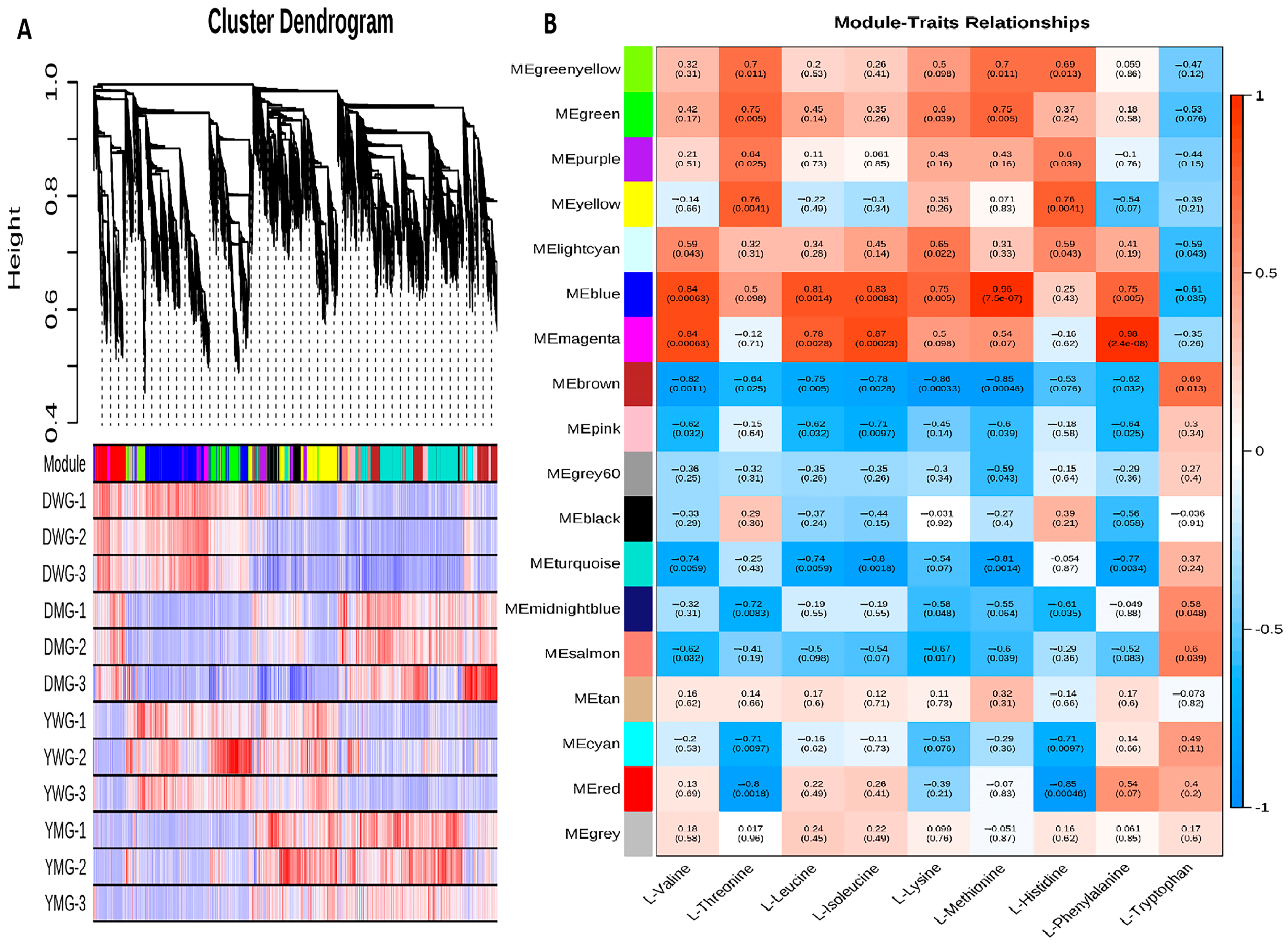
2.8. Weighted Gene Co-Expression Network Analysis (WGCNA) of AA Biosynthesis during Physiological Maturity
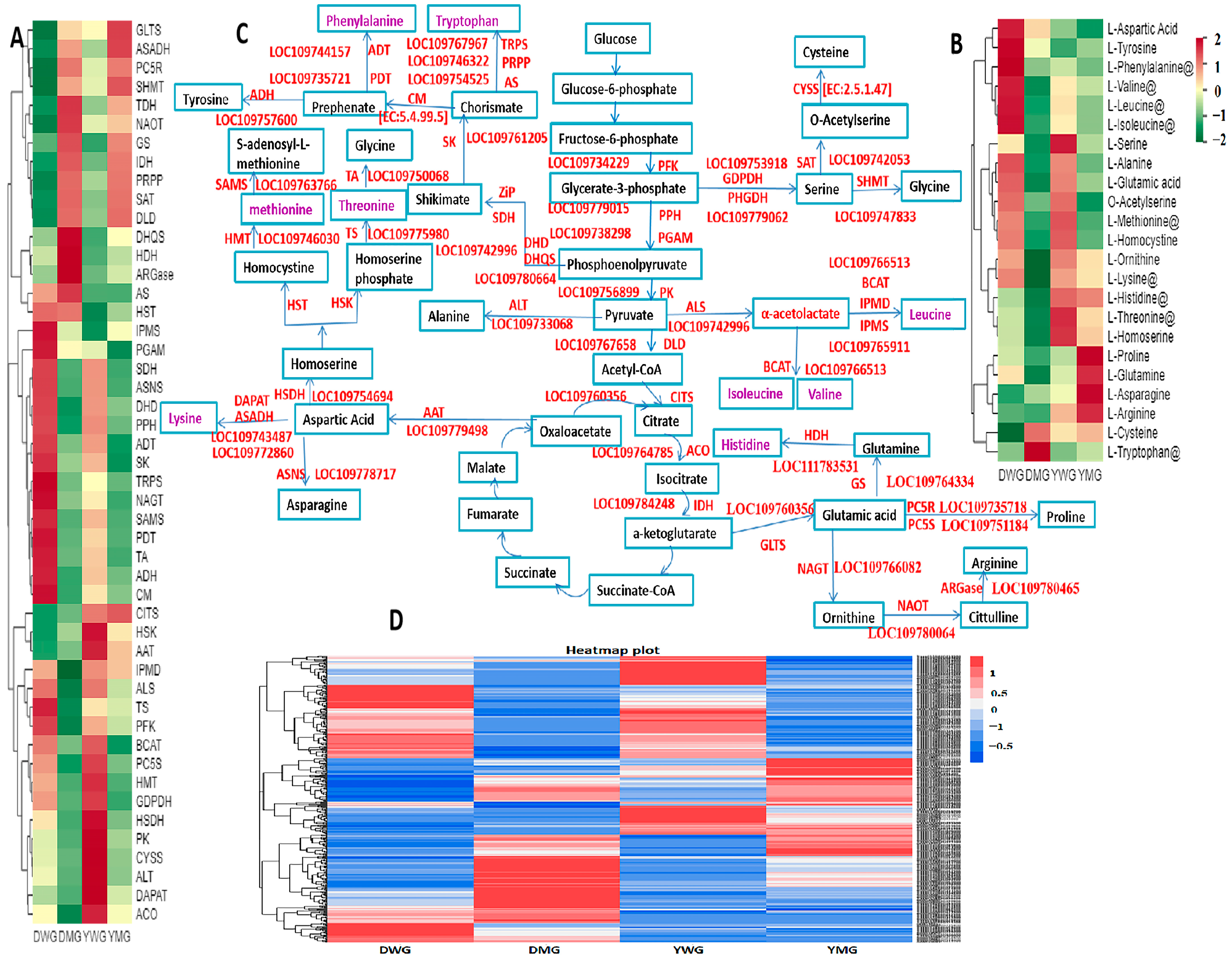
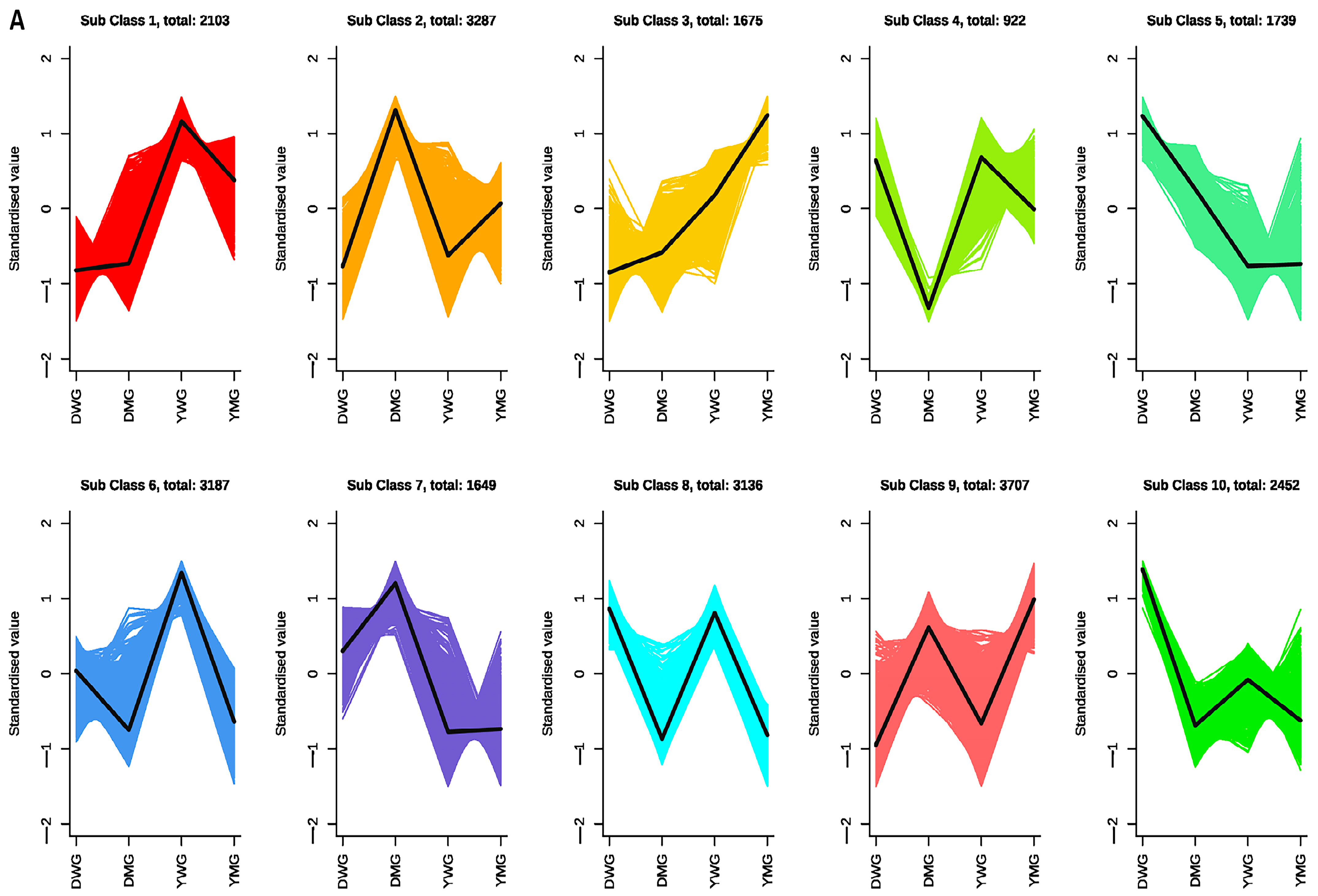
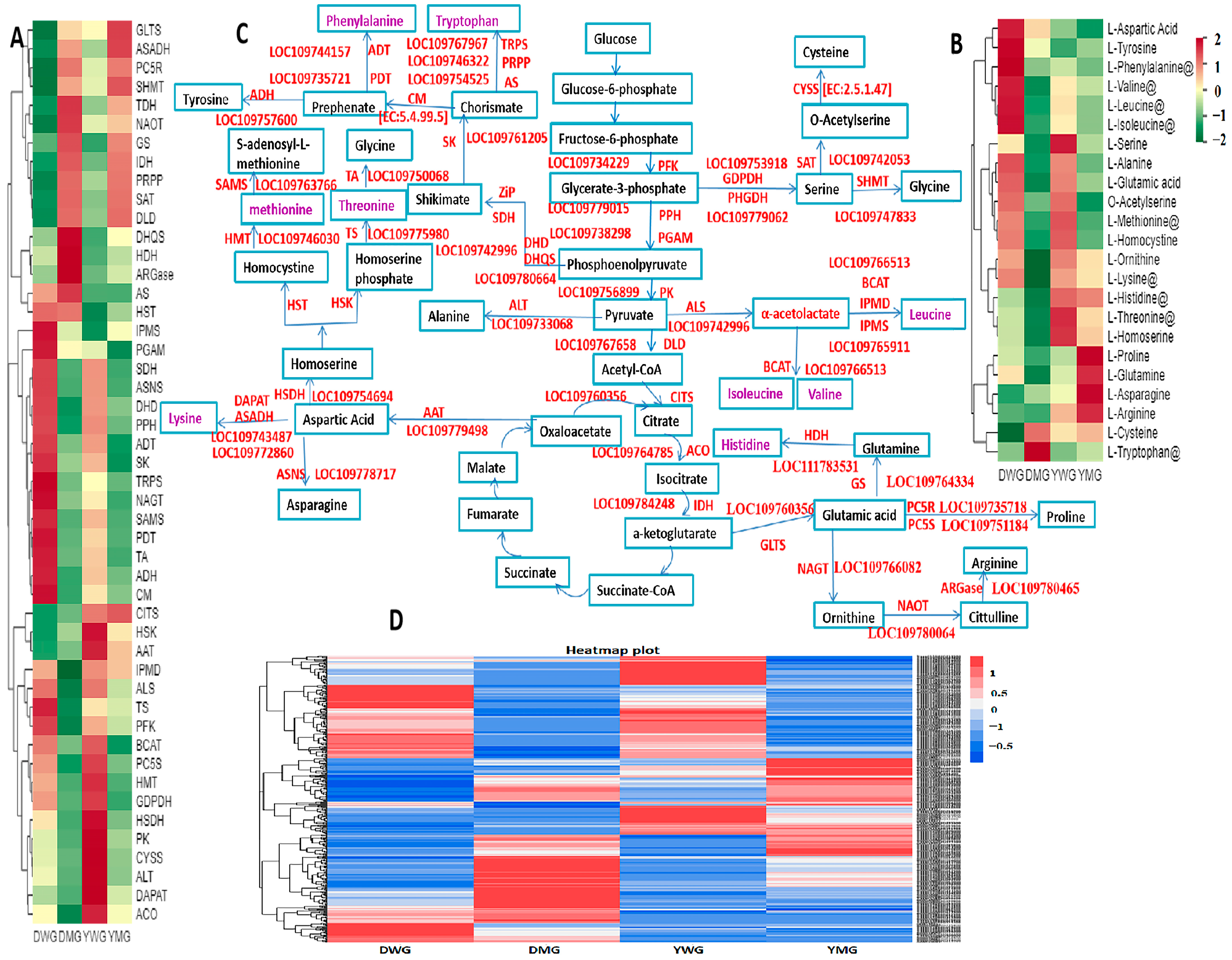
2.9. Identification of the Expression Patterns and Metabolic Pathways of AA Biosynthesis Genes in YHW Lines at Different Stages of Physiological Maturity
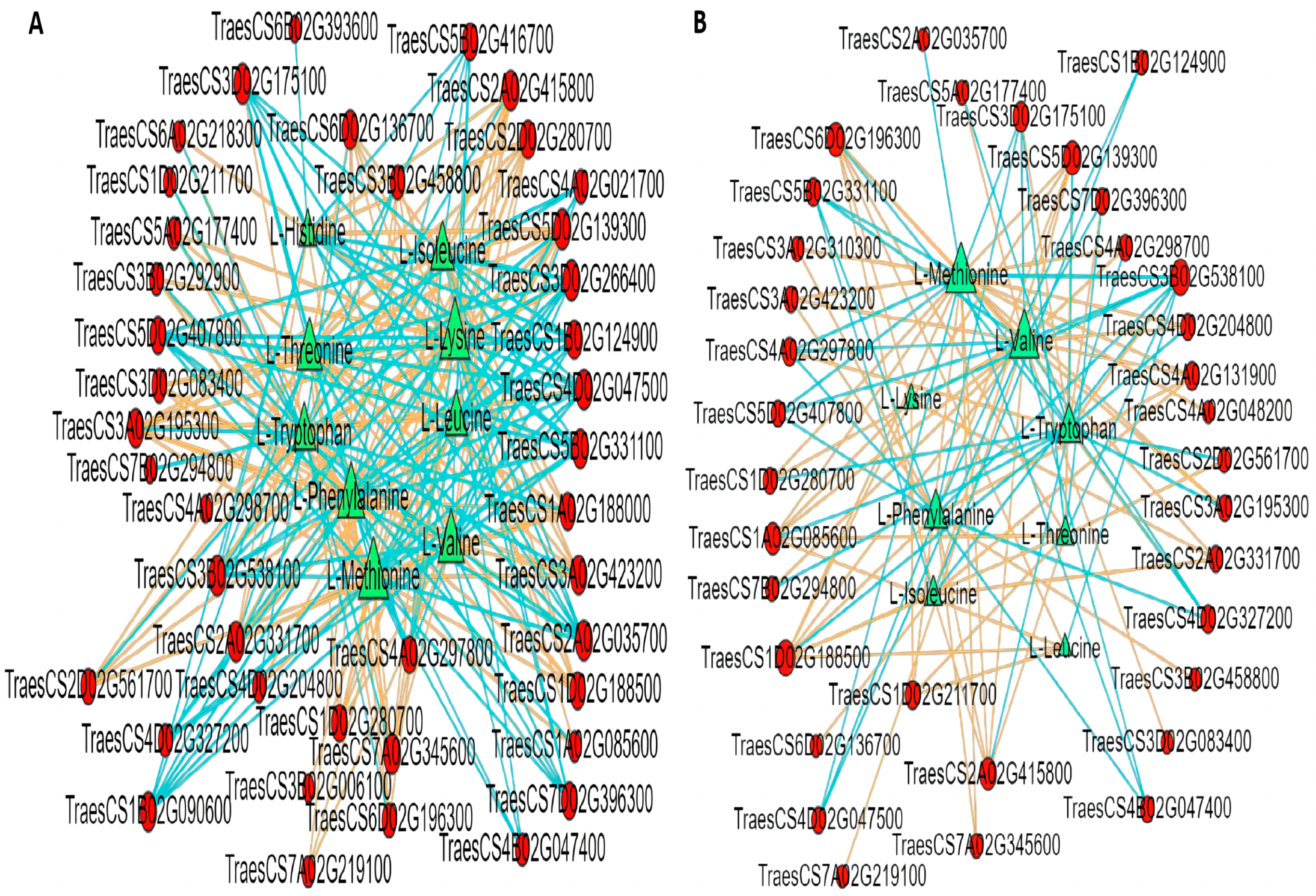
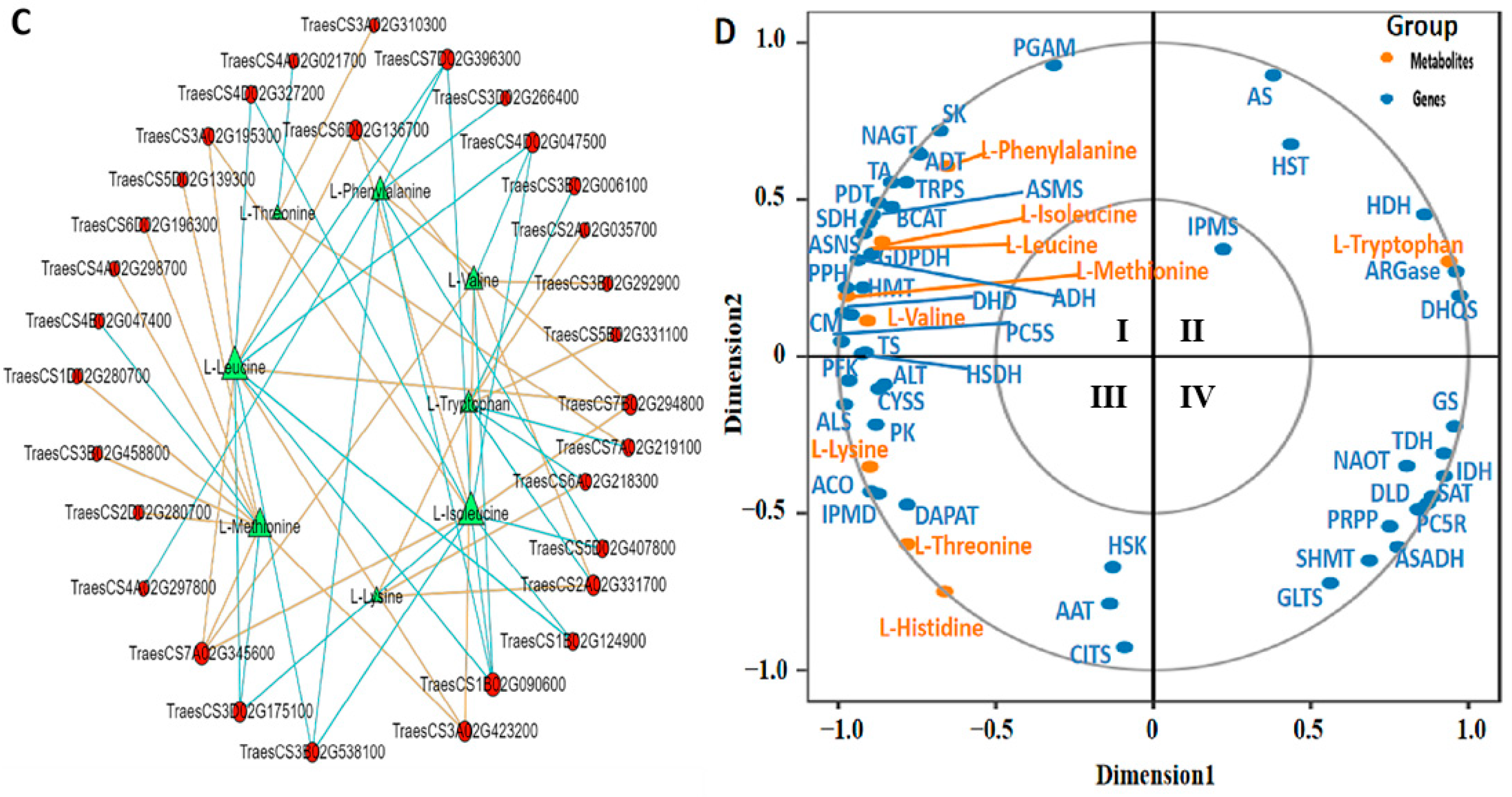
2.10. Correlation Analysis of EAAs and Transcriptome of YHW Grains at Different Levels of Physiological Maturity
3. Discussion
4. Materials and Methods
4.1. Plant Material and Samples
4.2. Measurement of Grain Characteristics
4.3. Metabolite Extraction and Profiling
4.4. Transcriptome Sequencing and Data Analysis
4.5. RT-qPCR
4.6. Co-Expression Network Construction
4.7. CCA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shivakumar, N.; Kashyap, S.; Kishore, S.; Thomas, T.; Varkey, A.; Devi, S.; Preston, T.; Jahoor, F.; Sheshshayee, M.S.; Kurpad, A.V. Protein-quality evaluation of complementary foods in Indian children. Am. J. Clin. Nutr. 2019, 109, 1319–1327. [Google Scholar] [CrossRef]
- Zhong, Y.; Xu, D.; Hebelstrup, K.H.; Yang, D.; Cai, J.; Wang, X.; Zhou, Q.; Cao, W.; Dai, T.; Jiang, D. Nitrogen topdressing timing modifies free amino acids profiles and storage protein gene expression in wheat grain. BMC Plant Biol. 2018, 18, 353. [Google Scholar] [CrossRef]
- Tsochatzis, E.; Papageorgiou, M.; Kalogiannis, S. Validation of a HILIC UHPLC-MS/MS method for amino acid profiling in Triticum species wheat flours. Foods 2019, 8, 514. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, Y.; Jiang, Y. Amino acids in rice grains and their regulation by polyamines and phytohormones. Plants 2022, 11, 1581. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, Y.; Huang, Y.; Zhao, X.; Dong, Z.; Jin, W.; Huang, W. Amino acid permease 6 regulates grain protein content in maize. CROP J. 2022, 10, 1536–1544. [Google Scholar] [CrossRef]
- Garg, A.; Kirchler, T.; Fillinger, S.; Wanke, F.; Stadelhofer, B.; Stahl, M.; Chaban, C. Targeted manipulation of bZIP53 DNA- binding properties influences Arabidopsis metabolism and growth. J. Exp. Bot. 2019, 70, 5659–5671. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, Y.; Sun, Y.; Xie, Z.; Luo, Y.; Long, Q.; Feng, J.; Liu, X.; Wang, B.; He, D.; et al. Natural variations of OsAUX5, a target gene of OsWRKY78, control the neutral essential amino acid content in rice grains. Mol. Plant. 2023, 16, 322–336. [Google Scholar] [CrossRef]
- Ma, D.; Xu, B.; Feng, J.; Hu, H.; Tang, J.; Yin, G.; Xie, Y.; Wang, C. Dynamic metabolomics and transcriptomics analyses for characterization of phenolic compounds and their biosynthetic characteristics in wheat grain. Front. Nutr. 2022, 9, 844337. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Liu, J.; Huang, T.; Zhang, X.; Xie, H.; Guo, Y.; Wang, Q.; Zhang, P.; Qin, P. Grain color formation and analysis of correlated genes by metabolome and transcriptome in different wheat lines at maturity. Front. Nutr. 2023, 10, 1112497. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liu, B.; Yao, Y.; Guo, Z.; Jia, H.; Kong, L.; Zhang, A.; Ma, W.; Ni, Z.; Xu, S.; et al. Wheat genomic study for genetic improvement of traits in China. Sci. China Life Sci. 2022, 65, 1718–1775. [Google Scholar] [CrossRef]
- Myer, R.O.; Brendemuhl, J.H.; Barnett, R.D. Crystalline lysine and threonine supplementation of soft red winter wheat or triticale, low-protein diets for growing-finishing swine. J. Anim. Sci. 1996, 74, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Fang, C.; Zhang, Y.; Fernie, A.R.; Luo, J. Plant metabolism paves the way for breeding crops with high nutritional value and stable yield. Sci. China Life Sci. 2021, 64, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, L.; Hu, J.; Pang, Y.; Hu, L.; Xiao, G.; Ma, X.; Kong, X.; Jia, J.; Wang, H.; et al. Dissecting genetic loci affecting grain morphological traits to improve grain weight via nested association mapping. Theor. Appl. Genet. 2019, 132, 3115–3128. [Google Scholar] [CrossRef]
- Dong, Y.; Zeng, X.; Chen, X. Expedition and investigation of yunnan wheat (Triticum aestivum ssp. yunnanense king). Acta Agron. Sin. 1981, 7, 145–146. [Google Scholar]
- Li, J.; Jiang, Y.; Yao, F.; Long, L.; Wang, Y.; Wu, Y.; Li, H.; Wang, J.; Jiang, Q.; Kang, H.; et al. Genome-wide association study reveals the genetic architecture of stripe rust resistance at the adult plant stage in Chinese endemic wheat. Front. Plant Sci. 2020, 11, 625. [Google Scholar] [CrossRef]
- Qi, L.J.; Hu, X.X.; Zhou, G.Y.; Wang, S.; Li, J.M.; Lu, W.; Wu, L.N.; Lu, M.B.; Sun, L.J.; Yang, X.L.; et al. Analysis of wheat protein quality in main wheat producing areas of China from 2004 to 2011. Sci. Agric. Sin. 2012, 45, 4242–4251. [Google Scholar] [CrossRef]
- Peng, Y.; Zhao, Y.; Yu, Z.; Zeng, J.; Xu, D.; Dong, J.; Ma, W. Wheat quality formation and its regulatory mechanism. Front. Plant Sci. 2022, 13, 834654. [Google Scholar] [CrossRef]
- Yancey, P.H.; Siebenaller, J.F. Co-evolution of proteins and solutions: Protein adaptation versus cytoprotective micromolecules and their roles in marine organisms. J. Exp. Biol. 2015, 218, 1880–1896. [Google Scholar] [CrossRef]
- Church, D.D.; Hirsch, K.R.; Park, S.; Kim, I.Y.; Gwin, J.A.; Pasiakos, S.M.; Wolfe, R.R.; Ferrando, A.A. Essential amino acids and protein synthesis: Insights into maximizing the muscle and whole-body response to feeding. Nutrients 2020, 12, 3717. [Google Scholar] [CrossRef]
- Navdeep, S.C. Amino acid metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040584. [Google Scholar] [CrossRef]
- Galili, G.; Amir, R.; Fernie, A.R. The regulation of essential amino acid synthesis and accumulation in plants. Annu. Rev. Plant Biol. 2016, 67, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wu, P.; Tian, J. Genetic analysis of amino acid content in wheat grain. J. Genet. 2014, 93, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, D.; Zhang, C.; Sreenivasulu, N.; Sun, S.S.; Liu, Q. Lysine biofortification of crops to promote sustained human health in the 21st century. J. Exp. Bot. 2022, 73, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Chu, H.D.; Le, N.Q. Improving Nutritional Quality of Plant Proteins through Genetic Engineering. Curr. Genom. 2016, 17, 220–229. [Google Scholar] [CrossRef]
- Mir, N.A.; Riar, C.S.; Singh, S. Nutritional constituents of pseudo cereals and their potential use in food systems: A review. Trends Food Sci. Technol. 2018, 75, 170–180. [Google Scholar] [CrossRef]
- Hu, H.; Fei, X.; He, B.; Chen, X.; Ma, L.; Han, P.; Luo, Y.; Liu, Y.; Wei, A. UHPLC-MS/MS Profile Combined with RNA-Seq Reveals the Amino Acid Metabolism in Zanthoxylum bungeanum Leaves under Drought Stress. Front. Nutr. 2022, 9, 921742. [Google Scholar] [CrossRef]
- Fini, M.; Torricelli, P.; Giavaresi, G.; Carpi, A.; Nicolini, A.; Giardino, R. Effect of L-lysine and L-arginine on primary osteoblast cultures from normal and osteopenic rats. Biomed. Pharmacother. 2001, 55, 213–220. [Google Scholar] [CrossRef]
- Sacksteder, K.A.; Biery, B.J.; Morrell, J.C.; Goodman, B.K.; Geisbrecht, B.V.; Cox, R.P.; Gould, S.J.; Geraghty, M.T. Identification of the alpha-aminoadipic semialdehyde synthase gene, which is defective in familial hyperlysinemia. Am. J. Hum. Genet. 2000, 66, 1736–1743. [Google Scholar] [CrossRef]
- Tang, Q.; Tan, P.; Ma, N.; Ma, X. Physiological Functions of Threonine in Animals: Beyond Nutrition Metabolism. Nutrients 2021, 13, 2592. [Google Scholar] [CrossRef]
- Karau, A.; Grayson, I. Amino acids in human and animal nutrition. Adv. Biochem. Eng. Biotechnol. 2014, 143, 189–228. [Google Scholar] [CrossRef]
- Xu, D.; Lin, Q.; Wu, W.; Wu, Y.; Liang, Y. Revealing the antiaging effects of cereal- and food oil-d erived active substances by a Caenor habditis elegans model. Food Funct. 2021, 12, 3296–3306. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xu, M.; Wang, W.; Galili, G. Fortifying horticultural crops with essential amino acids: A review. Int. J. Mol. Sci. 2017, 18, 1306. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Li, D.; Luo, J.; Xiao, Y.; Liu, H.; Pan, Q.; Zhang, X.; Jin, M.; Zhao, M.; Yan, J. The genetic architecture of amino acids dissection by association and linkage analysis in maize. Plant Biotechnol. J. 2017, 15, 1250–1263. [Google Scholar] [CrossRef] [PubMed]
- Iwaniuk, P.; Konecki, R.; Kaczynski, P.; Rysbekova, A.; Łozowicka, B. Influence of seven levels of chemical/biostimulator protection on amino acid profile and yield traits in wheat. CROP J. 2022, 10, 198–1206. [Google Scholar] [CrossRef]
- Iwaniuk, P.; Łuniewski, S.; Kaczyński, P.; Łozowicka, B. The Influence of Humic Acids and Nitrophenols on Metabolic Compounds and Pesticide Behavior in Wheat under Biotic Stress. Agronomy 2023, 13, 1378. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, G. Molecular prospective on the wheat grain development. Crit. Rev. Biotechnol. 2023, 43, 38–49. [Google Scholar] [CrossRef]
- Nadaud, I.; Girousse, C.; Debiton, C.; Chambon, C.; Bouzidi, M.F.; Martre, P.; Branlard, G. Proteomic and morphological analysis of early stages of wheat grain development. Proteomics 2010, 10, 2901–2910. [Google Scholar] [CrossRef]
- Zhang, S.; Ghatak, A.; Bazargani, M.M.; Bajaj, P.; Varshney, R.K.; Chaturvedi, P.; Jiang, D.; Weckwerth, W. Spatial distribution of proteins and metabolites in developing wheat grain and their differential regulatory response during the grain filling process. Plant J. 2021, 107, 669–687. [Google Scholar] [CrossRef]
- Chen, F.; Yu, Y.; Xia, X.; He, Z. Prevalence of a novel puroindo-line b allele in Yunnan endemic wheats (Triticum aestivum ssp. yunnanense King). Euphytica 2007, 156, 39–46. [Google Scholar] [CrossRef]
- Tegeder, M.; Ward, J.M. Molecular Evolution of Plant AAP and LHT Amino Acid Transporters. Front. Plant Sci. 2012, 3, 21. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, M.; Ruan, J. Integrated transcriptome and metabolic analyses reveals novel insights into free amino acid metabolism in Huangjinya tea cultivar. Front. Plant Sci. 2017, 8, 291. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Tiwari, M.; Singh, S.P.; Singh, R.; Singh, S.; Shirke, P.A.; Trivedi, P.K.; Misra, P. Sterol glycosyltransferases required for adaptation of Withania somnifera at high temperature. Physiol. Plant. 2017, 160, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Dickman, M.B.; Becker, D.F. Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic. Biol. Med. 2008, 44, 671–681. [Google Scholar] [CrossRef]
- Li, W.; Wen, L.; Chen, Z.; Zhang, Z.; Pang, X.; Deng, Z.; Liu, T.; Guo, Y. Study on metabolic variation in whole grains of four proso millet varieties reveals metabolites important for antioxidant properties and quality traits. Food Chem. 2021, 357, 129791. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, M.; Gao, C.; Fu, F.-F.; Wang, T.; El-Kassaby, Y.A.; Wang, G. Amino acid metabolism reprogramming in response to changing growth environment in ginkgo biloba leaves. LWT 2021, 144, 111276. [Google Scholar] [CrossRef]
- D’Este, M.; Alvarado-Morales, M.; Angelidaki, I. Amino acids production focusing on fermentation technologies—A review. Biotechnol. Adv. 2018, 36, 14–25. [Google Scholar] [CrossRef]
- Duan, W.K.; Shao, W.L.; Lin, W.D.; Yuan, L.; Lu, Q.W.; Chen, L.X.; Zagorchev, L.; Li, J.M. Integrated metabolomics and transcriptomics reveal the differences in fruit quality of the red and white Fragaria pentaphylla morphs. Food Biosci. 2021, 40, 100896. [Google Scholar] [CrossRef]
- Cramer, G.R.; Van Sluyter, S.C.; Hopper, D.W.; Pascovici, D.; Keighley, T.; Haynes, P.A. Proteomic nalysis indicates massive changes in metabolism prior to the inhibition of growth and photosynthesis of grapevine (Vitis vinifera L.) in response to water deficit. BMC Plant Biol. 2013, 13, 49. [Google Scholar] [CrossRef]
- Saavedra, E.; Encalada, R.; Vázquez, C.; Olivos-García, A.; Michels, P.A.M.; Moreno-Sánchez, R. Control and regulation of the pyrophosphate-dependent glucose metabolism in Entamoeba histolytica. Mol. Biochem. Parastt. 2019, 229, 75–87. [Google Scholar] [CrossRef]
- Xu, Y.; Magwanga, R.O.; Yang, X.; Jin, D.; Cai, X.; Hou, Y.; Wei, Y.; Zhou, Z.; Wang, K.; Liu, F. Genetic regulatory networks for salt-alkali stress in Gossypium hirsutum with differing morphological characteristics. BMC Genom. 2020, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hu, X.; Shi, T.; Yin, H.; Sun, D.; Hao, Y.; Xia, X.; Luo, J.; Fernie, A.R.; He, Z.; et al. Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnol. J. 2020, 18, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Hou, H.; Ma, X.; Sun, S.; Wang, H.; Kong, L. Metabolomics and gene expression analysis reveal the accumulation patterns of phenylpropanoids and flavonoids in different colored-grain wheats (Triticum aestivum L.). Food Res. Int. 2020, 138, 109711. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Hu, H.; Luo, Y.; Shi, Q.; Wei, A. Widely targeted metabolomic profiling combined with transcriptome analysis provides new insights into amino acid biosynthesis in green and red pepper fruits. Food Res. Int. 2022, 160, 111718. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from Rna-Seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for Rna-Seq data with Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Lou, H.; Yang, Y.; Zheng, S.; Ma, Z.; Chen, W.; Yu, C.; Song, L.; Wu, J. Identification of key genes contributing to amino acid biosynthesis in Torreya grandis using transcriptome and metabolome analysis. Food Chem. 2022, 379, 132078. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Molecular Weight (Da) | Formula | Class |
|---|---|---|---|
| L-Valine | 1.17 × 102 | C5H11NO2 | Essential amino acid |
| L-Threonine | 1.19 × 102 | C4H9NO3 | Essential amino acid |
| L-Leucine | 1.31 × 102 | C6H13NO2 | Essential amino acid |
| L-Isoleucine | 1.31 × 102 | C6H13NO2 | Essential amino acid |
| L-Lysine | 1.46 × 102 | C6H14N2O2 | Essential amino acid |
| L-Methionine | 1.49 × 102 | C5H11NO2S | Essential amino acid |
| L-Histidine | 1.55 × 102 | C6H9N3O2 | Essential amino acid |
| L-Phenylalanine | 1.65 × 102 | C9H11NO2 | Essential amino acid |
| L-Tryptophan | 2.04 × 102 | C11H12N2O2 | Essential amino acid |
| L-Alanine | 8.90 × 101 | C3H7NO2 | Amino acids |
| L-Serine | 1.05 × 102 | C3H7NO3 | Amino acids |
| L-Proline | 1.15 × 102 | C5H9NO2 | Amino acids |
| L-Homoserine | 1.19 × 102 | C4H9NO3 | Amino acids |
| L-Cysteine | 1.21 × 102 | C3H7NO2S | Amino acids |
| L-Norleucine | 1.31 × 102 | C6H13NO2 | Amino acids |
| L-Asparagine | 1.32 × 102 | C4H8N2O3 | Amino acids |
| L-Ornithine | 1.32 × 102 | C5H12N2O2 | Amino acids |
| L-Aspartic acid | 1.33 × 102 | C4H7NO4 | Amino acids |
| L-Glutamine | 1.46 × 102 | C5H10N2O3 | Amino acids |
| L-Glutamic acid | 1.47 × 102 | C5H9NO4 | Amino acids |
| L-Homomethionine | 1.63 × 102 | C6H13NO2S | Amino acids |
| L-Arginine | 1.74 × 102 | C6H14N4O2 | Amino acids |
| L-Tyrosine | 1.81 × 102 | C9H11NO3 | Amino acids |
| L-Homocystine | 2.68 × 102 | C8H16N2O4S2 | Amino acids |
| N-Methylglycine | 8.90 × 101 | C3H7NO2 | Amino acid derivatives |
| L-Azetidine-2-carboxylic acid | 1.01 × 102 | C4H7NO2 | Amino acid derivatives |
| Methyl 3-aminopropanoate | 1.03 × 102 | C4H9NO2 | Amino acid derivatives |
| N,N-Dimethylglycine | 1.03 × 102 | C4H9NO2 | Amino acid derivatives |
| N-Acetyl-L-glycine | 1.17 × 102 | C4H7NO3 | Amino acid derivatives |
| N-Methyl-α-aminoisobutyric acid | 1.17 × 102 | C5H11NO2 | Amino acid derivatives |
| 5-Oxoproline | 1.29 × 102 | C5H7NO3 | Amino acid derivatives |
| 5-Oxo-L-proline | 1.29 × 102 | C5H7NO3 | Amino acid derivatives |
| Cycloleucine | 1.29 × 102 | C6H11NO2 | Amino acid derivatives |
| N-Acetyl-beta-alanine | 1.31 × 102 | C5H9NO3 | Amino acid derivatives |
| Trans-4-hydroxy-L-proline | 1.31 × 102 | C5H9NO3 | Amino acid derivatives |
| S-Methyl-L-cysteine | 1.35 × 102 | C4H9NO2S | Amino acid derivatives |
| L-Cyclopentylglycine | 1.43 × 102 | C7H13NO2 | Amino acid derivatives |
| N-Methyl-trans-4-hydroxy-L-proline | 1.45 × 102 | C6H11NO3 | Amino acid derivatives |
| L-Threo-3-methylaspartate | 1.47 × 102 | C5H9NO4 | Amino acid derivatives |
| O-Acetylserine | 1.47 × 102 | C5H9NO4 | Amino acid derivatives |
| (2S,3R,4S)-4-Hydroxyisoleucine | 1.47 × 102 | C6H13NO3 | Amino acid derivatives |
| 4-Hydoxy-L-isoleucine | 1.47 × 102 | C6H13NO3 | Amino acid derivatives |
| N-Acetyl-L-threonine | 1.61 × 102 | C6H11NO4 | Amino acid derivatives |
| N-Methyl-L-glutamate | 1.61 × 102 | C6H11NO4 | Amino acid derivatives |
| 3-Hydroxy-3-methylpentane-1,5-dioic acid | 1.62 × 102 | C6H10O5 | Amino acid derivatives |
| 4-Hydroxy-L-glutamic acid | 1.63 × 102 | C5H9NO5 | Amino acid derivatives |
| L-Methionine methyl ester | 1.63 × 102 | C6H13NO2S | Amino acid derivatives |
| L-Methionine sulfoxide | 1.65 × 102 | C5H11NO3S | Amino acid derivatives |
| L-Glycyl-L-proline | 1.72 × 102 | C7H12N2O3 | Amino acid derivatives |
| N-Acetyl-L-leucine | 1.73 × 102 | C8H15NO3 | Amino acid derivatives |
| N-Alpha-acetyl-L-asparagine | 1.74 × 102 | C6H10N2O4 | Amino acids derivatives |
| N-α-Acetyl-L-ornithine | 1.74 × 102 | C7H14N2O3 | Amino acid derivatives |
| N-Acetyl-L-aspartic acid | 1.75 × 102 | C6H9NO5 | Amino acid derivatives |
| O-Phospho-L-serine | 1.85 × 102 | C3H8NO6P | Amino acid derivatives |
| N-Acetyl-L-glutamine | 1.88 × 102 | C7H12N2O4 | Amino acid derivatives |
| N-Glycyl-L-leucine | 1.88 × 102 | C8H16N2O3 | Amino acid derivatives |
| L-Glycyl-L-isoleucine | 1.88 × 102 | C8H16N2O3 | Amino acid derivatives |
| N6-Acetyl-L-lysine | 1.88 × 102 | C8H16N2O3 | Amino acid derivatives |
| Homoarginine | 1.88 × 102 | C7H16N4O2 | Amino acid derivatives |
| N-Monomethyl-L-arginine | 1.88 × 102 | C7H16N4O2 | Amino acid derivatives |
| Trimethyllysine | 1.88 × 102 | C9H20N2O2 | Amino acid derivatives |
| N-Acetyl-L-glutamic acid | 1.89 × 102 | C7H11NO5 | Amino acid derivatives |
| N-Acetyl-L-methionine | 1.91 × 102 | C7H13NO3S | Amino acid derivatives |
| L-Tyrosine methyl ester | 1.95 × 102 | C10H13NO3 | Amino acid derivatives |
| 3,4-Dihydroxy-L-phenylalanine (L-dopa) | 1.97 × 102 | C9H11NO4 | Amino acid derivatives |
| NG,NG-Dimethyl-L-arginine | 2.02 × 102 | C8H18N4O2 | Amino acid derivatives |
| N-Acetyl-L-phenylalanine | 2.07 × 102 | C11H13NO3 | Amino acid derivatives |
| 6-Hydroxydopaquinone | 2.11 × 102 | C9H9NO5 | Amino acid derivatives |
| N-Acetyl-L-arginine | 2.16 × 102 | C8H16N4O3 | Amino acid derivatives |
| 5-L-Glutamyl-L-amino acid | 2.18 × 102 | C8H14N2O5 | Amino acid derivatives |
| 5-Hydroxy-L-tryptophan | 2.20 × 102 | C11H12N2O3 | Amino acid derivatives |
| Glycylphenylalanine | 2.22 × 102 | C11H14N2O3 | Amino acid derivatives |
| L-Glycyl-L-phenylalanine | 2.22 × 102 | C11H14N2O3 | Amino acid derivatives |
| N-Acetyl-L-tyrosine | 2.23 × 102 | C11H13NO4 | Amino acid derivatives |
| L-Prolyl-L-leucine | 2.28 × 102 | C11H20N2O3 | Amino acid derivatives |
| L-Valyl-L-leucine | 2.30 × 102 | C11H22N2O3 | Amino acid derivatives |
| L-Lysine-butanoic acid | 2.34 × 102 | C10H22N2O4 | Amino acid derivatives |
| L-Alanyl-L-phenylalanine | 2.36 × 102 | C12H16N2O3 | Amino acid derivatives |
| L-Leucyl-L-leucine | 2.44 × 102 | C12H24N2O3 | Amino acid derivatives |
| N-Acetyl-L-tryptophan | 2.46 × 102 | C13H14N2O3 | Amino acid derivatives |
| L-Isoleucyl-L-aspartate | 2.46 × 102 | C10H18N2O5 | Amino acid derivatives |
| γ-Glutamyl-L-valine | 2.46 × 102 | C10H18N2O5 | Amino acid derivatives |
| L-γ-Glutamyl-L-leucine | 2.60 × 102 | C11H20N2O5 | Amino acid derivatives |
| Glycyl-tryptophan | 2.61 × 102 | C13H15N3O3 | Amino acid derivatives |
| L-Prolyl-L-phenylalanine | 2.62 × 102 | C14H18N2O3 | Amino acid derivatives |
| Phenylacetyl-L-glutamine | 2.64 × 102 | C13H16N2O4 | Amino acid derivatives |
| L-Valyl-L-phenylalanine | 2.64 × 102 | C14H20N2O3 | Amino acid derivatives |
| γ-Glutamylmethionine | 2.78 × 102 | C10H18N2O5S | Amino acid derivatives |
| L-Aspartyl-L-phenylalanine | 2.80 × 102 | C13H16N2O5 | Amino acid derivatives |
| γ-Glutamylphenylalanine | 2.94 × 102 | C14H18N2O5 | Amino acid derivatives |
| Glutathione reduced form | 3.07 × 102 | C10H17N3O6S | Amino acid derivatives |
| γ-Glutamyltyrosine | 3.10 × 102 | C14H18N2O6 | Amino acid derivatives |
| L-Phenylalanyl-L-phenylalanine | 3.12 × 102 | C18H20N2O3 | Amino acid derivatives |
| S-(Methyl)glutathione | 3.21 × 102 | C11H19N3O6S | Amino acid derivatives |
| Tryptophan glutamic acid | 3.33 × 102 | C16H19N3O5 | Amino acid derivatives |
| S-(5′-Adenosy)-L-homocysteine | 3.84 × 102 | C14H20N6O5S | Amino acid derivatives |
| Oxiglutatione | 6.12 × 102 | C20H32N6O12S2 | Amino acid derivatives |
| L-Alanyl-L-alanine | 1.60 × 102 | C6H12N2O3 | Oligopeptides |
| Cyclo(L-Ala-L-Pro) | 1.68 × 102 | C8H12N2O2 | Oligopeptides |
| Cyclo(D-Leu-L-Pro) | 2.10 × 102 | C11H18N2O2 | Oligopeptides |
| Cyclo(D-Phe-L-Pro) | 2.44 × 102 | C14H16N2O2 | Oligopeptides |
| Cyclo(Pro-Phe) | 2.44 × 102 | C14H16N2O2 | Oligopeptides |
| γ-Glu-Cys | 2.50 × 102 | C8H14N2O5S | Oligopeptides |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Zhang, P.; Zhang, X.; Wang, Q.; Liu, J.; Li, L.; Cheng, S.; Qin, P. Integrated Metabolome and Transcriptome Analyses Reveal Amino Acid Biosynthesis Mechanisms during the Physiological Maturity of Grains in Yunnan Hulled Wheat (Triticum aestivum ssp. yunnanense King). Int. J. Mol. Sci. 2023, 24, 13475. https://doi.org/10.3390/ijms241713475
Zhang C, Zhang P, Zhang X, Wang Q, Liu J, Li L, Cheng S, Qin P. Integrated Metabolome and Transcriptome Analyses Reveal Amino Acid Biosynthesis Mechanisms during the Physiological Maturity of Grains in Yunnan Hulled Wheat (Triticum aestivum ssp. yunnanense King). International Journal of Molecular Sciences. 2023; 24(17):13475. https://doi.org/10.3390/ijms241713475
Chicago/Turabian StyleZhang, Chuanli, Ping Zhang, Xuesong Zhang, Qianchao Wang, Junna Liu, Li Li, Shunhe Cheng, and Peng Qin. 2023. "Integrated Metabolome and Transcriptome Analyses Reveal Amino Acid Biosynthesis Mechanisms during the Physiological Maturity of Grains in Yunnan Hulled Wheat (Triticum aestivum ssp. yunnanense King)" International Journal of Molecular Sciences 24, no. 17: 13475. https://doi.org/10.3390/ijms241713475
APA StyleZhang, C., Zhang, P., Zhang, X., Wang, Q., Liu, J., Li, L., Cheng, S., & Qin, P. (2023). Integrated Metabolome and Transcriptome Analyses Reveal Amino Acid Biosynthesis Mechanisms during the Physiological Maturity of Grains in Yunnan Hulled Wheat (Triticum aestivum ssp. yunnanense King). International Journal of Molecular Sciences, 24(17), 13475. https://doi.org/10.3390/ijms241713475