
Dynamics of the Apo µ-Opioid Receptor in Complex with Gi Protein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
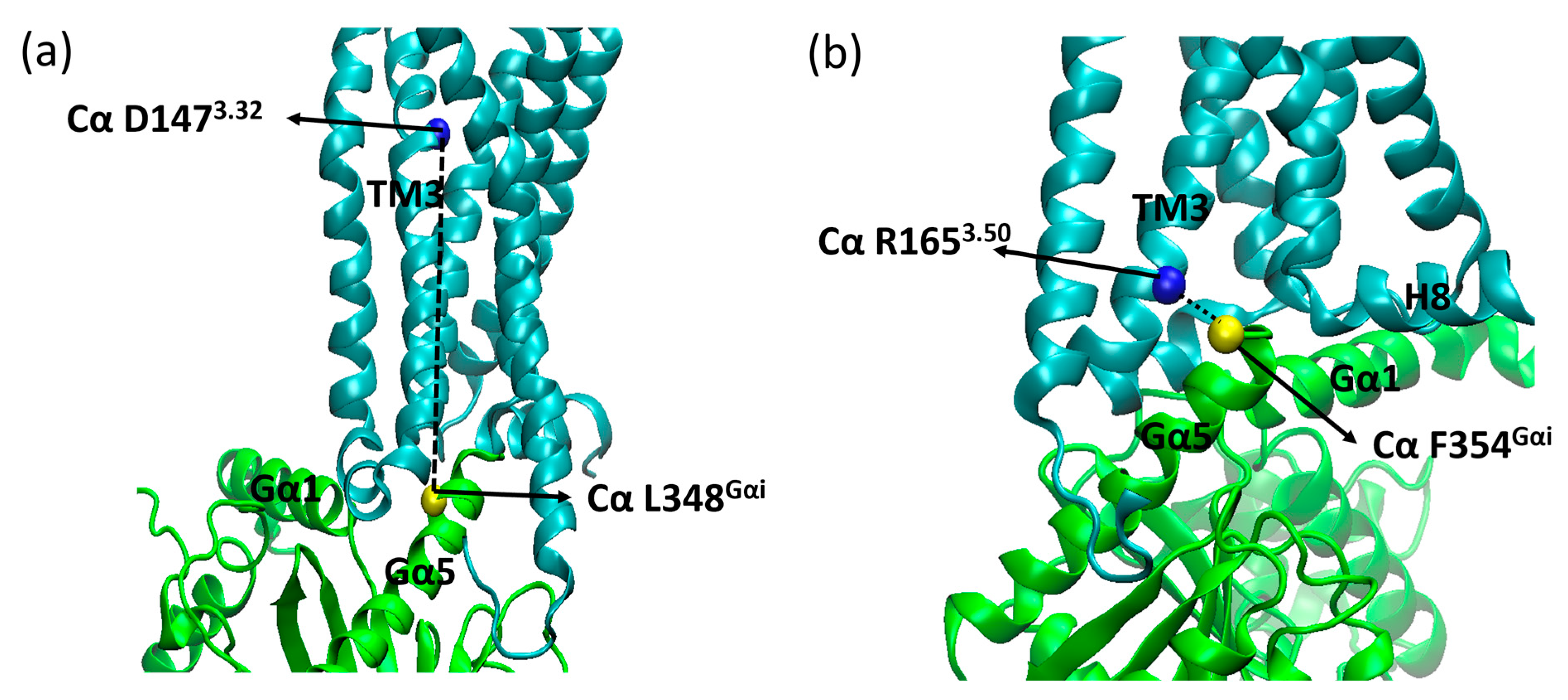
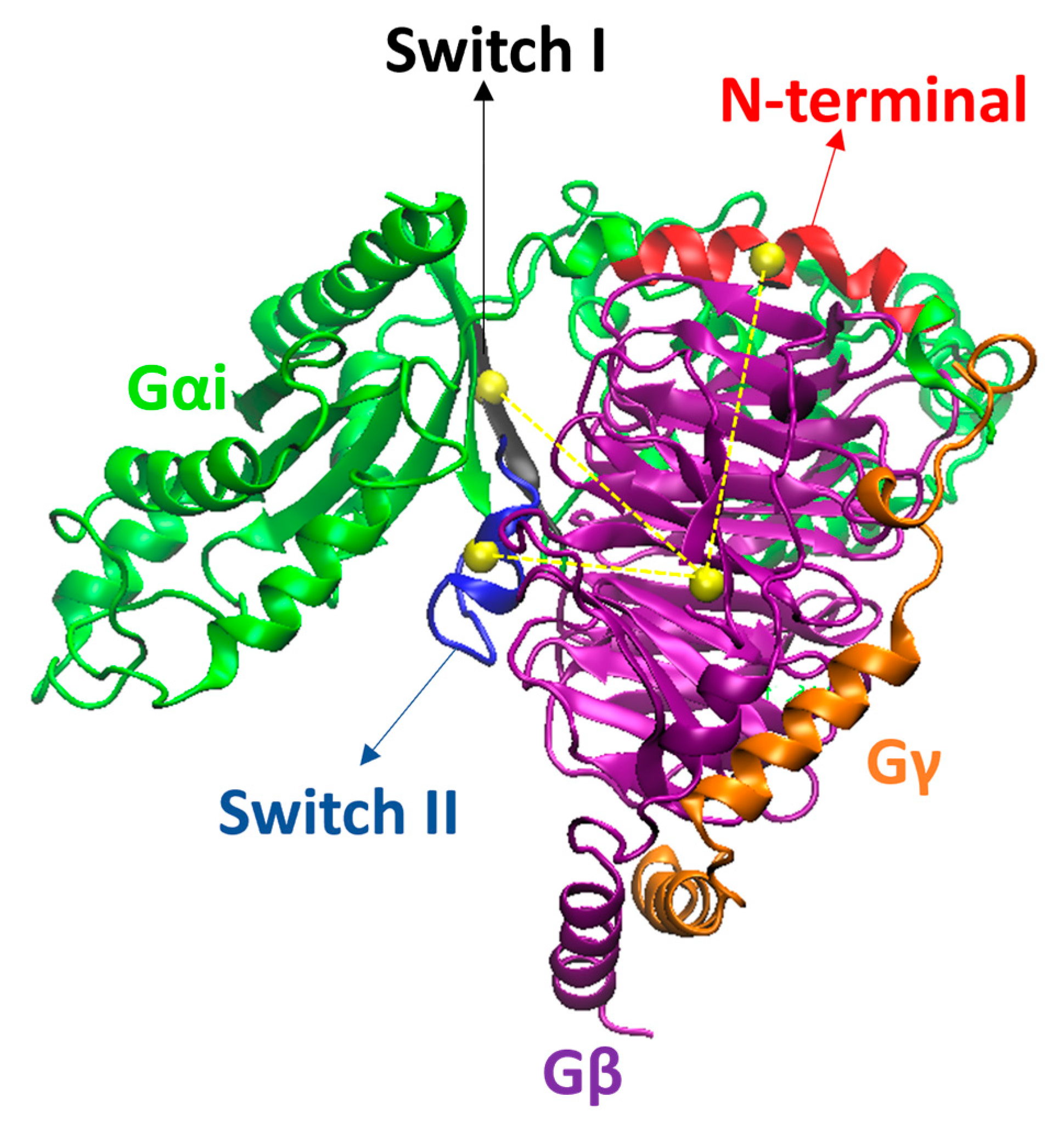
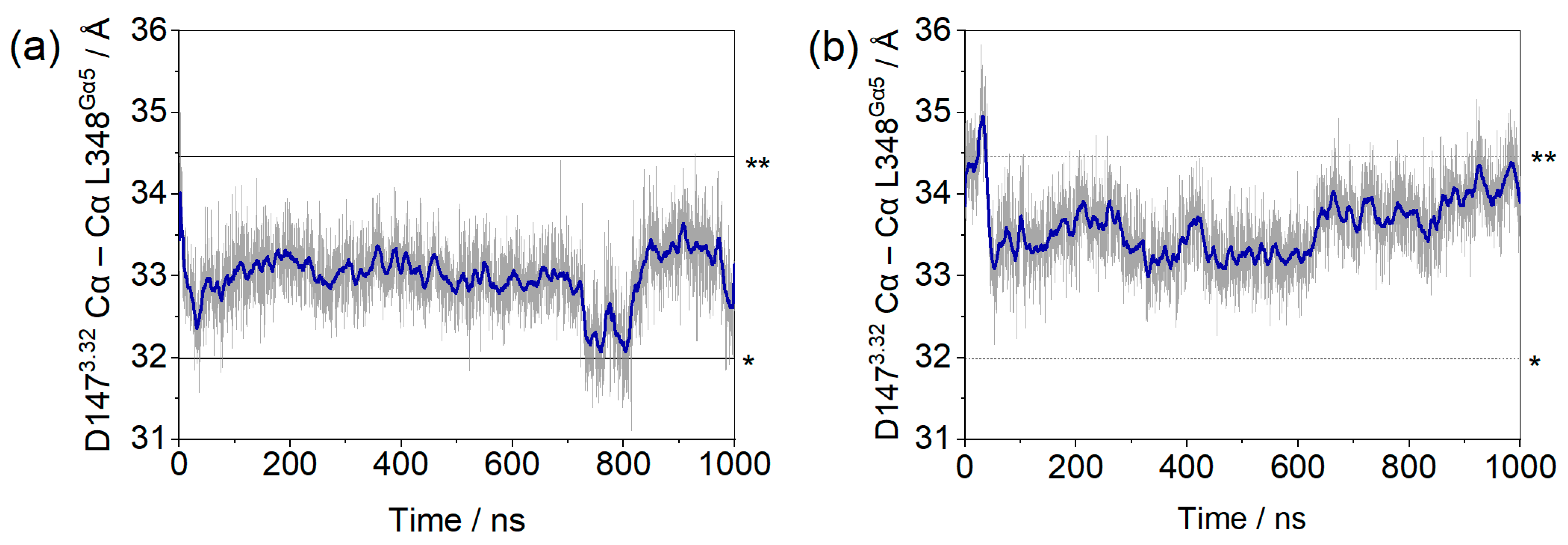
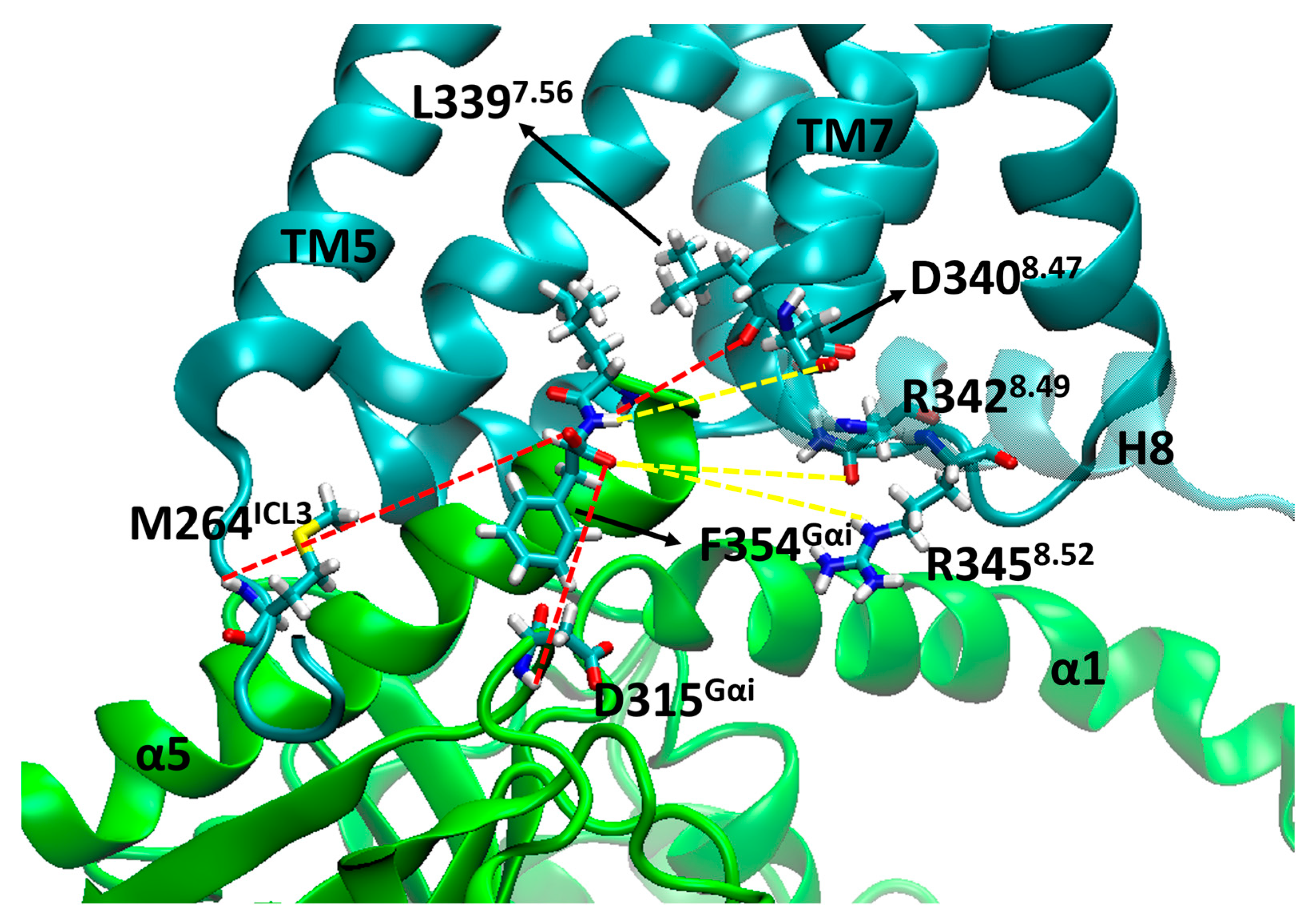
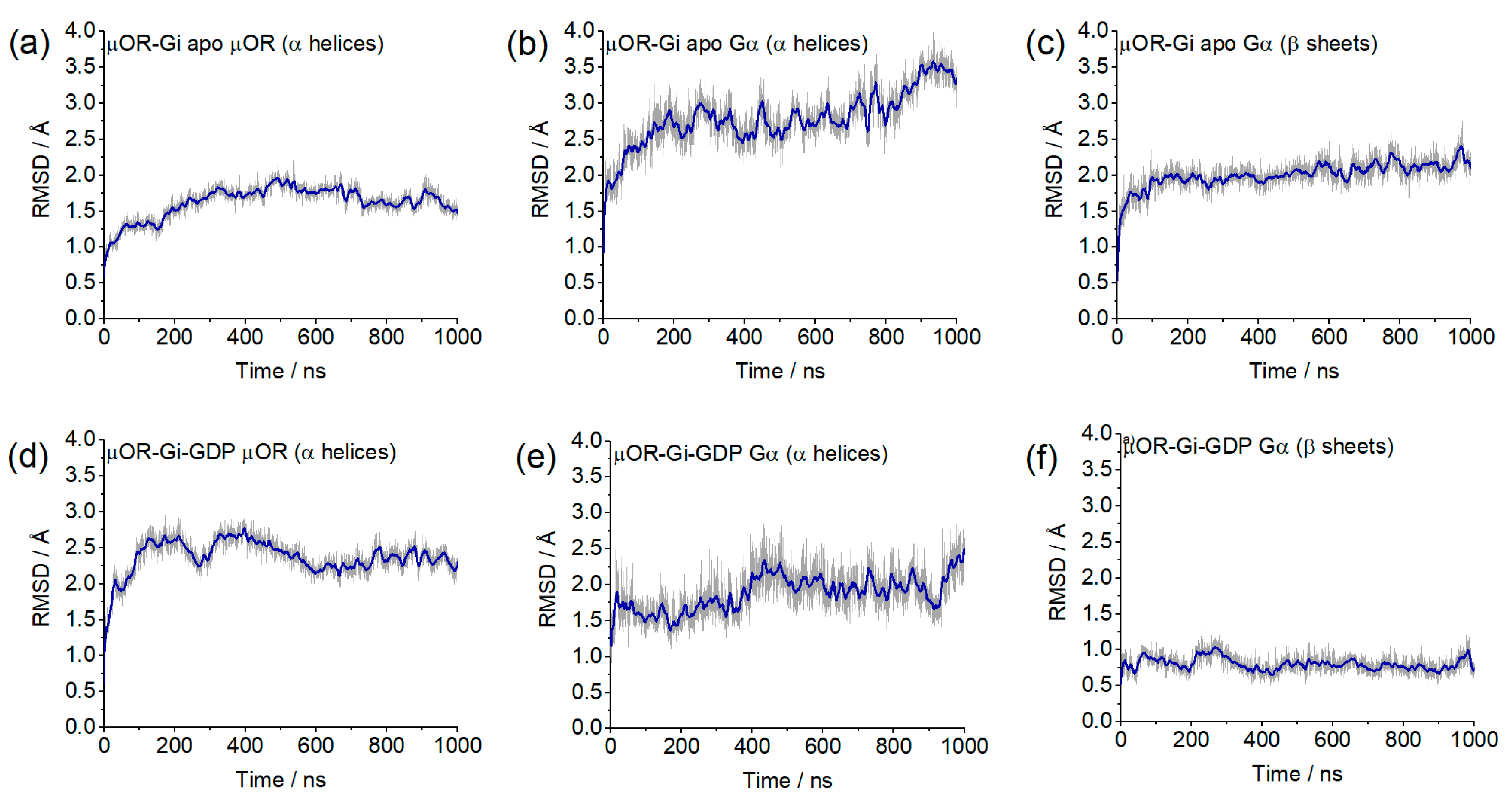
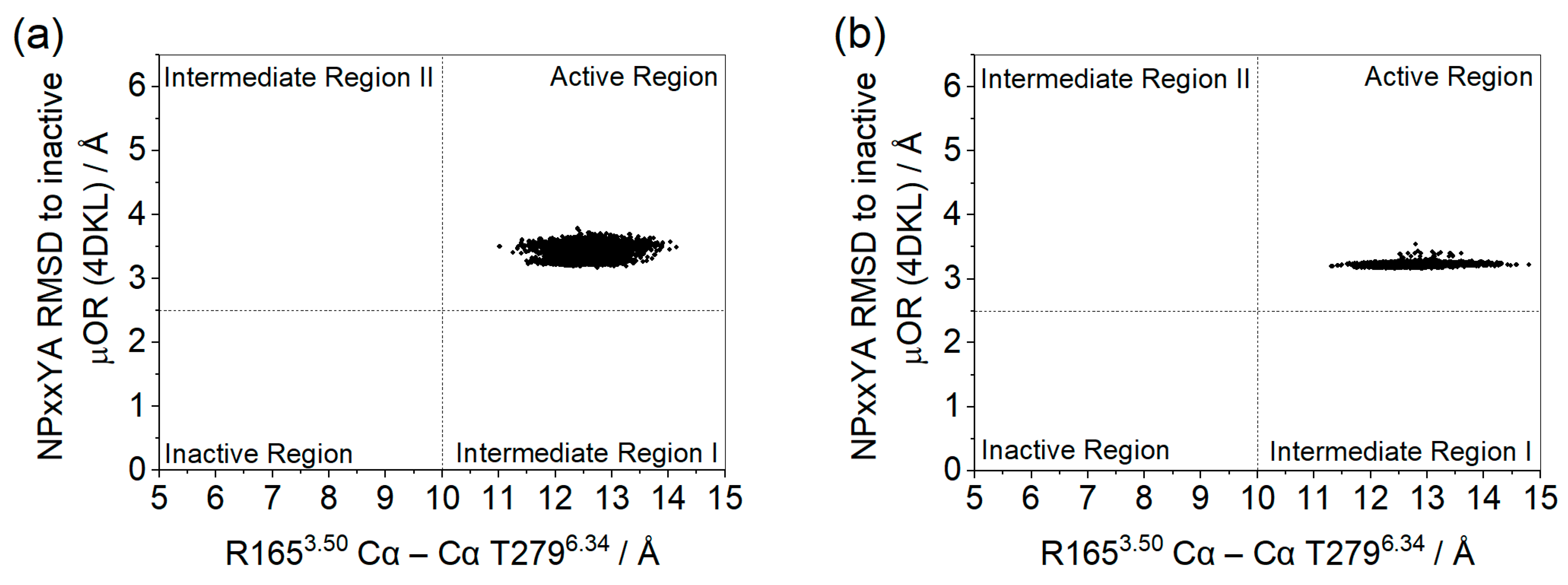
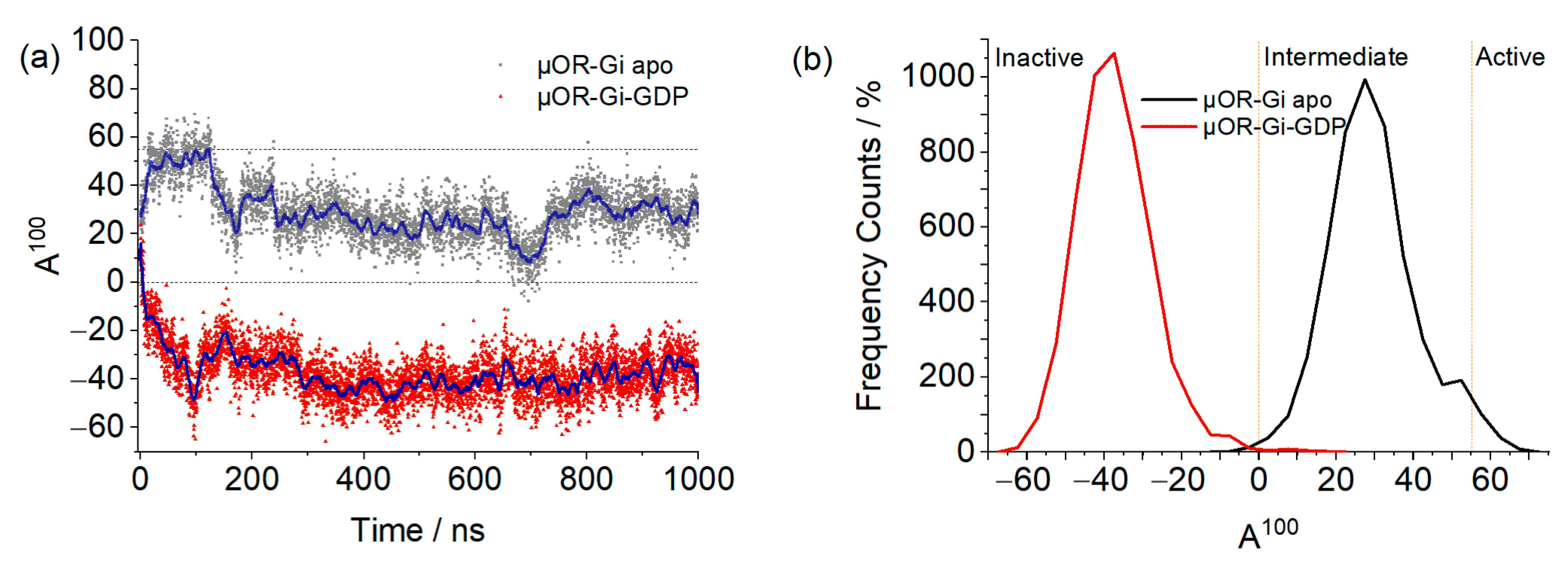
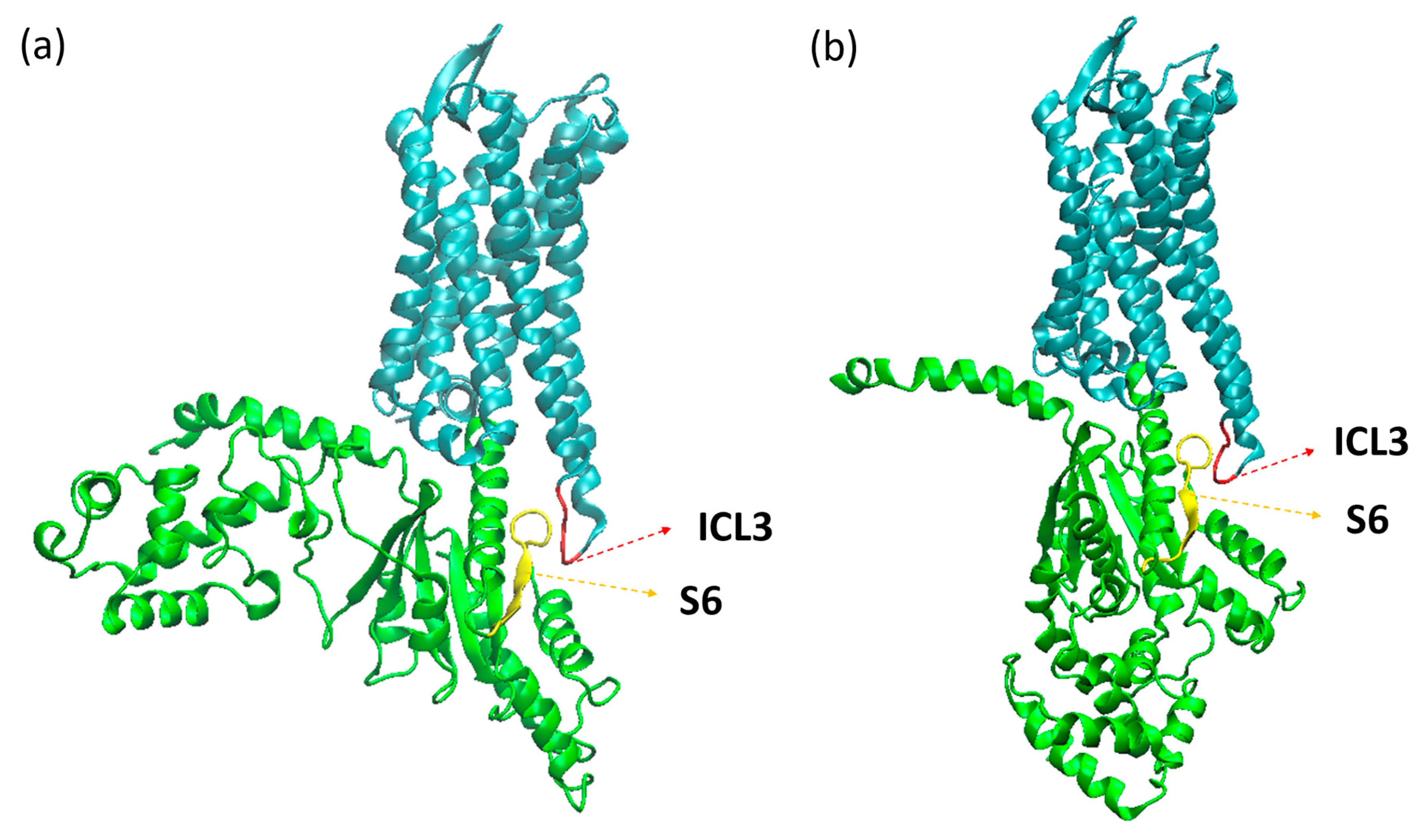
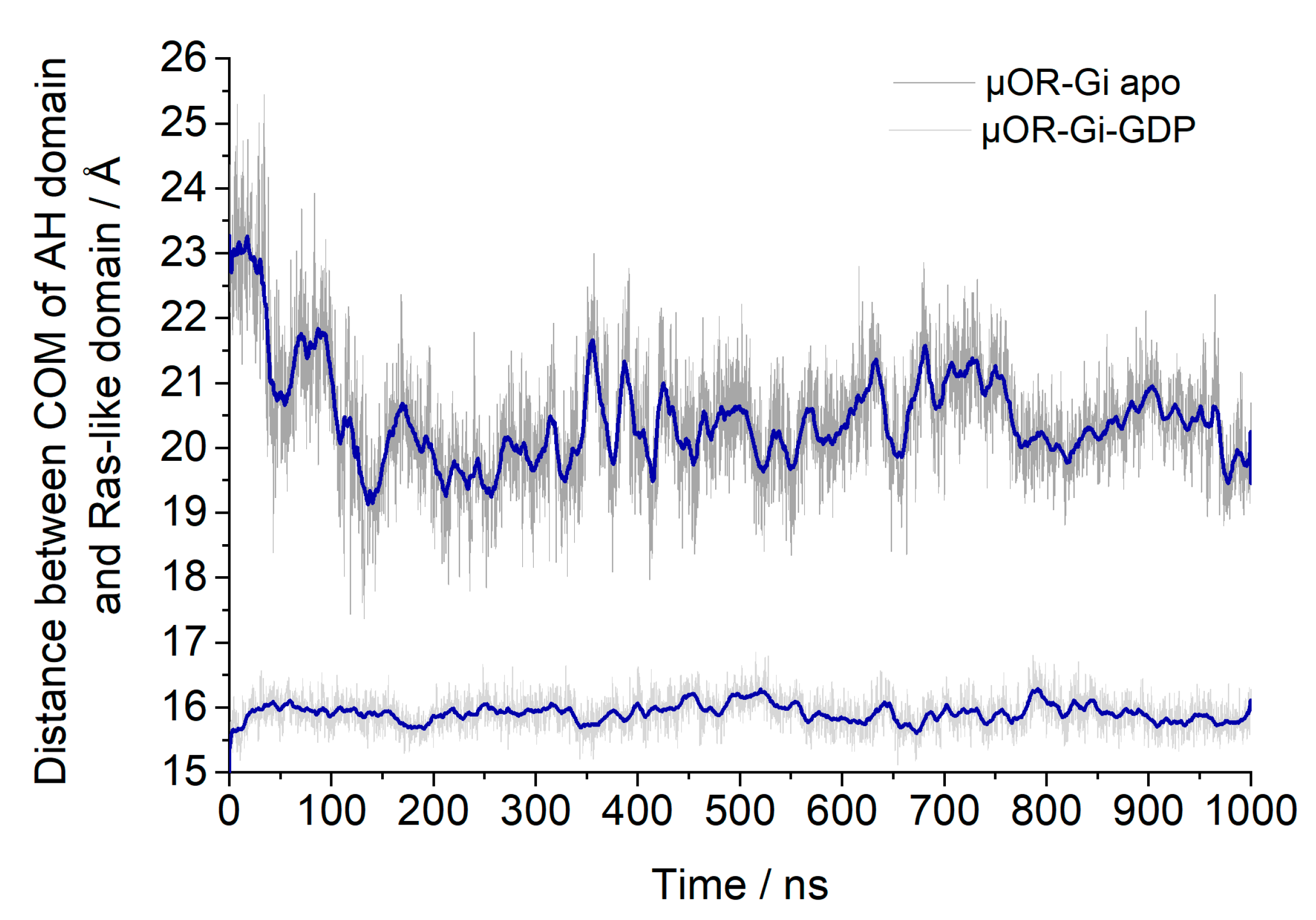
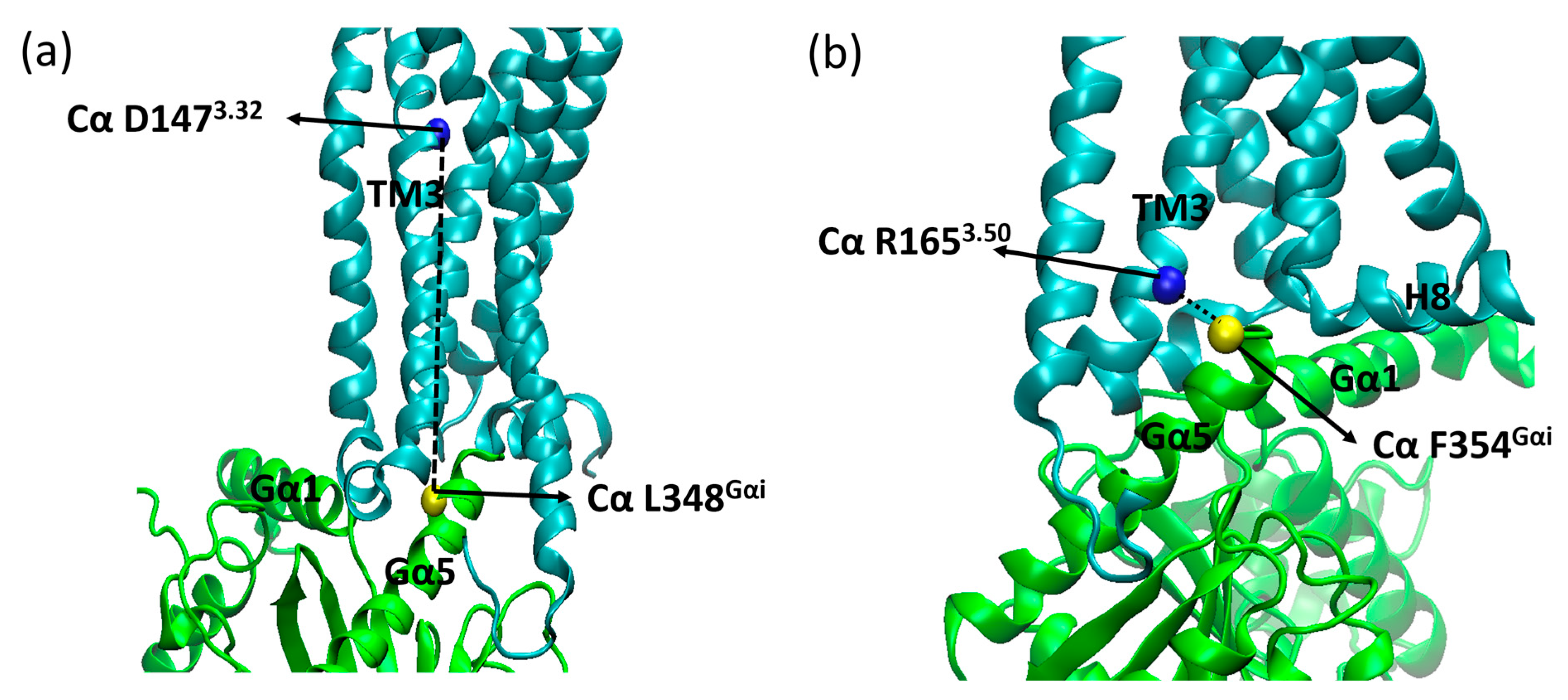
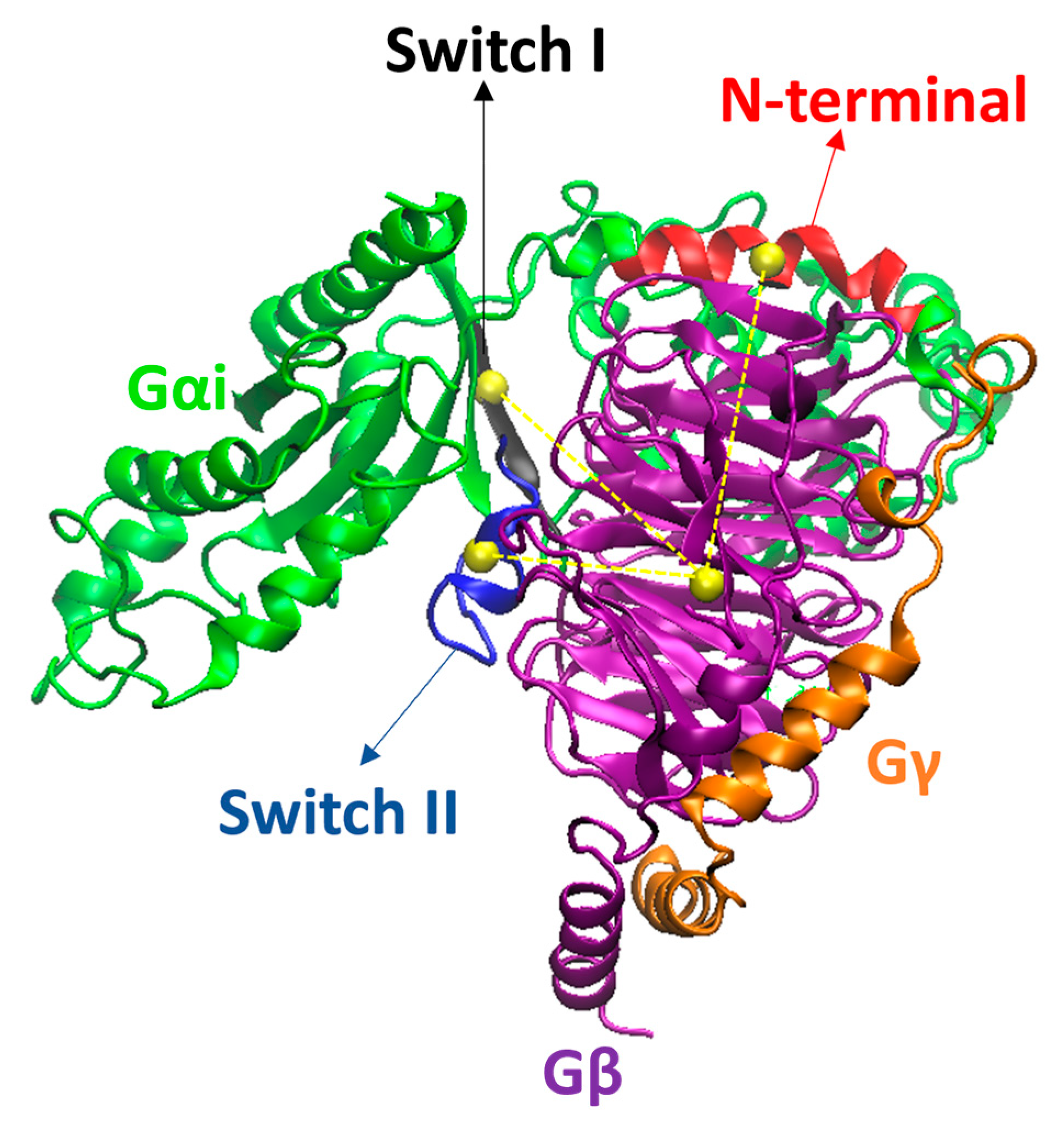
2. Results and Discussion
3. Materials and Methods
3.1. µOR—G-Protein Complex without GDP
3.2. µOR—G-Protein Complex with GDP
3.3. GDP Parametrization
3.4. Membrane Building
3.5. Structures Topologies
3.6. Membrane Insertion and Solvation
3.7. Energy Minimization
3.8. Molecular Dynamics
3.9. Assessment of Complex Contacts
3.10. RMSD Calculations
3.11. Assessment of Receptor Activation States
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bjarnadóttir, T.K.; Gloriam, D.E.; Hellstrand, S.H.; Kristiansson, H.; Fredriksson, R.; Schiöth, H.B. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 2006, 88, 263–273. [Google Scholar] [CrossRef]
- Cai, X.; Wang, D.; Zhang, R.; Chen, Y.; Chen, J. The transmembrane domains of GPCR dimers as targets for drug development. Drug Discov. Today 2023, 28, 103419. [Google Scholar] [CrossRef]
- Mafi, A.; Kim, S.K.; Goddard, W.A., 3rd. The mechanism for ligand activation of the GPCR-G protein complex. Proc. Natl. Acad. Sci. USA 2022, 119, e2110085119. [Google Scholar] [CrossRef]
- Okude, J.; Ueda, T.; Kofuku, Y.; Sato, M.; Nobuyama, N.; Kondo, K.; Shiraishi, Y.; Mizumura, T.; Onishi, K.; Natsume, M.; et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor. Angew. Chem. Int. Ed. 2015, 54, 15771–15776. [Google Scholar] [CrossRef]
- Calebiro, D.; Koszegi, Z.; Lanoiselée, Y.; Miljus, T.; O’Brien, S. G protein-coupled receptor-G protein interactions: A single-molecule perspective. Physiol. Rev. 2021, 101, 857–906. [Google Scholar] [CrossRef]
- Rovati, G.E.; Capra, V.; Neubig, R.R. The highly conserved DRY motif of class A G protein-coupled receptors: Beyond the ground state. Mol. Pharmacol. 2007, 71, 959–964. [Google Scholar] [CrossRef]
- Liu, C.; Hu, J.; Fan, W.; Zhu, P.; Cao, B.; Zheng, S.; Xia, Z.; Zhu, Y.; Zhao, A. Heterotrimeric G-protein gamma subunits regulate ABA signaling in response to drought through interacting with PP2Cs and SnRK2s in mulberry (Morus alba L.). Plant Physiol. Biochem. 2021, 161, 210–221. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Baltoumas, F.A.; Theodoropoulou, M.C.; Hamodrakas, S.J. Interactions of the alpha-subunits of heterotrimeric G-proteins with GPCRs, effectors and RGS proteins: A critical review and analysis of interacting surfaces, conformational shifts, structural diversity and electrostatic potentials. J. Struct. Biol. 2013, 182, 209–218. [Google Scholar] [CrossRef]
- Kamato, D.; Thach, L.; Bernard, R.; Chan, V.; Zheng, W.; Kaur, H.; Brimble, M.; Osman, N.; Little, P.J. Structure, Function, Pharmacology, and Therapeutic Potential of the G Protein, Gα/q,11. Front. Cardiovasc. Med. 2015, 2, 14. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the µ-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into mu-opioid receptor activation. Nature 2015, 524, 315. [Google Scholar] [CrossRef]
- Faouzi, A.; Wang, H.; Zaidi, S.A.; DiBerto, J.F.; Che, T.; Qu, Q.; Robertson, M.J.; Madasu, M.K.; El Daibani, A.; Varga, B.R.; et al. Structure-based design of bitopic ligands for the µ-opioid receptor. Nature 2023, 613, 767–774. [Google Scholar] [CrossRef]
- Wang, H.; Hetzer, F.; Huang, W.; Qu, Q.; Meyerowitz, J.; Kaindl, J.; Hübner, H.; Skiniotis, G.; Kobilka, B.K.; Gmeiner, P. Structure-Based Evolution of G Protein-Biased µ-Opioid Receptor Agonists. Angew. Chem. Int. Ed. 2022, 61, e202200269. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular recognition of morphine and fentanyl by the human µ-opioid receptor. Cell 2022, 185, 4361–4375.e19. [Google Scholar] [CrossRef]
- Liu, P.; Jia, M.Z.; Zhou, X.E.; De Waal, P.W.; Dickson, B.M.; Liu, B.; Hou, L.; Yin, Y.T.; Kang, Y.Y.; Shi, Y.; et al. The structural basis of the dominant negative phenotype of the Gαi1β1γ2 G203A/A326S heterotrimer. Acta Pharmacol. Sin. 2016, 37, 1259–1272. [Google Scholar] [CrossRef]
- Liu, S.; Paknejad, N.; Zhu, L.; Kihara, Y.; Ray, M.; Chun, J.; Liu, W.; Hite, R.K.; Huang, X.Y. Differential activation mechanisms of lipid GPCRs by lysophosphatidic acid and sphingosine 1-phosphate. Nat. Commun. 2022, 13, 731. [Google Scholar] [CrossRef]
- Maeda, S.; Koehl, A.; Matile, H.; Hu, H.; Hilger, D.; Schertler, G.F.; Manglik, A.; Skiniotis, G.; Dawson, R.J.; Kobilka, B.K. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 2018, 9, 3712. [Google Scholar] [CrossRef]
- Wall, M.A.; Coleman, D.E.; Lee, E.; Iñiguez-Lluhi, J.A.; Posner, B.A.; Gilman, A.G.; Sprang, S.R. The structure of the G protein heterotrimer Giα1β1γ2. Cell 1995, 83, 1047–1058. [Google Scholar] [CrossRef]
- Lee, Y.; Basith, S.; Choi, S. Recent Advances in Structure-Based Drug Design Targeting Class A G Protein-Coupled Receptors Utilizing Crystal Structures and Computational Simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Podlewska, S.; Bugno, R.; Kudla, L.; Bojarski, A.J.; Przewlocki, R. Molecular Modeling of micro Opioid Receptor Ligands with Various Functional Properties: PZM21, SR-17018, Morphine, and Fentanyl-Simulated Interaction Patterns Confronted with Experimental Data. Molecules 2020, 25, 4636. [Google Scholar] [CrossRef] [PubMed]
- Young, D.C. Computational Drug Design, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Rutkowska, E.; Bartuzi, D.; Targowska-Duda, K.M.; Matosiuk, D.; Selent, J. Computational methods for studying G protein-coupled receptors (GPCRs). Methods Cell Biol. 2016, 132, 359–399. [Google Scholar]
- Sena, D.M., Jr.; Cong, X.; Giorgetti, A.; Kless, A.; Carloni, P. Structural heterogeneity of the μ-opioid receptor’s conformational ensemble in the apo state. Sci. Rep. 2017, 8, 45761. [Google Scholar] [CrossRef]
- Mafi, A.; Kim, S.K.; Goddard, W.A. The G protein-first activation mechanism of opioid receptors by Gi protein and agonists. QRB Discov. 2021, 2, e9. [Google Scholar] [CrossRef] [PubMed]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Masureel, M.; Van Antwerpen, P.; Kobilka, B.K.; Govaerts, C. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol. 2016, 12, 35–39. [Google Scholar] [CrossRef]
- Luchini, A.; Vitiello, G. Mimicking the Mammalian Plasma Membrane: An Overview of Lipid Membrane Models for Biophysical Studies. Biomimetics 2020, 6, 3. [Google Scholar] [CrossRef]
- Qu, Q.; Huang, W.; Aydin, D.; Paggi, J.M.; Seven, A.B.; Wang, H.; Chakraborty, S.; Che, T.; DiBerto, J.F.; Robertson, M.J.; et al. Insights into distinct signaling profiles of the µOR activated by diverse agonists. Nat. Chem. Biol. 2023, 19, 423–430. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; et al. Structures of the entire human opioid receptor family. Cell 2023, 186, 413–427.e17. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Martinez-Rosell, G.; Provasi, D.; De Fabritiis, G.; Filizola, M. Dynamic and Kinetic Elements of mu-Opioid Receptor Functional Selectivity. Sci. Rep. 2017, 7, 11255. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, P.; Wifling, D.; Clark, T. Universal Activation Index for Class A GPCRs. J. Chem. Inf. Model. 2019, 59, 3938–3945. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, P.F.; Jarończyk, M.; Dobrowolski, J.C.; Sadlej, J. Molecular dynamics of fentanyl bound to μ-opioid receptor. J. Mol. Model. 2019, 25, 144. [Google Scholar] [CrossRef] [PubMed]
- Sena, D.M., Jr.; Cong, X.; Giorgetti, A. Ligand based conformational space studies of the μ-opioid receptor. Biochim. Et Biophys. Acta (BBA)—Gen. Subj. 2021, 1865, 129838. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Barca, G.M.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M. Amber 2021; University of California: San Francisco, CA, USA, 2021; p. 959. [Google Scholar]
- Ponder, J.W.; Case, D.A. Force Fields for Protein Simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Knight, C.J.; Hub, J.S. MemGen: A general web server for the setup of lipid membrane simulation systems. Bioinformatics 2015, 31, 2897–2899. [Google Scholar] [CrossRef]
- Grote, F.; Lyubartsev, A.P. Optimization of Slipids Force Field Parameters Describing Headgroups of Phospholipids. J. Phys. Chem. B 2020, 124, 8784–8793. [Google Scholar] [CrossRef]
- van Schaik, R.C.; Berendsen, H.J.; Torda, A.E.; van Gunsteren, W.F. A Structure Refinement Method Based on Molecular Dynamics in Four Spatial Dimensions. J. Mol. Biol. 1993, 234, 751–762. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; Van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bernetti, M.; Bussi, G. Pressure control using stochastic cell rescaling. J. Chem. Phys. 2020, 153, 114107. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins-Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Van Maaren, P.J.; Van Der Spoel, D. Molecular Dynamics Simulations of Water with Novel Shell-Model Potentials. J. Phys. Chem. B 2001, 105, 2618–2626. [Google Scholar] [CrossRef]
- Wang, D.; Sun, X.; Sadee, W. Different effects of opioid antagonists on μ-, δ-, and κ-opioid receptors with and without agonist pretreatment. J. Pharmacol. Exp. Ther. 2007, 321, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, M.R.P.d.; Bezerra, R.F.S.; Serafim, D.D.B.; Sena Junior, D.M. Dynamics of the Apo µ-Opioid Receptor in Complex with Gi Protein. Int. J. Mol. Sci. 2023, 24, 13430. https://doi.org/10.3390/ijms241713430
Lima MRPd, Bezerra RFS, Serafim DDB, Sena Junior DM. Dynamics of the Apo µ-Opioid Receptor in Complex with Gi Protein. International Journal of Molecular Sciences. 2023; 24(17):13430. https://doi.org/10.3390/ijms241713430
Chicago/Turabian StyleLima, Mira Raya Paula de, Rubem Francisco Silva Bezerra, David Denis Bento Serafim, and Diniz Maciel Sena Junior. 2023. "Dynamics of the Apo µ-Opioid Receptor in Complex with Gi Protein" International Journal of Molecular Sciences 24, no. 17: 13430. https://doi.org/10.3390/ijms241713430
APA StyleLima, M. R. P. d., Bezerra, R. F. S., Serafim, D. D. B., & Sena Junior, D. M. (2023). Dynamics of the Apo µ-Opioid Receptor in Complex with Gi Protein. International Journal of Molecular Sciences, 24(17), 13430. https://doi.org/10.3390/ijms241713430