

Human lncRNA SUGCT-AS1 Regulates the Proinflammatory Response of Macrophage
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
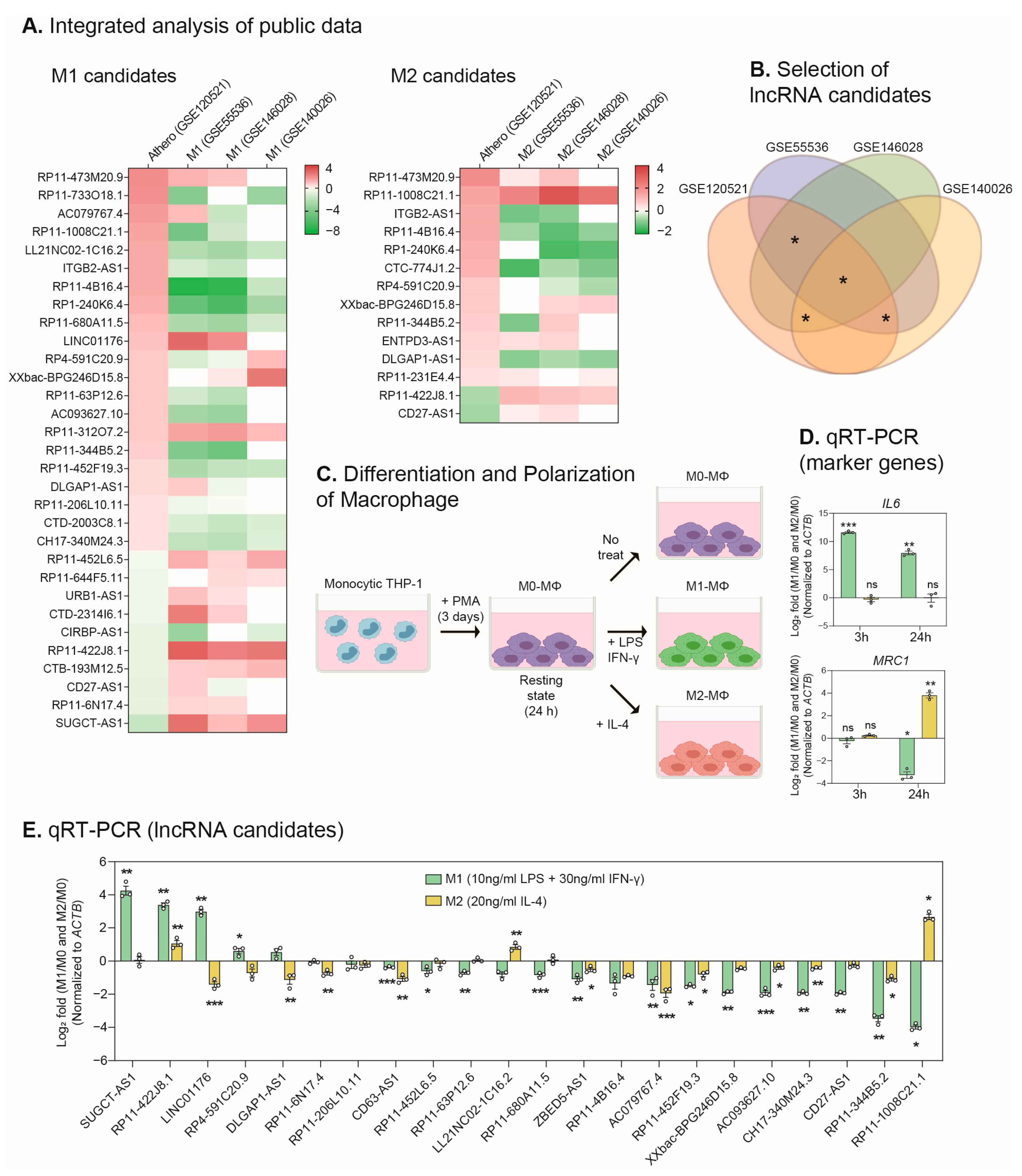
2.1. Identification of lncRNAs Involved in the Macrophage Polarization and the Progression of Atherosclerosis
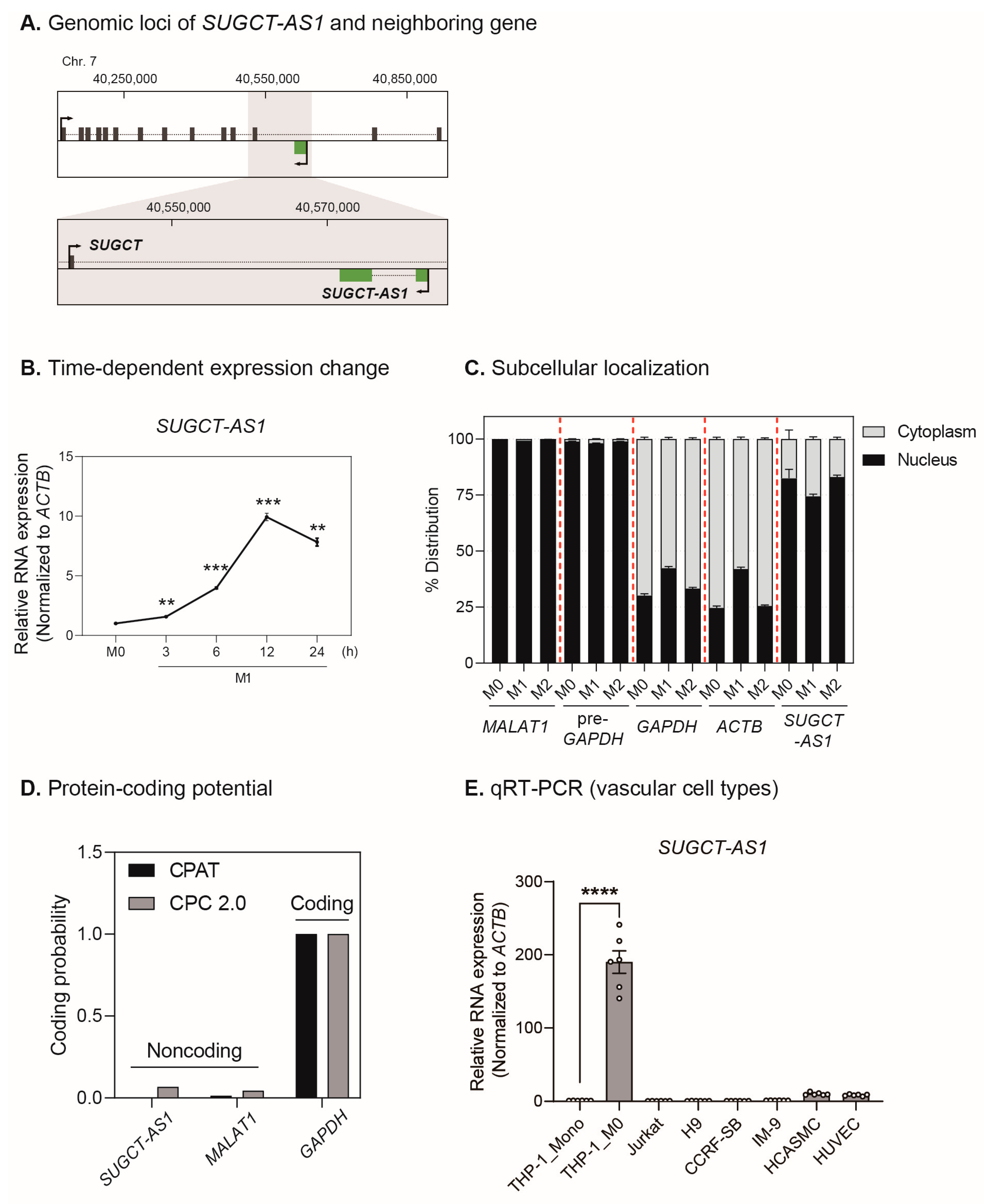
2.2. SUGCT-AS1 Is a Human Macrophage-Enriched Nuclear lncRNA
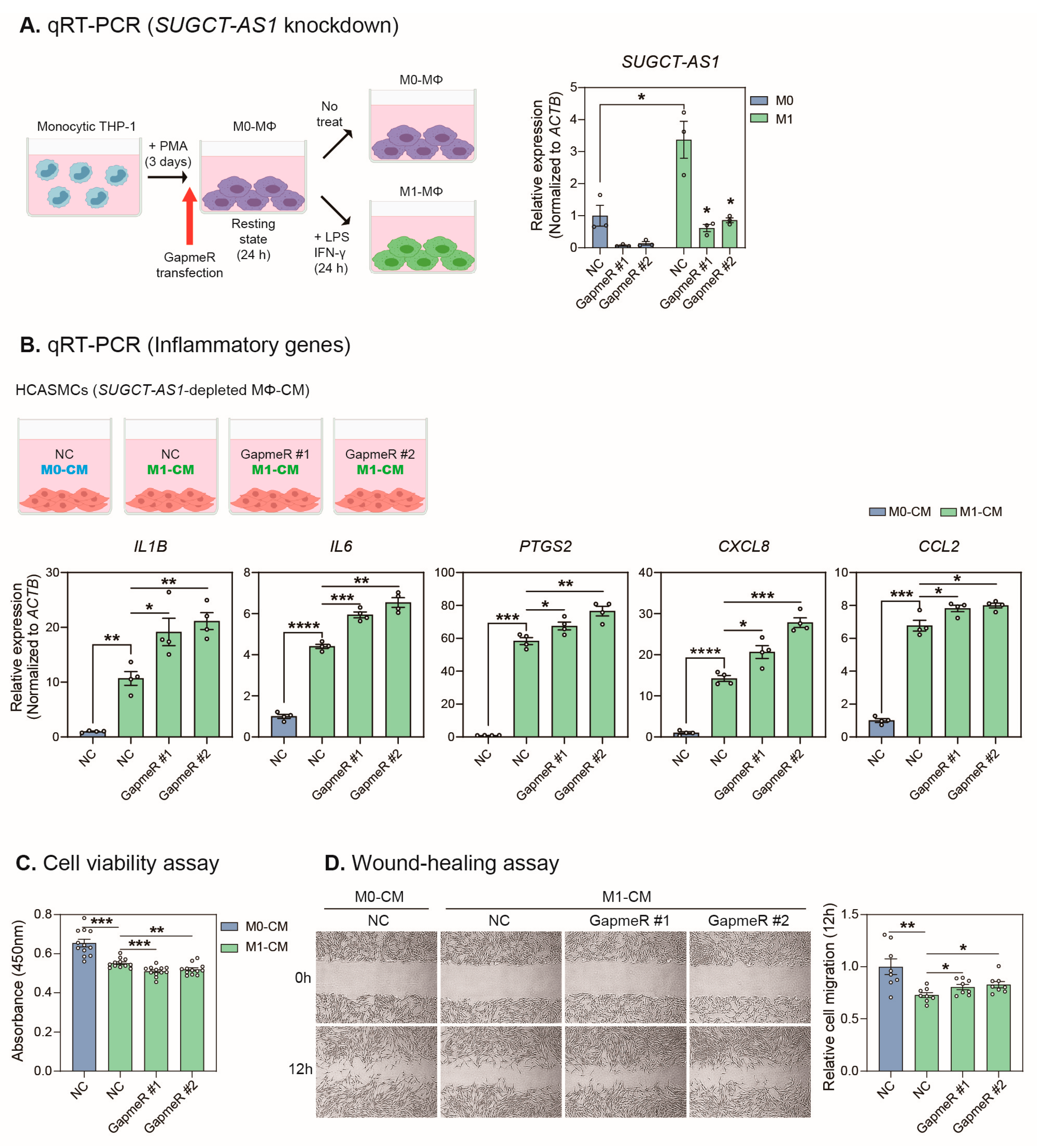
2.3. SUGCT-AS1-Depleted THP-1 Cells Induce a Pathogenic Phenotype in Vascular Smooth Muscle Cells
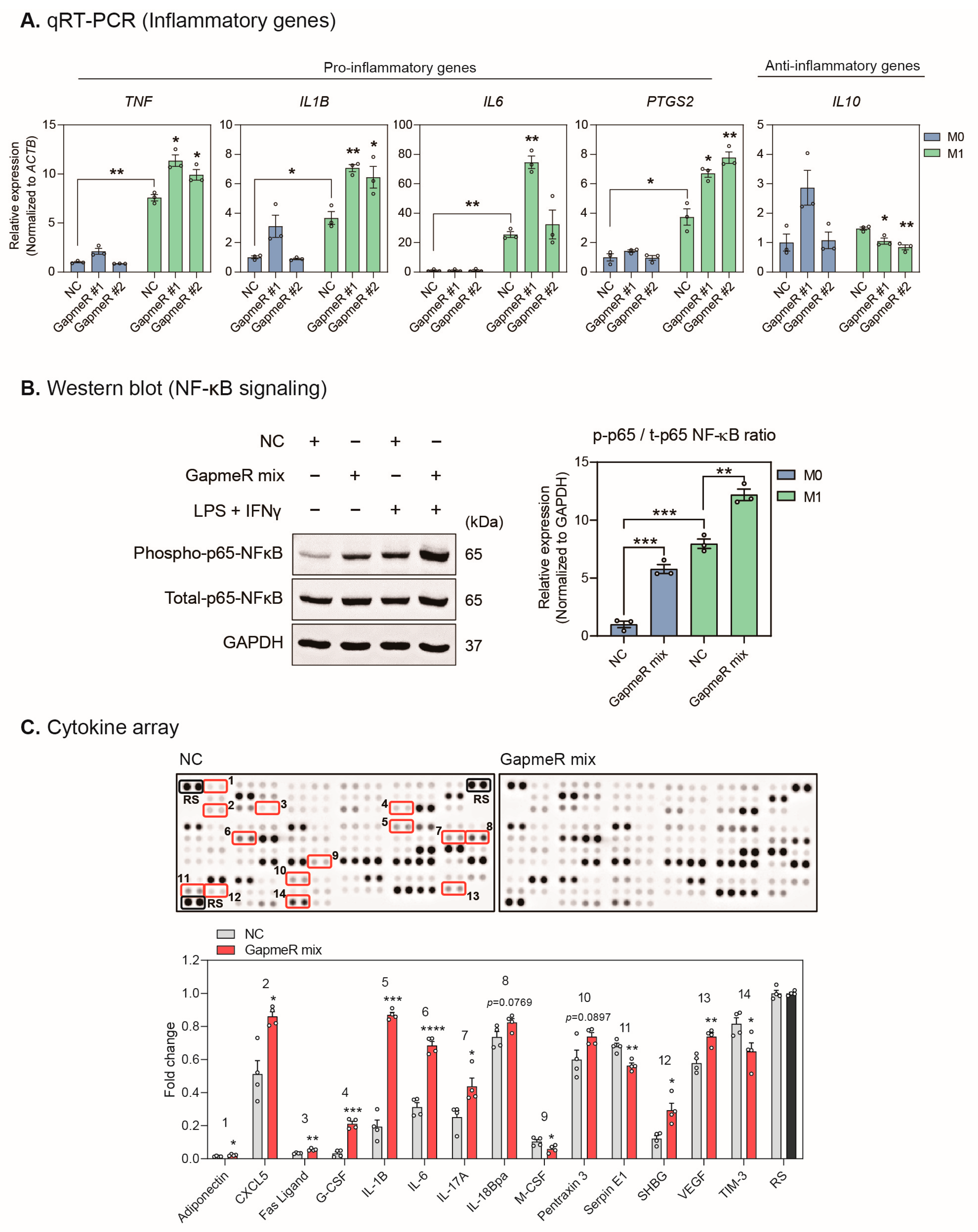
2.4. Depletion of SUGCT-AS1 Promotes the Secretion of Proinflammatory Cytokines in THP-1 Cells
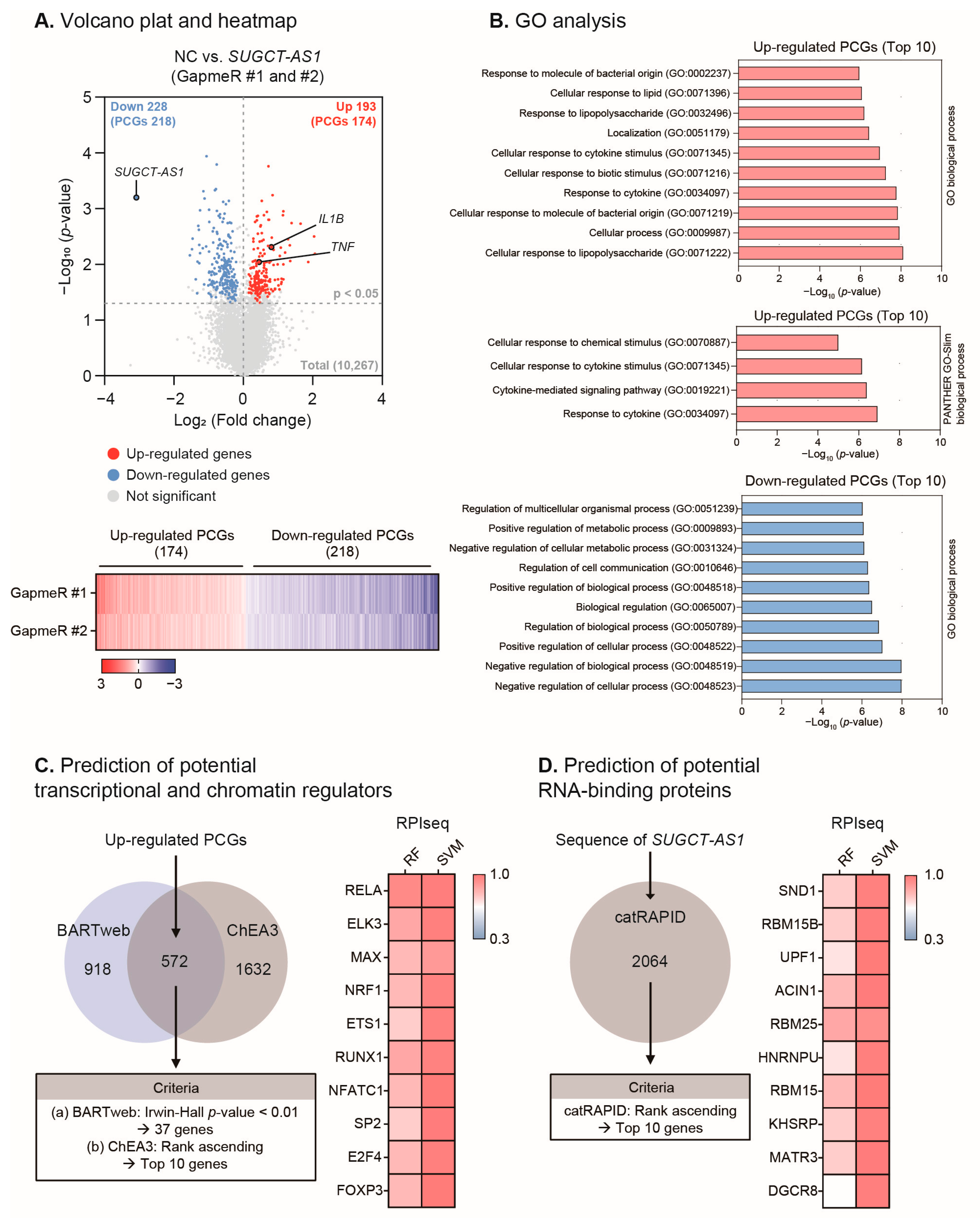
2.5. SUGCT-AS1 Alters the Expression of Genes Involved in Inflammation and the Response to Cytokines
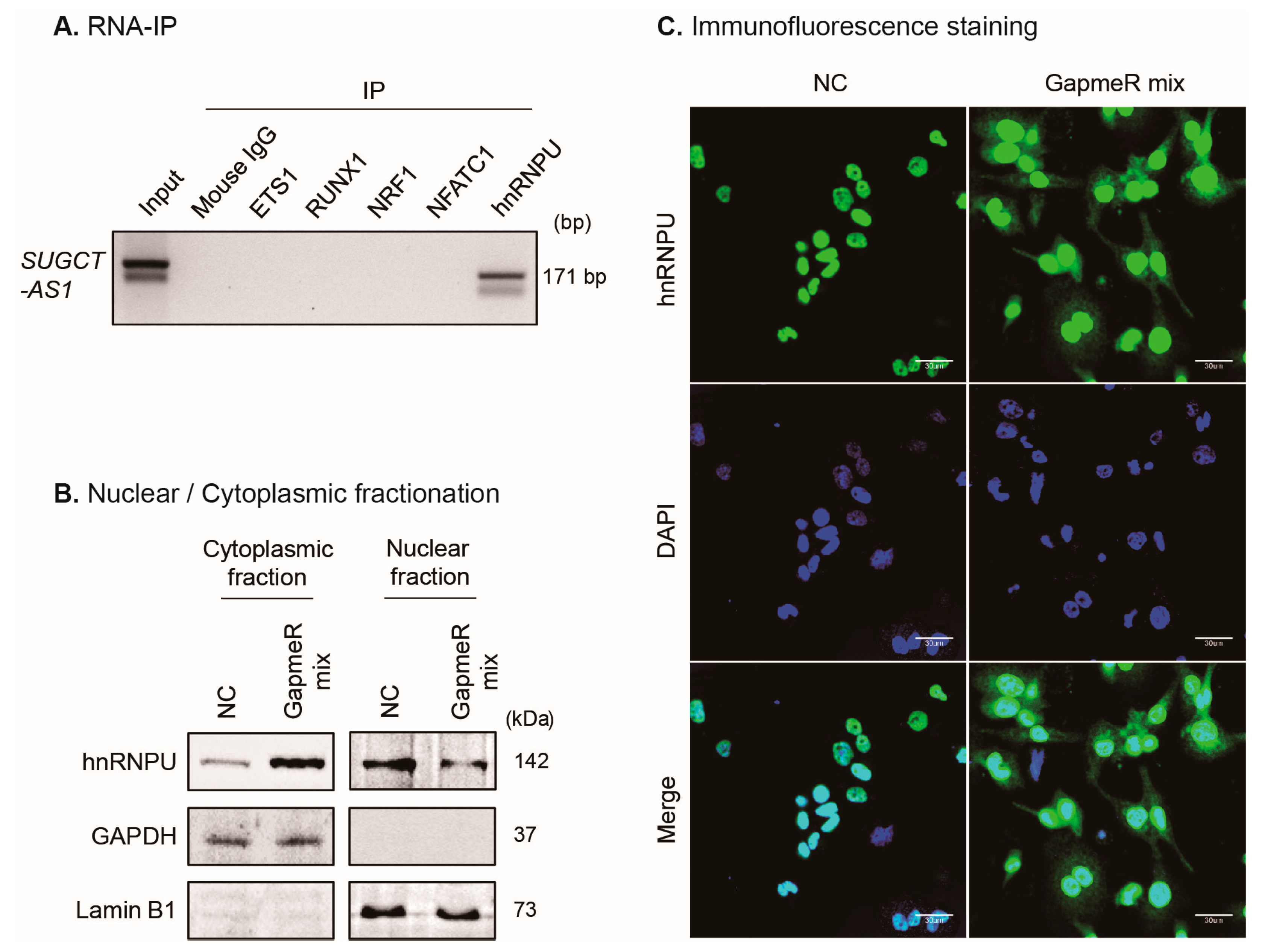
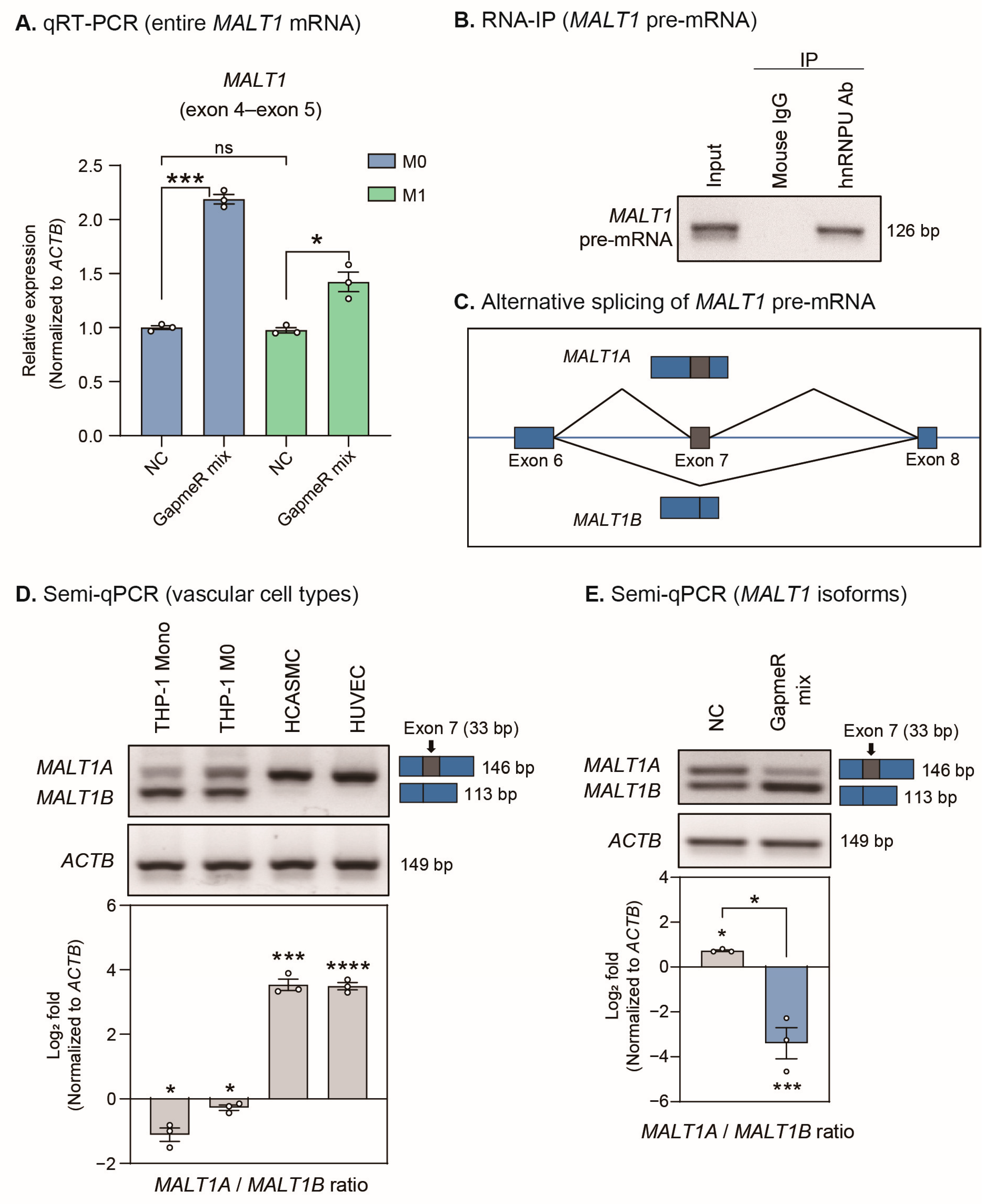
2.6. SUGCT-AS1 Regulates Alternative Splicing of MALT1 mRNA by Regulating Intracellular Translocation of hnRNPU
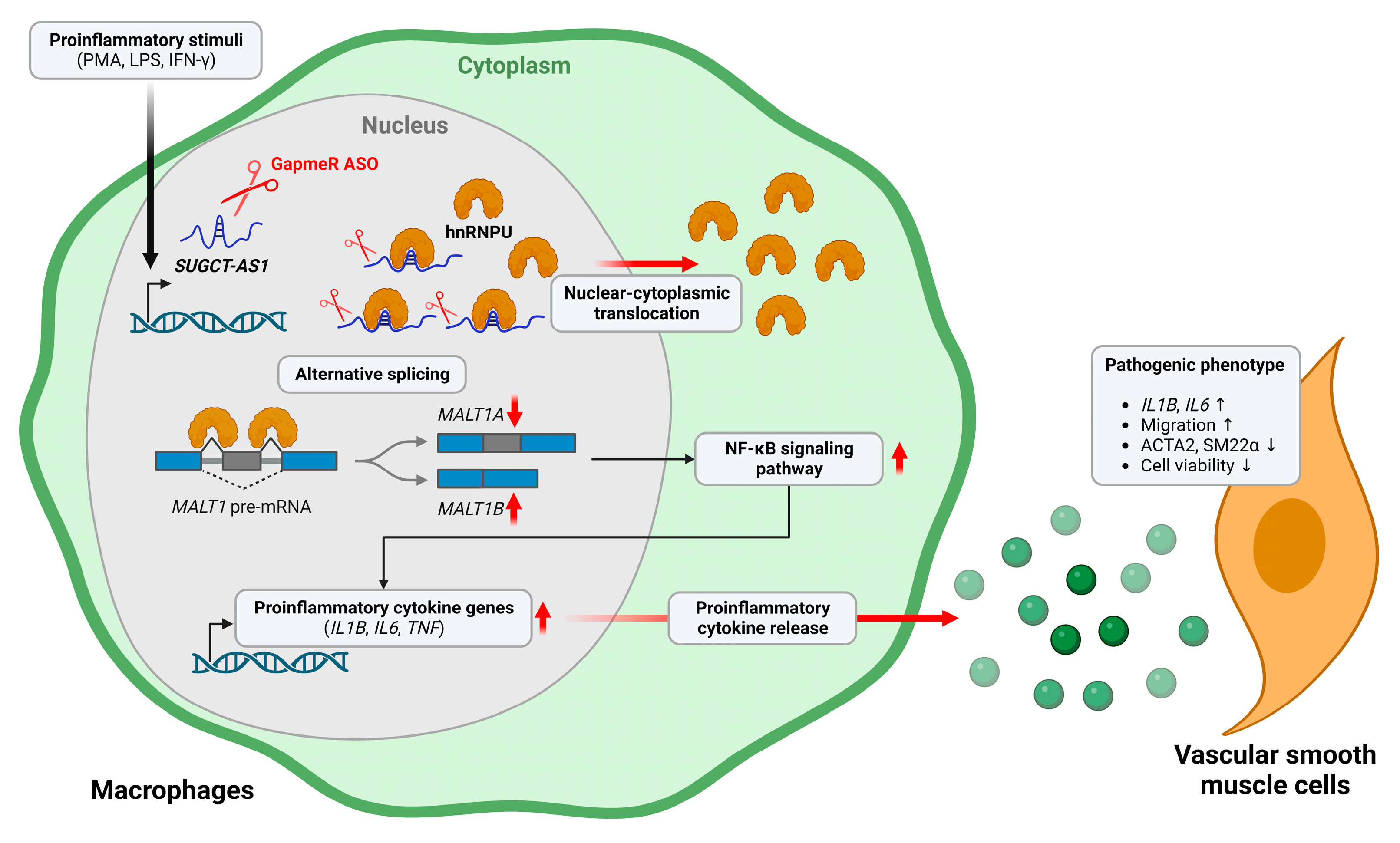
3. Discussion
4. Materials and Methods
4.1. Selection of lncRNAs Involved in Macrophage Polarization
4.2. Cell Culture
4.3. RNA Preparation and PCR
4.4. Cellular Fractionation
4.5. Cell Viability Assay
4.6. Wound Healing Assay
4.7. Western Blot Analysis
4.8. Human Cytokine Array
4.9. Suppression of lncRNA Expression
4.10. RNA Sequencing
4.11. Bioinformatics Analysis
4.12. RNA-Binding Protein Immunoprecipitation
4.13. Immunofluorescence Staining
4.14. Plasmid Construction
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Jorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Park, S.H. Regulation of Macrophage Activation and Differentiation in Atherosclerosis. J. Lipid Atheroscler. 2021, 10, 251–267. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Rua, R.; McGavern, D.B. Elucidation of monocyte/macrophage dynamics and function by intravital imaging. J. Leukoc. Biol. 2015, 98, 319–332. [Google Scholar] [CrossRef]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef]
- Park, S.; Lee, I.K. Progression of Multifaceted Immune Cells in Atherosclerotic Development. J. Lipid Atheroscler. 2019, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Li, G.; Seimon, T.A.; Kuriakose, G.; Ron, D.; Tabas, I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009, 9, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fu, Y.; Gu, X.; Xi, X.; Peng, X.; Wang, C.; Sun, Q.; Wang, X.; Qian, F.; Qin, Z.; et al. Macrophage-Enriched lncRNA RAPIA: A Novel Therapeutic Target for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1464–1478. [Google Scholar] [CrossRef]
- Simion, V.; Zhou, H.; Pierce, J.B.; Yang, D.; Haemmig, S.; Tesmenitsky, Y.; Sukhova, G.; Stone, P.H.; Libby, P.; Feinberg, M.W. LncRNA VINAS regulates atherosclerosis by modulating NF-kappaB and MAPK signaling. JCI Insight 2020, 5, e140627. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef]
- Frankish, A.; Carbonell-Sala, S.; Diekhans, M.; Jungreis, I.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Arnan, C.; Barnes, I.; et al. GENCODE: Reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 2023, 51, D942–D949. [Google Scholar] [CrossRef]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Fasolo, F.; Di Gregoli, K.; Maegdefessel, L.; Johnson, J.L. Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc. Res. 2019, 115, 1732–1756. [Google Scholar] [CrossRef]
- Simion, V.; Zhou, H.; Haemmig, S.; Pierce, J.B.; Mendes, S.; Tesmenitsky, Y.; Perez-Cremades, D.; Lee, J.F.; Chen, A.F.; Ronda, N.; et al. A macrophage-specific lncRNA regulates apoptosis and atherosclerosis by tethering HuR in the nucleus. Nat. Commun. 2020, 11, 6135. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, C.; Wang, Y.; Shi, J.; Zhang, X.; Li, W.; Nunez, S.; Foulkes, A.S.; Lin, J.; Hinkle, C.C.; et al. Deep RNA Sequencing Uncovers a Repertoire of Human Macrophage Long Intergenic Noncoding RNAs Modulated by Macrophage Activation and Associated with Cardiometabolic Diseases. J. Am. Heart Assoc. 2017, 6, e007431. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.D.; Ballantyne, M.D.; Miscianinov, V.; Pinel, K.; Hung, J.; Scanlon, J.P.; Iyinikkel, J.; Kaczynski, J.; Tavares, A.S.; Bradshaw, A.C.; et al. The Human-Specific and Smooth Muscle Cell-Enriched LncRNA SMILR Promotes Proliferation by Regulating Mitotic CENPF mRNA and Drives Cell-Cycle Progression Which Can Be Targeted to Limit Vascular Remodeling. Circ. Res. 2019, 125, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xue, C.; Shah, R.; Bermingham, K.; Hinkle, C.C.; Li, W.; Rodrigues, A.; Tabita-Martinez, J.; Millar, J.S.; Cuchel, M.; et al. Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circ. Res. 2015, 117, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Gurvich, O.L.; Puttonen, K.A.; Bailey, A.; Kailaanmaki, A.; Skirdenko, V.; Sivonen, M.; Pietikainen, S.; Parker, N.R.; Yla-Herttuala, S.; Kekarainen, T. Transcriptomics uncovers substantial variability associated with alterations in manufacturing processes of macrophage cell therapy products. Sci. Rep. 2020, 10, 14049. [Google Scholar] [CrossRef]
- Halaby, M.J.; Hezaveh, K.; Lamorte, S.; Ciudad, M.T.; Kloetgen, A.; MacLeod, B.L.; Guo, M.; Chakravarthy, A.; Medina, T.D.S.; Ugel, S.; et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci. Immunol. 2019, 4, eaax8189. [Google Scholar] [CrossRef]
- Simmonds, R.E. Transient up-regulation of miR-155-3p by lipopolysaccharide in primary human monocyte-derived macrophages results in RISC incorporation but does not alter TNF expression. Wellcome Open Res. 2019, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, M.; Chen, B.; Chen, Y.; Wang, X.; Rai, K.R.; Zhao, Z.; Liu, S.; Li, Y.; Xiao, M.; Chen, J.L. Identification of lncRNA-155 encoded by MIR155HG as a novel regulator of innate immunity against influenza A virus infection. Cell Microbiol. 2019, 21, e13036. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xue, Y.; Han, Y.; Lin, L.; Wu, C.; Xu, S.; Jiang, Z.; Xu, J.; Liu, Q.; Cao, X. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science 2014, 344, 310–313. [Google Scholar] [CrossRef]
- Zhang, X.; Li, D.Y.; Reilly, M.P. Long intergenic noncoding RNAs in cardiovascular diseases: Challenges and strategies for physiological studies and translation. Atherosclerosis 2019, 281, 180–188. [Google Scholar] [CrossRef]
- Liang, H.; Yu, M.; Yang, R.; Zhang, L.; Zhang, L.; Zhu, D.; Luo, H.; Hong, Y.; Yu, T.; Sun, J.; et al. A PTAL-miR-101-FN1 Axis Promotes EMT and Invasion-Metastasis in Serous Ovarian Cancer. Mol. Ther. Oncolytics 2020, 16, 53–62. [Google Scholar] [CrossRef]
- Hao, X.; Li, Y.; Huang, G.; Zeng, Y. Role of the N6-methyladenosine regulatory factor in reducing the risk of cardiovascular disease: Subtype diagnosis following aerobic exercise-assisted weight loss. Am. J. Transl. Res. 2022, 14, 5363–5378. [Google Scholar]
- Li, T.; Wang, T.; Jing, J.; Sun, L. Expression Pattern and Clinical Value of Key m6A RNA Modification Regulators in Abdominal Aortic Aneurysm. J. Inflamm. Res. 2021, 14, 4245–4258. [Google Scholar] [CrossRef]
- Ye, J.; Beetz, N.; O’Keeffe, S.; Tapia, J.C.; Macpherson, L.; Chen, W.V.; Bassel-Duby, R.; Olson, E.N.; Maniatis, T. hnRNP U protein is required for normal pre-mRNA splicing and postnatal heart development and function. Proc. Natl. Acad. Sci. USA 2015, 112, E3020–E3029. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.L.; Lai, T.C.; Lin, S.R.; Lin, S.W.; Chen, Y.C.; Pu, C.M.; Lee, I.T.; Tsai, J.S.; Lee, C.W.; Chen, Y.L. Conditioned medium from adipose-derived stem cells attenuates ischemia/reperfusion-induced cardiac injury through the microRNA-221/222/PUMA/ETS-1 pathway. Theranostics 2021, 11, 3131–3149. [Google Scholar] [CrossRef] [PubMed]
- Riddell, A.; McBride, M.; Braun, T.; Nicklin, S.A.; Cameron, E.; Loughrey, C.M.; Martin, T.P. RUNX1: An emerging therapeutic target for cardiovascular disease. Cardiovasc. Res. 2020, 116, 1410–1423. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Sun, L.; Wang, G.; Chen, B.; Luo, F. RUNX1: A Regulator of NF-kB Signaling in Pulmonary Diseases. Curr. Protein Pept. Sci. 2018, 19, 172–178. [Google Scholar] [CrossRef]
- Cui, M.; Atmanli, A.; Morales, M.G.; Tan, W.; Chen, K.; Xiao, X.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Nrf1 promotes heart regeneration and repair by regulating proteostasis and redox balance. Nat. Commun. 2021, 12, 5270. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, F.; Gentile, A.; Gauvrit, S.; Stainier, D.Y.R.; Bensimon-Brito, A. Nfatc1 Promotes Interstitial Cell Formation During Cardiac Valve Development in Zebrafish. Circ. Res. 2020, 126, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.K.; Brown, M.; Wilhide, M.; He, S.; Ren, X. NF-kappaB in cardiovascular disease: Diverse and specific effects of a “general” transcription factor? Cardiovasc. Toxicol. 2005, 5, 183–202. [Google Scholar] [CrossRef]
- Siomi, M.C.; Eder, P.S.; Kataoka, N.; Wan, L.; Liu, Q.; Dreyfuss, G. Transportin-mediated nuclear import of heterogeneous nuclear RNP proteins. J. Cell Biol. 1997, 138, 1181–1192. [Google Scholar] [CrossRef]
- Roth, S.; Khalaila, I. The effect of O-GlcNAcylation on hnRNP A1 translocation and interaction with transportin1. Exp. Cell Res. 2017, 350, 210–217. [Google Scholar] [CrossRef]
- Meininger, I.; Griesbach, R.A.; Hu, D.; Gehring, T.; Seeholzer, T.; Bertossi, A.; Kranich, J.; Oeckinghaus, A.; Eitelhuber, A.C.; Greczmiel, U.; et al. Alternative splicing of MALT1 controls signalling and activation of CD4(+) T cells. Nat. Commun. 2016, 7, 11292. [Google Scholar] [CrossRef]
- Delekta, P.C.; Apel, I.J.; Gu, S.; Siu, K.; Hattori, Y.; McAllister-Lucas, L.M.; Lucas, P.C. Thrombin-dependent NF-kappaB activation and monocyte/endothelial adhesion are mediated by the CARMA3.Bcl10.MALT1 signalosome. J. Biol. Chem. 2010, 285, 41432–41442. [Google Scholar] [CrossRef]
- Lim, Y.H.; Ryu, J.; Kook, H.; Kim, Y.K. Identification of Long Noncoding RNAs Involved in Differentiation and Survival of Vascular Smooth Muscle Cells. Mol. Ther. Nucleic Acids 2020, 22, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.T.; Heward, J.A.; Donnelly, L.E.; Jones, S.W.; Lindsay, M.A. Catalog of Differentially Expressed Long Non-Coding RNA following Activation of Human and Mouse Innate Immune Response. Front. Immunol. 2017, 8, 1038. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Turner, D.; Mesirov, J.P. igv.js: An embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 2023, 39, btac830. [Google Scholar] [CrossRef] [PubMed]
- Kutukculer, N.; Seeholzer, T.; O’Neill, T.J.; Grass, C.; Aykut, A.; Karaca, N.E.; Durmaz, A.; Cogulu, O.; Aksu, G.; Gehring, T.; et al. Human immune disorder associated with homozygous hypomorphic mutation affecting MALT1B splice variant. J. Allergy Clin. Immunol. 2021, 147, 775–778.e778. [Google Scholar] [CrossRef]
- Vargova, K.; Curik, N.; Burda, P.; Basova, P.; Kulvait, V.; Pospisil, V.; Savvulidi, F.; Kokavec, J.; Necas, E.; Berkova, A.; et al. MYB transcriptionally regulates the miR-155 host gene in chronic lymphocytic leukemia. Blood 2011, 117, 3816–3825. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214. [Google Scholar] [CrossRef]
- Jones, A.N.; Grass, C.; Meininger, I.; Geerlof, A.; Klostermann, M.; Zarnack, K.; Krappmann, D.; Sattler, M. Modulation of pre-mRNA structure by hnRNP proteins regulates alternative splicing of MALT1. Sci. Adv. 2022, 8, eabp9153. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Zhang, M.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. Nuclear to cytoplasmic translocation of heterogeneous nuclear ribonucleoprotein U enhances TLR-induced proinflammatory cytokine production by stabilizing mRNAs in macrophages. J. Immunol. 2012, 188, 3179–3187. [Google Scholar] [CrossRef]
- Yao, Z.; Duan, S.; Hou, D.; Wang, W.; Wang, G.; Liu, Y.; Wen, L.; Wu, M. B23 acts as a nucleolar stress sensor and promotes cell survival through its dynamic interaction with hnRNPU and hnRNPA1. Oncogene 2010, 29, 1821–1834. [Google Scholar] [CrossRef]
- Creamer, K.M.; Kolpa, H.J.; Lawrence, J.B. Nascent RNA scaffolds contribute to chromosome territory architecture and counter chromatin compaction. Mol. Cell 2021, 81, 3509–3525.e3505. [Google Scholar] [CrossRef]
- Herman, A.B.; Tsitsipatis, D.; Gorospe, M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol. Cell 2022, 82, 2252–2266. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Ouyang, X.P.; Yu, X.H.; Novak, P.; Zhou, L.; He, P.P.; Yin, K. N6-Adenosine Methylation (m(6)A) RNA Modification: An Emerging Role in Cardiovascular Diseases. J. Cardiovasc. Transl. Res. 2021, 14, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, Y.K. Discovery and Functional Prediction of Long Non-Coding RNAs Common to Ischemic Stroke and Myocardial Infarction. J. Lipid Atheroscler. 2020, 9, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Y.-H.; Yoon, G.; Ryu, Y.; Jeong, D.; Song, J.; Kim, Y.S.; Ahn, Y.; Kook, H.; Kim, Y.-K. Human lncRNA SUGCT-AS1 Regulates the Proinflammatory Response of Macrophage. Int. J. Mol. Sci. 2023, 24, 13315. https://doi.org/10.3390/ijms241713315
Lim Y-H, Yoon G, Ryu Y, Jeong D, Song J, Kim YS, Ahn Y, Kook H, Kim Y-K. Human lncRNA SUGCT-AS1 Regulates the Proinflammatory Response of Macrophage. International Journal of Molecular Sciences. 2023; 24(17):13315. https://doi.org/10.3390/ijms241713315
Chicago/Turabian StyleLim, Yeong-Hwan, Gwangho Yoon, Yeongseo Ryu, Dahee Jeong, Juhyun Song, Yong Sook Kim, Youngkeun Ahn, Hyun Kook, and Young-Kook Kim. 2023. "Human lncRNA SUGCT-AS1 Regulates the Proinflammatory Response of Macrophage" International Journal of Molecular Sciences 24, no. 17: 13315. https://doi.org/10.3390/ijms241713315
APA StyleLim, Y.-H., Yoon, G., Ryu, Y., Jeong, D., Song, J., Kim, Y. S., Ahn, Y., Kook, H., & Kim, Y.-K. (2023). Human lncRNA SUGCT-AS1 Regulates the Proinflammatory Response of Macrophage. International Journal of Molecular Sciences, 24(17), 13315. https://doi.org/10.3390/ijms241713315