

Circulating miRNA Expression Is Inversely Correlated with Tumor Tissue or Sentinel Lymph Nodes in Estrogen Receptor-Positive Early Breast Cancer Patients

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Patients
2.2. RNA-Sequencing
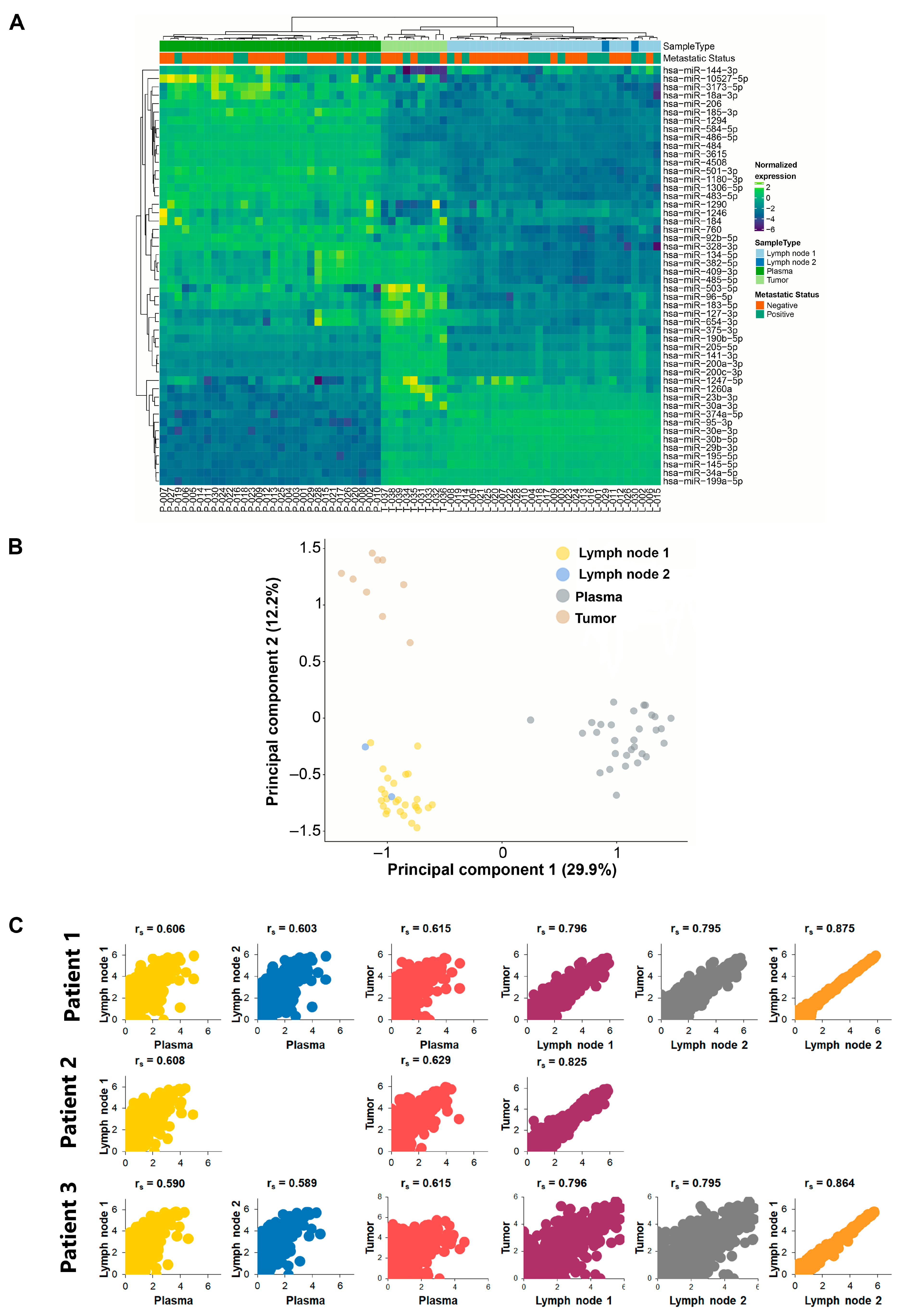
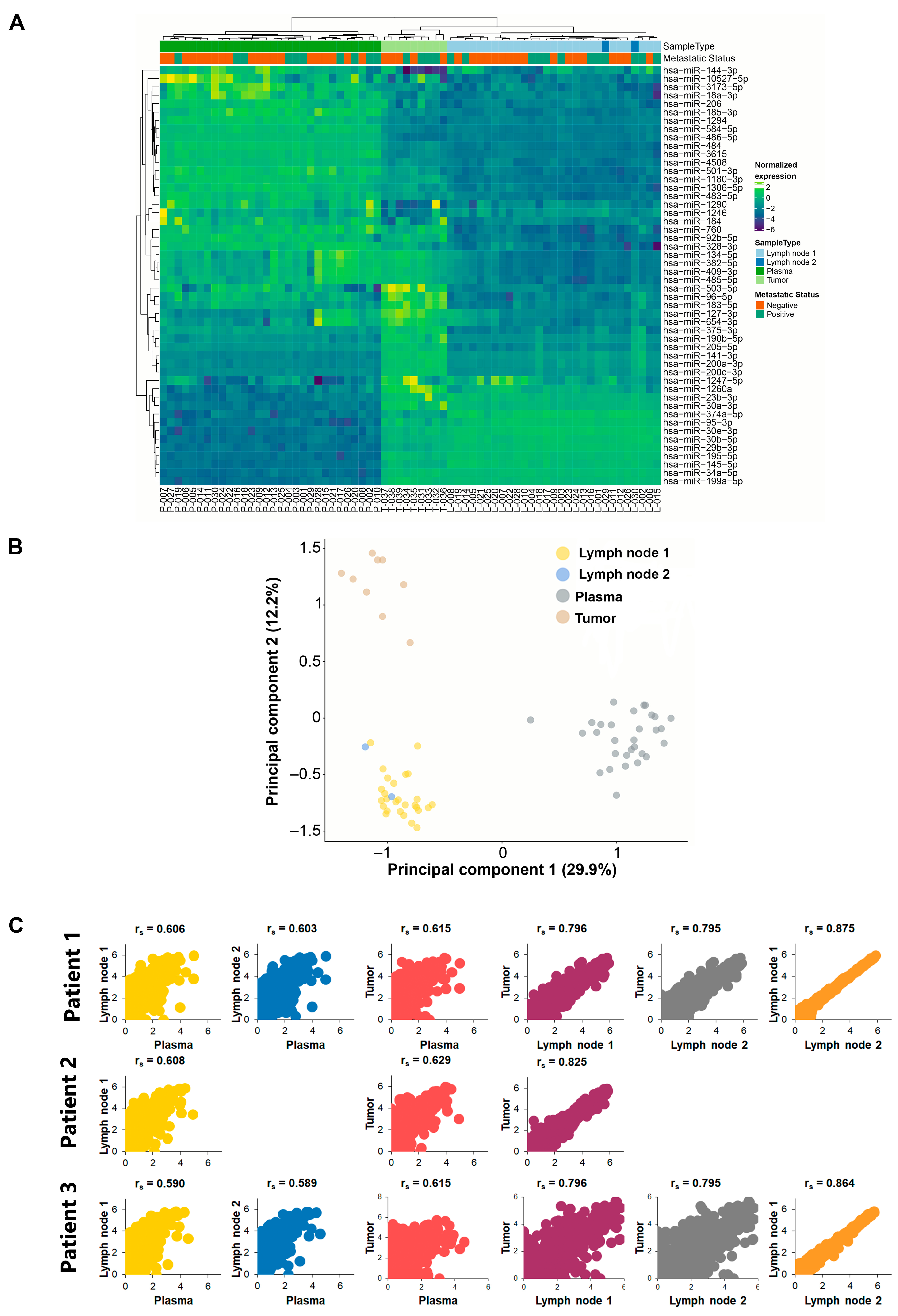
2.3. Unsupervised Clustering Analysis
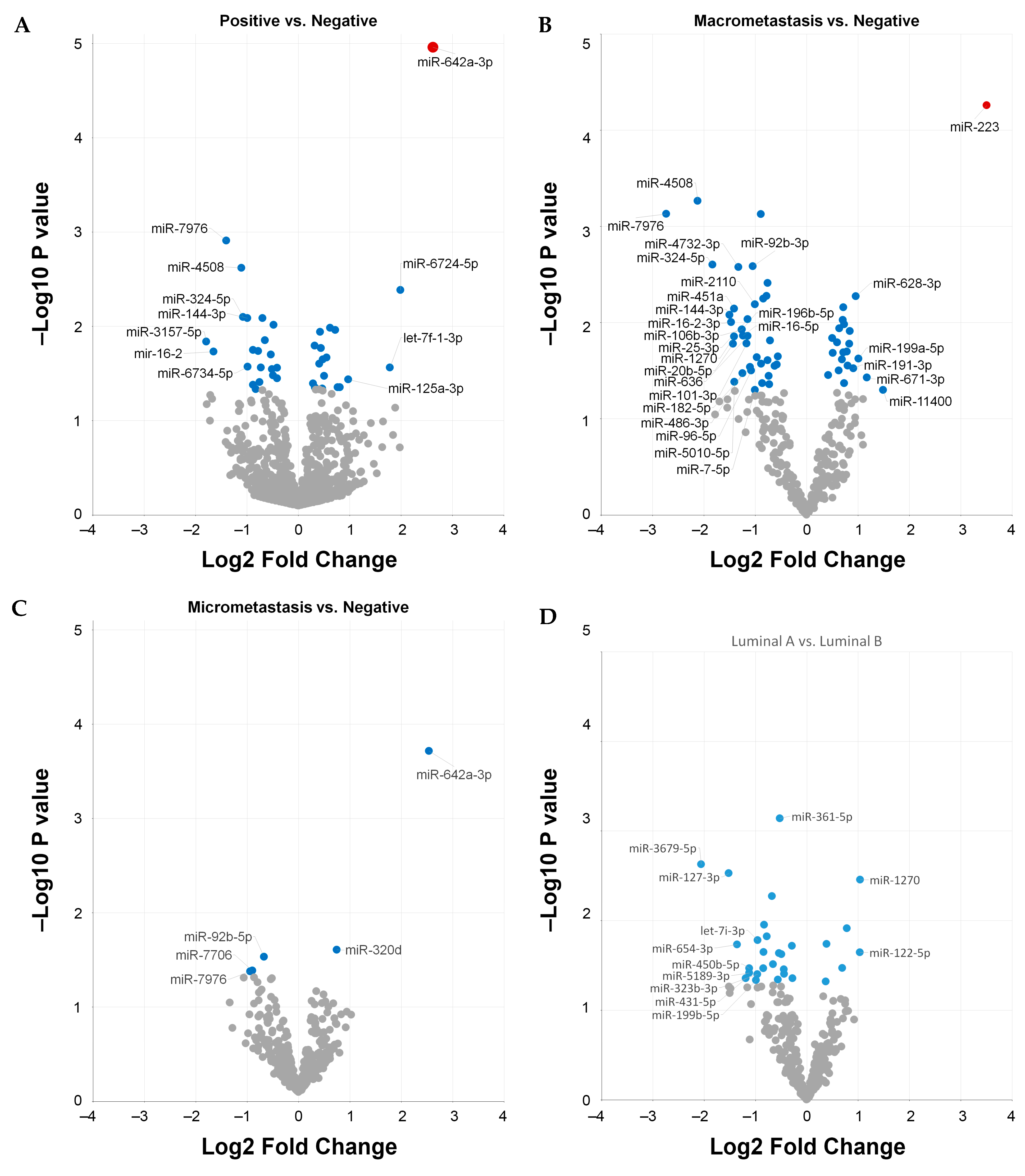
2.4. Differentially Expressed Circulating miRNAs
2.5. Biological Significance and Enriched Analysis of Circulating miRNAs
2.6. Clinicopathological Correlation with Circulating miRNAs
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Sample Processing
4.3. RNA Isolation
4.4. RNA-Sequencing
4.5. Differential Expression Analysis
4.6. Biological Significance Analysis, Gene Ontology (GO) and Enrichment Analysis
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, R.; Ahmed, S.A. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl. Res. 2011, 157, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Nicoloso, M.S.; Spizzo, R.; Shimizu, M.; Rossi, S.; Calin, G.A. MicroRNAs--the micro steering wheel of tumour metastases. Nat. Rev. Cancer 2009, 9, 293–302. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2018, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Pozniak, T.; Shcharbin, D.; Bryszewska, M. Circulating microRNAs in Medicine. Int. J. Mol. Sci. 2022, 23, 3996. [Google Scholar] [CrossRef] [PubMed]
- Escuin, D.; Lopez-Vilaro, L.; Bell, O.; Mora, J.; Moral, A.; Perez, J.I.; Arqueros, C.; Ramon, Y.C.T.; Lerma, E.; Barnadas, A. MicroRNA-1291 Is Associated With Locoregional Metastases in Patients With Early-Stage Breast Cancer. Front. Genet. 2020, 11, 562114. [Google Scholar] [CrossRef] [PubMed]
- Gravgaard, K.H.; Lyng, M.B.; Laenkholm, A.V.; Sokilde, R.; Nielsen, B.S.; Litman, T.; Ditzel, H.J. The miRNA-200 family and miRNA-9 exhibit differential expression in primary versus corresponding metastatic tissue in breast cancer. Breast Cancer Res. Treat. 2012, 134, 207–217. [Google Scholar] [CrossRef]
- Escuin, D.; Lopez-Vilaro, L.; Mora, J.; Bell, O.; Moral, A.; Perez, I.; Arqueros, C.; Garcia-Valdecasas, B.; Ramon, Y.C.T.; Lerma, E.; et al. Circulating microRNAs in Early Breast Cancer Patients and Its Association With Lymph Node Metastases. Front. Oncol. 2021, 11, 627811. [Google Scholar] [CrossRef]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Valastyan, S. Roles of microRNAs and other non-coding RNAs in breast cancer metastasis. J. Mammary Gland. Biol. Neoplasia 2012, 17, 23–32. [Google Scholar] [CrossRef]
- Shek, L.L.; Godolphin, W. Model for breast cancer survival: Relative prognostic roles of axillary nodal status, TNM stage, estrogen receptor concentration, and tumor necrosis. Cancer Res. 1988, 48, 5565–5569. [Google Scholar] [PubMed]
- Chan, M.; Liaw, C.S.; Ji, S.M.; Tan, H.H.; Wong, C.Y.; Thike, A.A.; Tan, P.H.; Ho, G.H.; Lee, A.S. Identification of circulating microRNA signatures for breast cancer detection. Clin. Cancer Res. 2013, 19, 4477–4487. [Google Scholar] [CrossRef] [PubMed]
- Monzo, M.; Santasusagna, S.; Moreno, I.; Martinez, F.; Hernandez, R.; Munoz, C.; Castellano, J.J.; Moreno, J.; Navarro, A. Exosomal microRNAs isolated from plasma of mesenteric veins linked to liver metastases in resected patients with colon cancer. Oncotarget 2017, 8, 30859–30869. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, Y.; Xu, G.; Geng, B.; Cui, Q. Transcriptome analysis reveals non-identical microRNA profiles between arterial and venous plasma. Oncotarget 2017, 8, 28471–28480. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Li, M.; Wang, H.; Yin, Z.; Chen, L.; Zhou, Y.; Han, Y.; Cui, Q.; Zhou, Y.; Xue, L. Transcriptome analysis of arterial and venous circulating miRNAs during hypertension. Sci. Rep. 2021, 11, 3469. [Google Scholar] [CrossRef]
- Santasusagna, S.; Moreno, I.; Navarro, A.; Martinez Rodenas, F.; Hernandez, R.; Castellano, J.J.; Munoz, C.; Monzo, M. Prognostic Impact of miR-200 Family Members in Plasma and Exosomes from Tumor-Draining versus Peripheral Veins of Colon Cancer Patients. Oncology 2018, 95, 309–318. [Google Scholar] [CrossRef]
- Tamrazi, A.; Sundaresan, S.; Gulati, A.; Tan, F.J.; Wadhwa, V.; Bartlett, B.R.; Diaz, L.A.J. Endovascular image-guided sampling of tumor-draining veins provides an enriched source of oncological biomarkers. Front. Oncol. 2023, 13, 916196. [Google Scholar] [CrossRef]
- Damascelli, B.; Ticha, V.; Repetti, E.; Dorji, T. Beyond Standard Practice in Liquid Biopsy: Selective Venous Sampling. J. Vasc. Interv. Radiol. 2021, 32, 668–671. [Google Scholar] [CrossRef]
- Shi, Y.; Jia, Y.; Zhao, W.; Zhou, L.; Xie, X.; Tong, Z. Histone deacetylase inhibitors alter the expression of molecular markers in breast cancer cells via microRNAs. Int. J. Mol. Med. 2018, 42, 435–442. [Google Scholar] [CrossRef]
- Cao, J.; Shao, H.; Hu, J.; Jin, R.; Feng, A.; Zhang, B.; Li, S.; Chen, T.; Jeungpanich, S.; Topatana, W.; et al. Identification of invasion-metastasis associated MiRNAs in gallbladder cancer by bioinformatics and experimental validation. J. Transl. Med. 2022, 20, 188. [Google Scholar] [CrossRef]
- Cho, Y.E.; Vorn, R.; Chimenti, M.; Crouch, K.; Shaoshuai, C.; Narayanaswamy, J.; Harken, A.; Schmidt, R.; Gill, J.; Lee, H. Extracellular vesicle miRNAs in breast milk of obese mothers. Front. Nutr. 2022, 9, 976886. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, L.; Li, T.; Yang, J.; Wei, Y. The role of miRNA-223 in cancer: Function, diagnosis and therapy. Gene 2017, 616, 1–7. [Google Scholar] [CrossRef]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Su, F.; Lin, L.; Liu, Y.; Huang, J.D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [Google Scholar] [CrossRef]
- Huang, R.; Song, X.; Wang, C.M. MiR-223 regulates CDDP resistance in pancreatic cancer via targeting FoxO3a. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7892–7898. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, J.; Yi, L.; Wang, Y.; Dong, Z.; Liu, Z.; Ou-yang, S.; Wu, H.; Zhong, Z.; Yin, Z.; et al. MiR-223-3p targeting SEPT6 promotes the biological behavior of prostate cancer. Sci. Rep. 2014, 4, 7546. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, M.; Nakabayashi, K.; Yoshidome, K.; Kaneko, T.; Iwase, T.; Akiyama, F.; Kato, Y.; Tsuda, H.; Ueda, S.; Sato, K.; et al. One-step nucleic acid amplification for intraoperative detection of lymph node metastasis in breast cancer patients. Clin. Cancer Res. 2007, 13, 4807–4816. [Google Scholar] [CrossRef] [PubMed]
- Webber, C.; Gospodarowicz, M.; Sobin, L.H.; Wittekind, C.; Greene, F.L.; Mason, M.D.; Compton, C.; Brierley, J.; Groome, P.A. Improving the TNM classification: Findings from a 10-year continuous literature review. Int. J. Cancer 2014, 135, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Elston, C.W.; Ellis, I.O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Appierto, V.; Callari, M.; Cavadini, E.; Morelli, D.; Daidone, M.G.; Tiberio, P. A lipemia-independent NanoDrop((R))-based score to identify hemolysis in plasma and serum samples. Bioanalysis 2014, 6, 1215–1226. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- R_Development_Core_Team. R: A Language and Environment for Statistical Computing; The R Foundation for Statistical Computing: Vienna, Austria, 2011; ISBN 3-900051-07-0. Available online: http://www.R-project.org/ (accessed on 6 August 2021).
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef]
- Gene Ontology, C. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Variable | N (%) | |
|---|---|---|
| Patients | 30 (100) | |
| Age, years | Mean + SD | 63.9 ± 7.6 |
| Median (range) | 62.4 (52.6–83.8) | |
| Tumor stage | IA | 16 (53.3) |
| IIA | 9 (30) | |
| IIB | 5 (16.7) | |
| Tumor grade | I | 10 (33.3) |
| II | 20 (66.7) | |
| Node status | Negative | 18 (60) |
| Positive | 12 (40) | |
| Micrometastasis | 7 (23.3) | |
| Macrometastasis | 5 (16.7) | |
| Tumor focality | Unifocal | 18 (60) |
| Multifocal | 10 (33.3) | |
| Multicentric | 2 (6.7) | |
| Estrogen receptor | ER+ PR+ | 27 (90) |
| ER+ PR− | 3 (10) | |
| Molecular subtype | Luminal A | 20 (66.7) |
| Luminal B | 10 (33.3) | |
| Ki67 | <14% | 21 (70) |
| ≥14% | 9 (30) | |
| Surgery | Mastectomy | 8 (26.7) |
| Lumpectomy | 22 (73.3) | |
| Lymphovascular invasion | Negative | 29 (96.7) |
| Positive | 1 (3.3) |
| Mature miRNA | Target Genes | Targets | Log FC | p-Value |
|---|---|---|---|---|
| hsa-miR-192-5p | DHFR, WNK1, RB1, ACVR2B, AKT1 | 41 | −1.439 | 0.001 |
| hsa-miR-376c-3p | BCL2, BMI1, RUNX2, NR5A2, GRB2 | 11 | −2.432 | 0.001 |
| hsa-miR-192-5p | DHFR, WNK1, RB1, ACVR2B, AKT1 | 41 | −1.439 | 0.001 |
| hsa-miR-193b-5p | STMN1 | 1 | −1.572 | 0.003 |
| hsa-miR-99a-5p | FGFR3, AKT1, CAPNS1, FKBP5, MTOR | 15 | −0.967 | 0.005 |
| hsa-miR-885-5p | CASP3, CDK2, CTNNB1, IGF1R, MCM5 | 5 | −1.910 | 0.008 |
| hsa-miR-9-5p | NTRK3, PRDM1, REST, FOXO1, NFKB1 | 82 | 1.456 | 0.009 |
| hsa-miR-15b-3p | CCND3, IGF1R, RECK, TRIM31, SIRT4 | 5 | −0.647 | 0.010 |
| hsa-miR-92b-3p | CDKN1C, ITGA6, ITGAV, SMAD3, SMAD7 | 13 | −0.624 | 0.010 |
| hsa-miR-425-5p | CCND1, BTK, FGFR3, SMAD2, PTEN | 9 | −0.484 | 0.010 |
| hsa-let-7a-5p | NF2, TRIM71, HMGA2, CASP3, DICER1 | 55 | −0.437 | 0.011 |
| hsa-miR-320d | GNAI1, RBFOX2 | 2 | 0.616 | 0.013 |
| hsa-miR-194-5p | ACVR2B, BMI1, BMP1, CDH2, CDKN1B | 23 | −0.826 | 0.013 |
| hsa-miR-486-5p | ARHGAP5, CDK4, DOCK3, FBN1, FOXO1 | 14 | −0.491 | 0.015 |
| hsa-miR-629-5p | HNF4A, TRIM33 | 2 | 0.471 | 0.015 |
| hsa-miR-125b-5p | ERBB2, ERBB3, BAK1, NTRK3, SMO | 122 | 0.599 | 0.018 |
| hsa-miR-92a-3p | ITGA5, BMPR2, CD69, CDH1, DNMT1 | 38 | −0.498 | 0.018 |
| hsa-miR-27a-3p | SP1, SP3, SP4, RUNX1, FOXO1 | 67 | 0.362 | 0.018 |
| hsa-miR-223-3p | FOXO3, FOXO1, FBXW7, IGFR1, MEF2C | 67 | 0.812 | 0.018 |
| hsa-miR-625-3p | MAP2K6 | 1 | −0.623 | 0.019 |
| hsa-miR-96-5p | FOXO1, IRS1, AQP5, CELSR2, ODF2 | 31 | −0.848 | 0.021 |
| hsa-miR-122-5p | CCNG1, BCL2L2, ADAM17, NDRG3, ADAM10 | 67 | −1.286 | 0.021 |
| hsa-miR-378a-3p | RUNX1, CDK6, CYP19A1, GLI3, GRB2 | 19 | −0.514 | 0.022 |
| hsa-miR-25-3p | FBXW7, ATP2A2, CDH1, CDKN1C, CYP2B6 | 29 | −0.648 | 0.023 |
| hsa-miR-483-3p | CDK4, CCN2, IGF1, SMAD4, RASGRF1 | 10 | −1.207 | 0.023 |
| hsa-miR-642a-3p | GRK2, CACNA1B, CDC25B, CDKN1A, FOXN3 | 82 | 0.796 | 0.023 |
| hsa-miR-34a-5p | E2F3, VEGFA, BCL2, MYCN, SIRT1 | 127 | −0.980 | 0.024 |
| hsa-miR-7-5p | IRS2, IRS1, EGFR, RAF1, PAK1 | 46 | −0.737 | 0.031 |
| hsa-let-7b-5p | HMGA2, CDK6, CDC25A, ACTG1, ACVR1 | 40 | −0.402 | 0.031 |
| hsa-miR-30c-5p | UBE2I, BCL9, CAMK2D, CASP3, RUNX2 | 39 | −0.475 | 0.033 |
| hsa-miR-885-3p | BMPR1A, CD274 | 2 | −1.663 | 0.034 |
| hsa-miR-24-3p | CDKN2A, ACVR1B, DHFR, MYC, E2F2 | 84 | 0.301 | 0.034 |
| hsa-miR-365a-3p | HDAC4, ACVR1, BAX, CCND1, BCL2 | 10 | −1.078 | 0.035 |
| hsa-miR-20b-5p | ESR1, BRCA1, CDKN1A, EFNB2, EPHB4 | 20 | −0.860 | 0.036 |
| hsa-miR-126-3p | SPPL2A, ZSWIM1, PIGS, ZNF257, ZNF543 | 44 | 0.316 | 0.038 |
| hsa-miR-101-5p | PRKDC, ATM, EZH2, FOS, STMN1 | 12 | −1.359 | 0.040 |
| hsa-miR-23b-3p | PLAU, MET, CA2, RUNX2, CCNG1 | 31 | 0.338 | 0.041 |
| hsa-miR-942-5p | CDKN1A, GSK3B, IFI27, NFKBIA, SFRP4 | 6 | −0.348 | 0.043 |
| hsa-miR-451a | MIF, ADAM10, AKT1, BCL2, CDKN2D | 22 | −0.707 | 0.043 |
| hsa-miR-339-3p | FOXO1, MCL1, NFKB1 | 3 | 0.483 | 0.044 |
| hsa-miR-140-3p | CD38, COL4A1, FN1, GPC1, ITGA6 | 9 | −0.389 | 0.044 |
| hsa-miR-485-3p | NTRK3, MAT1A, NFYB, PPARGC1A, SLC40A1 | 6 | −0.993 | 0.045 |
| hsa-miR-324-5p | GLI1, SMO, ETS1, SP1, MTFR1 | 6 | −0.860 | 0.047 |
| hsa-let-7c-5p | MYC, HMGA2, TGFBR1, BCL2L1, CASP3 | 28 | 0.506 | 0.048 |
| hsa-miR-155-5p | AGTR1, RHOA, ETS1, MEIS1, FOXO3 | 112 | 0.432 | 0.049 |
| GO ID | GO Term | Counts | q-Value |
|---|---|---|---|
| BIOLOGICAL PROCESS | |||
| 0048732 | Gland development | 30/130 | 1.7 × 10−18 |
| 0048608 | Reproductive structure development | 30/130 | 1.7 × 10−18 |
| 0061458 | Reproductive system development | 30/130 | 1.7 × 10−18 |
| 1901652 | Response to peptide | 29/130 | 6.62 × 10−16 |
| 0050678 | Regulation of epithelial cell proliferation | 25/130 | 6.73 × 10−15 |
| 0048545 | Response to steroid hormone | 25/130 | 8.22 × 10−15 |
| 0070482 | Response to oxygen levels | 25/130 | 1.67 × 10−14 |
| 0050673 | Epithelial cell proliferation | 25/130 | 1.17 × 10−13 |
| 0071900 | Regulation of protein serine/threonine kinase activity | 26/130 | 1.72 × 10−13 |
| 0097305 | Response to alcohol | 19/130 | 1.1 × 10−12 |
| 0045785 | Positive regulation of cell adhesion | 23/130 | 1.16 × 10−12 |
| CELLULAR COMPONENT | |||
| 0005667 | Transcription factor complex | 19/131 | 1.14 × 10−11 |
| 0044798 | Nuclear transcription factor complex | 14/131 | 1.25 × 10−10 |
| 0090575 | RNA polymerase II transcription factor complex | 13/131 | 3.02 × 10−10 |
| 0000785 | Chromatin | 18/131 | 7.87 × 10−07 |
| 0045121 | Membrane raft | 14/131 | 1.51 × 10−06 |
| 0098857 | Membrane microdomain | 14/131 | 1.51 × 10−06 |
| 0098589 | Membrane region | 14/131 | 2 × 10−06 |
| 0000790 | Nuclear chromatin | 13/131 | 8.06 × 10−06 |
| 0044454 | Nuclear chromosome part | 15/131 | 2.58 × 10−05 |
| 1990909 | WNT signalosome | 4/131 | 2.58 × 10−05 |
| MOLECULAR FUNCTION | |||
| 0000978 | RNA polymerase II proximal promoter sequence-specific DNA binding | 22/130 | 4.96 × 10−10 |
| 0000987 | Proximal promoter sequence-specific DNA binding | 22/130 | 5.91 × 10−10 |
| 0019902 | Phosphatase binding | 15/130 | 5.91 x−10 |
| 0001228 | DNA-binding transcription activator activity, RNA polymerase II-specific | 19/130 | 1 × 10−08 |
| 0031625 | Ubiquitin protein ligase binding | 16/130 | 1.49 × 10−08 |
| 0019903 | Protein phosphatase binding | 12/130 | 1.87 × 10−08 |
| 0044389 | Ubiquitin-like protein ligase binding | 16/130 | 2.48 × 10−08 |
| 0003682 | Chromatin binding | 19/130 | 3.39 × 10−08 |
| 0001085 | RNA polymerase II transcription factor binding | 10/130 | 4.99 × 10−07 |
| 0033613 | Activating transcription factor binding | 8/130 | 8.48 × 10−07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escuin, D.; López-Vilaró, L.; Bell, O.; Mora, J.; García-Valdecasas, B.; Moral, A.; Clos, M.; Boronat, L.; Arqueros, C.; Barnadas, A. Circulating miRNA Expression Is Inversely Correlated with Tumor Tissue or Sentinel Lymph Nodes in Estrogen Receptor-Positive Early Breast Cancer Patients. Int. J. Mol. Sci. 2023, 24, 13293. https://doi.org/10.3390/ijms241713293
Escuin D, López-Vilaró L, Bell O, Mora J, García-Valdecasas B, Moral A, Clos M, Boronat L, Arqueros C, Barnadas A. Circulating miRNA Expression Is Inversely Correlated with Tumor Tissue or Sentinel Lymph Nodes in Estrogen Receptor-Positive Early Breast Cancer Patients. International Journal of Molecular Sciences. 2023; 24(17):13293. https://doi.org/10.3390/ijms241713293
Chicago/Turabian StyleEscuin, Daniel, Laura López-Vilaró, Olga Bell, Josefina Mora, Bárbara García-Valdecasas, Antonio Moral, Montserrat Clos, Laia Boronat, Cristina Arqueros, and Agustí Barnadas. 2023. "Circulating miRNA Expression Is Inversely Correlated with Tumor Tissue or Sentinel Lymph Nodes in Estrogen Receptor-Positive Early Breast Cancer Patients" International Journal of Molecular Sciences 24, no. 17: 13293. https://doi.org/10.3390/ijms241713293
APA StyleEscuin, D., López-Vilaró, L., Bell, O., Mora, J., García-Valdecasas, B., Moral, A., Clos, M., Boronat, L., Arqueros, C., & Barnadas, A. (2023). Circulating miRNA Expression Is Inversely Correlated with Tumor Tissue or Sentinel Lymph Nodes in Estrogen Receptor-Positive Early Breast Cancer Patients. International Journal of Molecular Sciences, 24(17), 13293. https://doi.org/10.3390/ijms241713293