
Structural Variations Contribute to the Genetic Etiology of Autism Spectrum Disorder and Language Impairments

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Family Samples and Project Overview
2.2. Candidate CNV Identification
2.3. A Candidate Syndromic CNV in Individual FAM23-003
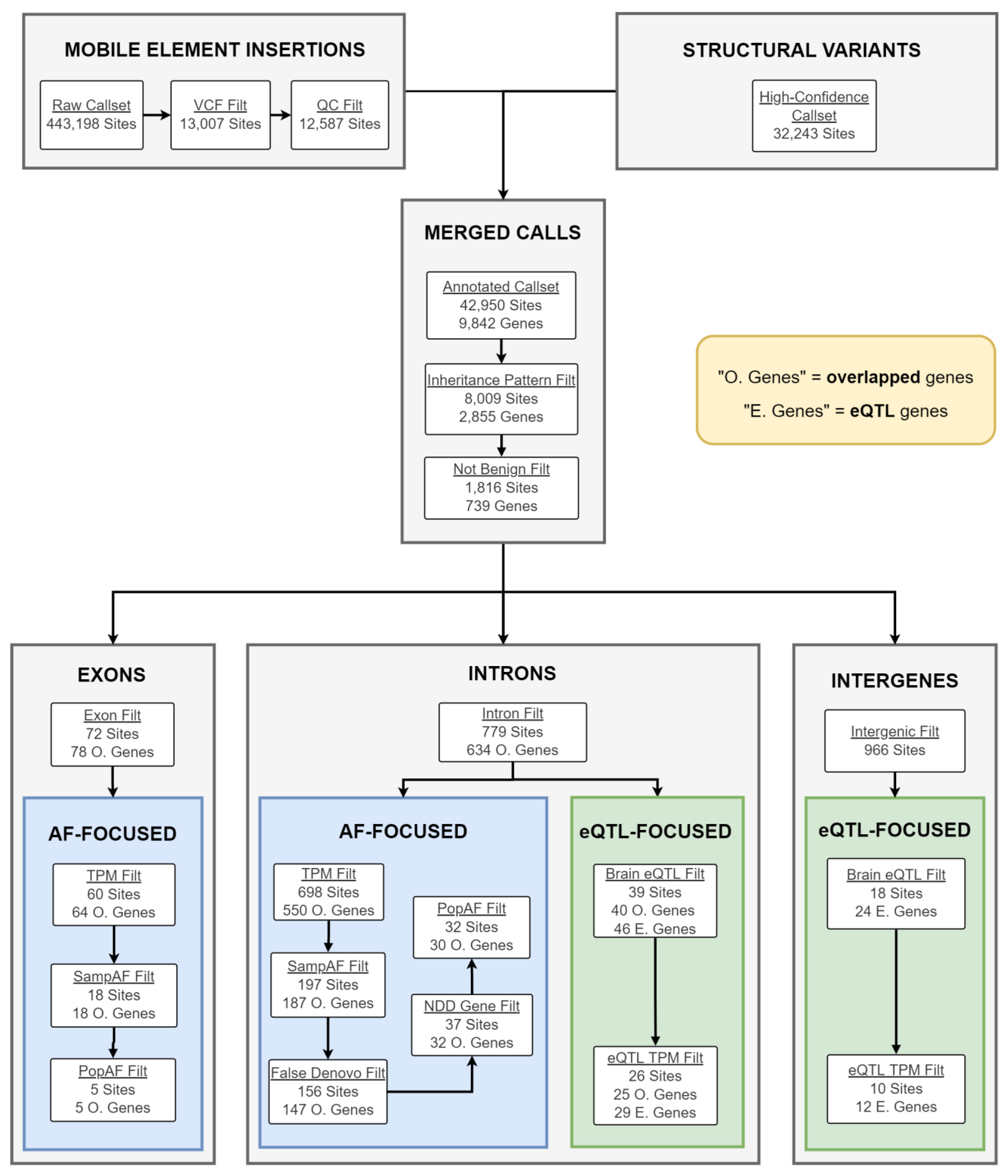
2.4. Candidate gSV/MEI Identification
2.5. Candidate Gene Analysis
2.6. GO Term and Pathway Enrichment Analysis
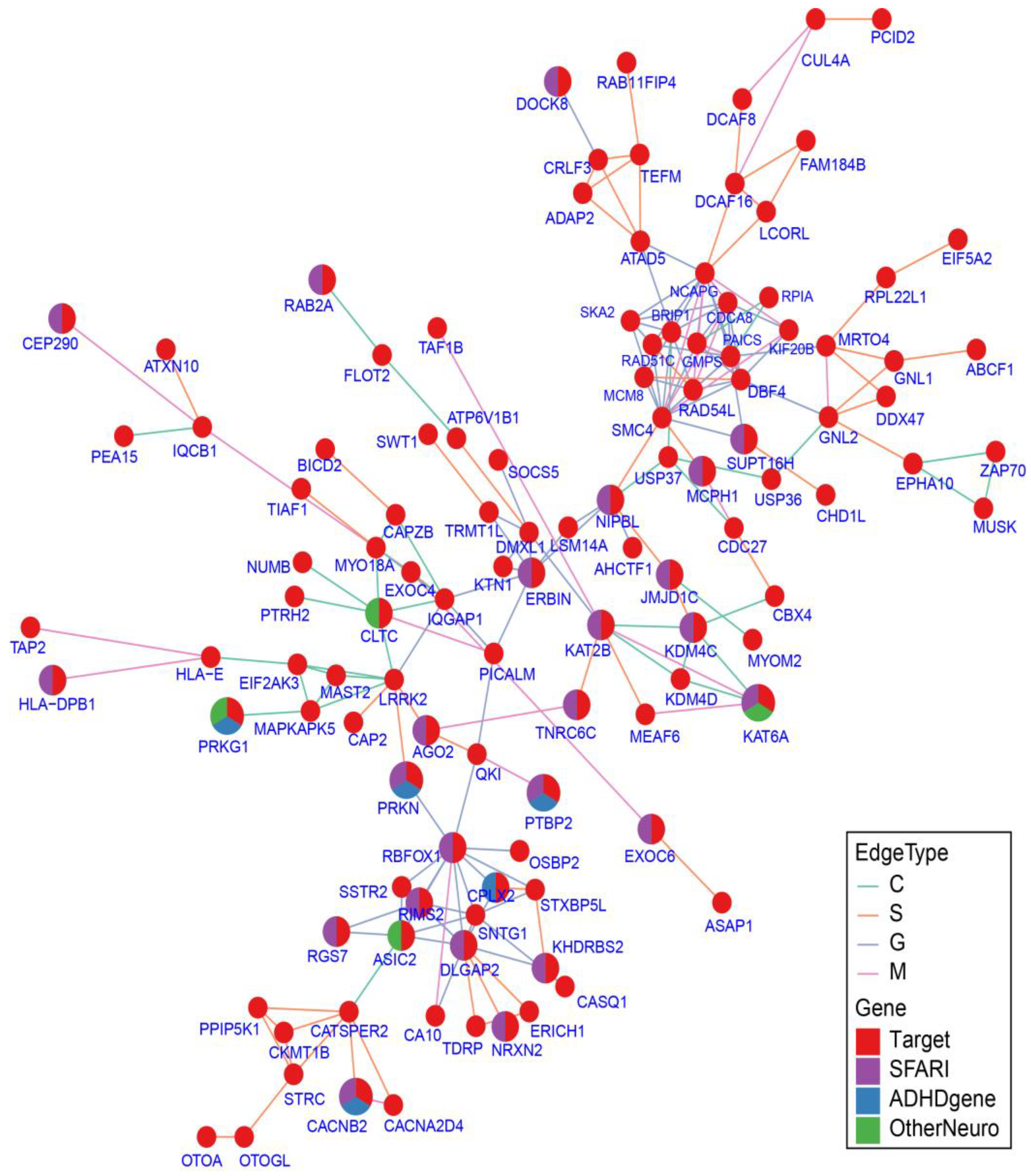
2.7. Protein–Protein Interaction Network Analysis
3. Discussion
4. Materials and Methods
4.1. Family Selection and Phenotyping
4.2. Microarray Genotyping and Quality Control
4.3. CNV Identification and Quality Control
4.4. CNV Merging, Annotation, and Segregation Analysis
4.5. CNV Filtration and Prioritization
4.6. Whole-Genome Sequencing Data and the gSV Call Set
4.7. Mobile Element Insertion Identification and Filtering
4.8. Merging gSV and MEI Call Sets
4.9. gSV/MEI Annotation, Segregation Analysis, and Filtering
4.10. gSV/MEI Candidate Prioritization
4.11. Pathogenicity Prediction
4.12. Gene Ontology and Pathway Enrichment Analyses, Protein–Protein Interaction Network Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, (DSM-5), 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Christensen, D.L.; Baio, J.; Van Naarden Braun, K.; Bilder, D.; Charles, J.; Constantino, J.N.; Daniels, J.; Durkin, M.S.; Fitzgerald, R.T.; Kurzius-Spencer, M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years–Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill. Summ. 2016, 65, 1–23. [Google Scholar] [CrossRef]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happe, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child. Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.E.; Law, J.K.; Yenokyan, G.; McGready, J.; Kaufmann, W.E.; Law, P.A. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch. Pediatr. Adolesc. Med. 2009, 163, 907–914. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 178, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, e472-86. [Google Scholar] [CrossRef]
- Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; Reeves, C.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Marshall, C.; Feuk, L.; Scherer, S.W. Copy-number variation in control population cohorts. Hum. Mol. Genet. 2007, 16, R168–R173. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.H., Jr.; Scherer, S.W. Copy-number variations associated with neuropsychiatric conditions. Nature 2008, 455, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Meyer-Lindenberg, A.; Steinberg, S.; Magnusdottir, B.; Morgen, K.; Arnarsdottir, S.; Bjornsdottir, G.; Walters, G.B.; Jonsdottir, G.A.; Doyle, O.M.; et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014, 505, 361–366. [Google Scholar] [CrossRef]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Noor, A.; Rosanio, G.; Chapman, T.R.; Barrera, D.J.; Lin, G.N.; Malhotra, D.; Watts, A.C.; et al. Frequency and Complexity of De Novo Structural Mutation in Autism. Am. J. Hum. Genet. 2016, 98, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Chen, H.I.; Chien, W.H.; Li, L.H.; Wu, Y.Y.; Chiu, Y.N.; Tsai, W.C.; Gau, S.S. High resolution analysis of rare copy number variants in patients with autism spectrum disorder from Taiwan. Sci. Rep. 2017, 7, 11919. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; O’Roak, B.J.; Karakoc, E.; Mohajeri, K.; Nelson, B.; Vives, L.; Jacquemont, S.; Munson, J.; Bernier, R.; Eichler, E.E. Transmission disequilibrium of small CNVs in simplex autism. Am. J. Hum. Genet. 2013, 93, 595–606. [Google Scholar] [CrossRef][Green Version]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, Y.; Payer, L.M.; Lords, H.; Steranka, J.P.; Burns, K.H.; Xing, J. Polymorphic mobile element insertions contribute to gene expression and alternative splicing in human tissues. Genome Biol. 2020, 21, 185. [Google Scholar] [CrossRef]
- Scott, A.J.; Chiang, C.; Hall, I.M. Structural variants are a major source of gene expression differences in humans and often affect multiple nearby genes. Genome Res. 2021, 31, 2249–2257. [Google Scholar] [CrossRef] [PubMed]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Kleiber, M.L.; Whitney, J.; Maile, M.S.; Hong, O.; Chapman, T.R.; Tan, S.; Tandon, P.; et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 2018, 360, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, C.W.; Hou, L.; Flax, J.F.; Hare, A.; Cheong, S.Y.; Fermano, Z.; Zimmerman-Bier, B.; Cartwright, C.; Azaro, M.A.; Buyske, S.; et al. A genome scan for loci shared by autism spectrum disorder and language impairment. Am. J. Psychiatry 2014, 171, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Brown, T.T. Chapter 72—Specific Language Impairment. In Neurobiology of Language; Hickok, G., Small, S.L., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 899–912. [Google Scholar] [CrossRef]
- NIH. Developmental Language Disorder. Available online: https://www.nidcd.nih.gov/health/developmental-language-disorder (accessed on 20 August 2023).
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Vieland, V.J.; Bassett, A.S.; Tallal, P.; Brzustowicz, L.M. A major susceptibility locus for specific language impairment is located on 13q21. Am. J. Hum. Genet. 2002, 71, 45–55. [Google Scholar] [CrossRef]
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Smith, B.J.; Vieland, V.J.; Tallal, P.; Brzustowicz, L.M. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum. Hered. 2004, 57, 10–20. [Google Scholar] [CrossRef]
- Bradford, Y.; Haines, J.; Hutcheson, H.; Gardiner, M.; Braun, T.; Sheffield, V.; Cassavant, T.; Huang, W.; Wang, K.; Vieland, V.; et al. Incorporating language phenotypes strengthens evidence of linkage to autism. Am. J. Med. Genet. 2001, 105, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, C.W.; Flax, J.F.; Fermano, Z.; Hare, A.; Hou, L.; Petrill, S.A.; Buyske, S.; Brzustowicz, L.M. Gene x gene interaction in shared etiology of autism and specific language impairment. Biol. Psychiatry 2012, 72, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Cao, X.; Mahaganapathy, V.; Azaro, M.; Gwin, C.; Wilson, S.; Buyske, S.; Bartlett, C.W.; Flax, J.F.; Brzustowicz, L.M.; et al. Common genetic risk factors in ASD and ADHD co-occurring families. Hum. Genet. 2023, 142, 217–230. [Google Scholar] [CrossRef]
- Li, Y.; Sun, C.; Guo, Y.; Qiu, S.; Li, Y.; Liu, Y.; Zhong, W.; Wang, H.; Cheng, Y.; Liu, Y. DIP2C polymorphisms are implicated in susceptibility and clinical phenotypes of autism spectrum disorder. Psychiatry Res. 2022, 316, 114792. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mandell, J.D.; Kumar, Y.; Sun, N.; Morris, M.T.; Arbelaez, J.; Nasello, C.; Dong, S.; Duhn, C.; Zhao, X.; et al. De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis. Cell Rep. 2018, 24, 3441–3454.e12. [Google Scholar] [CrossRef] [PubMed]
- Chaimowicz, C.; Ruffault, P.L.; Cheret, C.; Woehler, A.; Zampieri, N.; Fortin, G.; Garratt, A.N.; Birchmeier, C. Teashirt 1 (Tshz1) is essential for the development, survival and function of hypoglossal and phrenic motor neurons in mouse. Development 2019, 146, dev174045. [Google Scholar] [CrossRef] [PubMed]
- Mascheretti, S.; Riva, V.; Feng, B.; Trezzi, V.; Andreola, C.; Giorda, R.; Villa, M.; Dionne, G.; Gori, S.; Marino, C.; et al. The Mediation Role of Dynamic Multisensory Processing Using Molecular Genetic Data in Dyslexia. Brain Sci. 2020, 10, 993. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Kushima, I.; Mori, D.; Yoshimi, A.; Aleksic, B.; Nawa, Y.; Toyama, M.; Furuta, S.; Yu, Y.; Ishizuka, K.; et al. Rare genetic variants in the gene encoding histone lysine demethylase 4C (KDM4C) and their contributions to susceptibility to schizophrenia and autism spectrum disorder. Transl. Psychiatry 2020, 10, 421. [Google Scholar] [CrossRef]
- Maitra, S.; Khandelwal, N.; Kootar, S.; Sant, P.; Pathak, S.S.; Reddy, S.K.A.P.; Murty, U.S.; Chakravarty, S.; Kumar, A. Histone Lysine Demethylase JMJD2D/KDM4D and Family Members Mediate Effects of Chronic Social Defeat Stress on Mouse Hippocampal Neurogenesis and Mood Disorders. Brain Sci. 2020, 10, 833. [Google Scholar] [CrossRef]
- Saez, M.A.; Fernandez-Rodriguez, J.; Moutinho, C.; Sanchez-Mut, J.V.; Gomez, A.; Vidal, E.; Petazzi, P.; Szczesna, K.; Lopez-Serra, P.; Lucariello, M.; et al. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet. Med. 2016, 18, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jiang-Xie, L.F.; Liao, H.M.; Chen, C.H.; Chen, Y.T.; Ho, S.Y.; Lu, D.H.; Lee, L.J.; Liou, H.H.; Fu, W.M.; Gau, S.S. Autism-associated gene Dlgap2 mutant mice demonstrate exacerbated aggressive behaviors and orbitofrontal cortex deficits. Mol. Autism 2014, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Damianov, A.; Lin, C.H.; Fontes, M.; Parikshak, N.N.; Anderson, E.S.; Geschwind, D.H.; Black, D.L.; Martin, K.C. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron 2016, 89, 113–128. [Google Scholar] [CrossRef]
- Lee, C.T.; Bendriem, R.M.; Kindberg, A.A.; Worden, L.T.; Williams, M.P.; Drgon, T.; Mallon, B.S.; Harvey, B.K.; Richie, C.T.; Hamilton, R.S.; et al. Functional consequences of 17q21.31/WNT3-WNT9B amplification in hPSCs with respect to neural differentiation. Cell Rep. 2015, 10, 616–632. [Google Scholar] [CrossRef]
- Eising, E.; Carrion-Castillo, A.; Vino, A.; Strand, E.A.; Jakielski, K.J.; Scerri, T.S.; Hildebrand, M.S.; Webster, R.; Ma, A.; Mazoyer, B.; et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 2019, 24, 1065–1078. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Zhou, A.; Cao, X.; Mahaganapathy, V.; Azaro, M.; Gwin, C.; Wilson, S.; Buyske, S.; Bartlett, C.W.; Flax, J.F.; et al. MicroRNA and MicroRNA-Target Variants Associated with Autism Spectrum Disorder and Related Disorders. Genes 2022, 13, 1329. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Darvishi, K.; Shi, X.; Rajan, D.; Rigler, D.; Fitzgerald, T.; Lionel, A.C.; Thiruvahindrapuram, B.; Macdonald, J.R.; Mills, R.; et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011, 29, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Skaar, D.A.; Rayner-Evans, M.Y.; Konidari, I.; Whitehead, P.L.; Jaworski, J.M.; Cuccaro, M.L.; Pericak-Vance, M.A.; Gilbert, J.R. Identification of chromosome 7 inversion breakpoints in an autistic family narrows candidate region for autism susceptibility. Autism Res. 2009, 2, 258–266. [Google Scholar] [CrossRef]
- Tabet, A.C.; Verloes, A.; Pilorge, M.; Delaby, E.; Delorme, R.; Nygren, G.; Devillard, F.; Gerard, M.; Passemard, S.; Heron, D.; et al. Complex nature of apparently balanced chromosomal rearrangements in patients with autism spectrum disorder. Mol. Autism 2015, 6, 19. [Google Scholar] [CrossRef]
- Zhou, A.; Lin, T.; Xing, J. Evaluating nanopore sequencing data processing pipelines for structural variation identification. Genome Biol. 2019, 20, 237. [Google Scholar] [CrossRef]
- Guo, Y.; He, J.; Zhao, S.; Wu, H.; Zhong, X.; Sheng, Q.; Samuels, D.C.; Shyr, Y.; Long, J. Illumina human exome genotyping array clustering and quality control. Nat. Protoc. 2014, 9, 2643–2662. [Google Scholar] [CrossRef]
- Axiom™. Analysis Suite v4.0.1 User Guide. Publication Number 703307 Revision 7.
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef]
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Paila, U.; Chapman, B.A.; Kirchner, R.; Quinlan, A.R. GEMINI: Integrative Exploration of Genetic Variation and Genome Annotations. PLoS Comput. Biol. 2013, 9, e1003153. [Google Scholar] [CrossRef] [PubMed]
- Aguet, F.; Brown, A.A.; Castel, S.E.; Davis, J.R.; He, Y.; Jo, B.; Mohammadi, P.; Park, Y.; Parsana, P.; Segrè, A.V.; et al. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Aguet, F.; Barbeira, A.N.; Bonazzola, R.; Brown, A.; Castel, S.E.; Jo, B.; Kasela, S.; Kim-Hellmuth, S.; Liang, Y.; Oliva, M.; et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. bioRxiv 2019, 787903. [Google Scholar]
- Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, S.J.; Xu, Y.B.; Lisgo, S.N.; Harkin, L.F.; Copp, A.J.; Gerrelli, D.; Clowry, G.J.; Talbot, A.; Keogh, M.J.; Coxhead, J.; et al. HDBR Expression: A Unique Resource for Global and Individual Gene Expression Studies during Early Human Brain Development. Front. Neuroanat. 2016, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Mohiyuddin, M.; Mu, J.C.; Li, J.; Bani Asadi, N.; Gerstein, M.B.; Abyzov, A.; Wong, W.H.; Lam, H.Y. MetaSV: An accurate and integrative structural-variant caller for next generation sequencing. Bioinformatics 2015, 31, 2741–2744. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.J.; Lam, V.K.; Harris, D.N.; Chuang, N.T.; Scott, E.C.; Pittard, W.S.; Mills, R.E.; Genomes Project, C.; Devine, S.E. The Mobile Element Locator Tool (MELT): Population-scale mobile element discovery and biology. Genome Res. 2017, 27, 1916–1929. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sun, Z.; Wang, J.; Huang, H.; Kocher, J.P.; Wang, L. CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 2014, 30, 1006–1007. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bahler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alfoldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, Y.; Abdulkadir, M.; Deng, L.; Fernandez, T.V.; Garcia-Delgar, B.; Hagstrom, J.; Hoekstra, P.J.; King, R.A.; Koesterich, J.; et al. Whole-exome sequencing identifies genes associated with Tourette’s disorder in multiplex families. Mol. Psychiatry 2021, 26, 6937–6951. [Google Scholar] [CrossRef]
- Sharo, A.G.; Hu, Z.; Sunyaev, S.R.; Brenner, S.E. StrVCTVRE: A supervised learning method to predict the pathogenicity of human genome structural variants. Am. J. Hum. Genet. 2022, 109, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Danis, D.; Jacobsen, J.O.B.; Balachandran, P.; Zhu, Q.; Yilmaz, F.; Reese, J.; Haimel, M.; Lyon, G.J.; Helbig, I.; Mungall, C.J.; et al. SvAnna: Efficient and accurate pathogenicity prediction of coding and regulatory structural variants in long-read genome sequencing. Genome Med. 2022, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef]
- Hagberg, A.; Swart, P.; S Chult, D. Exploring Network Structure, Dynamics, and Function Using Networkx; Los Alamos National Lab (LANL): Los Alamos, NM, USA, 2008. [Google Scholar]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Krishnan, A.; Troyanskaya, O.G. GIANT 2.0: Genome-scale integrated analysis of gene networks in tissues. Nucleic Acids Res. 2018, 46, W65–W70. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Patients | Male | Female | Families | Dominant | Recessive/De Novo | |
|---|---|---|---|---|---|---|---|
| CNV | ASD | 147 | 119 | 28 | 109 | 0 | 109 |
| LI* | 196 | 146 | 50 | 109 | 7 | 109 | |
| RI* | 235 | 160 | 75 | 109 | 8 | 108 | |
| Cohort | 524 | 300 | 224 | 109 | - | - | |
| gSV/MEI | ASD | 83 | 65 | 18 | 73 | 0 | 73 |
| LI* | 117 | 86 | 31 | 73 | 7 | 73 | |
| RI* | 134 | 96 | 38 | 73 | 8 | 73 | |
| Cohort | 272 | 166 | 106 | 73 | - | - |
| Phenotype | Prioritized CNVs | Median Length (bp) | Deletion | Duplication | StrVCTVRE >0 | SvAnna >0 |
|---|---|---|---|---|---|---|
| ASD | 174 | 58,649 | 111 | 63 | 135 | 114 |
| LI* | 196 | 54,899 | 139 | 57 | 152 | 130 |
| RI* | 151 | 64,567 | 106 | 45 | 102 | 96 |
| Chr | Start | End | Genotype | Family | Genes | Known Phenotype (OMIM) | StrVCTVRE | pSV |
|---|---|---|---|---|---|---|---|---|
| 1 | 19,540,528 | 19,988,894 | CN1 | FAM96 | AKR7A2|AKR7A3|AKR7L|CAPZB|EMC1|EMC1-AS1|LOC100506730|LOC105378614|MICOS10|MICOS10-NBL1|MRTO4|NBL1|RPS14P3|SLC66A1 | Cerebellar atrophy, visual impairment, and psychomotor retardation | 0.707 | 9 |
| 1 | 19,545,053 | 19,596,156 | CN1 | FAM96 | AKR7L|EMC1|EMC1-AS1|MRTO4 | 0.673 | 2 | |
| 1 | 19,622,270 | 19,715,311 | CN0 | FAM96 | AKR7A2|CAPZB|SLC66A1 | 0.699 | 3 | |
| 1 | 19,733,320 | 19,988,894 | CN1 | FAM96 | CAPZB|LOC105378614|MICOS10|MICOS10-NBL1|NBL1|RPS14P3 | 0.627 | 4 | |
| 1 | 46,721,155 | 46,782,409 | CN1 | FAM19 | LRRC41|RAD54L|UQCRH | 0.752 | 2 | |
| 1 | 241,829,586 | 241,843,408 | CN1 | FAM5 | WDR64 | 0.594 | 2 | |
| 2 | 98,317,685 | 98,391,355 | CN1 | FAM88 | C2orf92|TMEM131|ZAP70 | Autoimmune disease, multisystem, infantile-onset, 2; Immunodeficiency 48 | 0.627 | 2 |
| 6 | 30,337,738 | 30,541,852 | CN1 | FAM2 | ABCF1|GNL1|HLA-E|LINC02569|PRR3 | 0.745 | 4 | |
| 7 | 87,436,780 | 87,514,832 | CN1 | FAM138 | DBF4|RUNDC3B|SLC25A40 | 0.652 | 3 | |
| 8 | 176,818 | 2518,930 | CN1 | FAM30 | ARHGEF10|CLN8|DLGAP2|DLGAP2-AS1|ERICH1|FAM87A|FBXO25|KBTBD11|KBTBD11-OT1|LOC101927752|LOC101927815|LOC101928058|LOC105377777|LOC286083|LOC401442|MIR3674|MIR596|MIR7160|MYOM2|RPL23AP53|TDRP|ZNF596 | Ceroid lipofuscinosis, neuronal, 8; Ceroid lipofuscinosis, neuronal, 8, Northern epilepsy variant | 0.732 | 7 |
| 11 | 94,681,989 | 94,732,407 | CN1 | FAM44 | CWC15|KDM4D | 0.694 | 2 | |
| 15 | 90794,757 | 90,950,358 | CN3 | FAM27 | CIB1|GABARAPL3|IQGAP1|NGRN|TTLL13P|ZNF774 | 0.561 | 1.2 | |
| 17 | 27,187,789 | 27,434,490 | CN3 | FAM21 | DHRS13|ERAL1|FLOT2|LOC101927018|MIR144|MIR451A|MIR451B|MIR4732|MYO18A|PHF12|PIPOX|SEZ6|TIAF1 | 0.725 | 1.6 | |
| 17 | 29,107,588 | 29,262,773 | CN3 | FAM21 | ADAP2|ATAD5|CRLF3|SUZ12P1|TEFM | 0.529 | 1.2 | |
| 17 | 56,584,205 | 57,229,716 | CN3 | FAM21 | MIR301A|MIR454|MTMR4|PPM1E|RAD51C|SEPTIN4|SEPTIN4-AS1|SKA2|TEX14|TRIM37 | 0.592 | 1.4 | |
| 17 | 56,717,956 | 57,017,420 | CN1 | FAM2 | PPM1E|RAD51C|TEX14 | Fanconi anemia, complementation group O; Breast-ovarian cancer, familial, susceptibility to, 3 | 0.581 | 2 |
| 17 | 57,567,551 | 57,875,554 | CN1 | FAM104 | CLTC|DHX40|LINC01476|PTRH2|VMP1 | Infantile-onset multisystem neurologic, endocrine, and pancreatic disease; Mental retardation, AD 56, | 0.813 | 2 |
| 22 | 31,190,639 | 31,375,585 | CN1 | FAM27 | LOC107985544|MORC2|MORC2-AS1|OSBP2|TUG1 | Charcot–Marie–Tooth disease, axonal, type 2Z | 0.742 | 2 |
| Chr | Start | End | Type | Gene | Sample AF | Pop AF | Tissue Count | Pipeline |
|---|---|---|---|---|---|---|---|---|
| 10 | 94,707,842 | 94,708,290 | INS | EXOC6 | 2.32% | 1.14% | - | ExonicAF |
| 17 | 78,356,535 | 78,356,813 | INS | RNF213 | 4.44% | 4.49% | - | ExonicAF |
| 22 | 36,561,302 | 36,561,477 | INS | APOL3 | 1.16% | 3.08% | - | ExonicAF |
| 3 | 155,656,737 | 155,656,738 | INS | GMPS | 0.39% | 0.04% | - | ExonicAF |
| 8 | 66,927,128 | 66,927,399 | INS | DNAJC5B | 0.39% | 0.26% | - | ExonicAF |
| 10 | 104,645,257 | 104,645,587 | DEL | AS3MT | 42.88% | - | 14 | IntronicEQTL |
| 4 | 152,340,722 | 152,341,553 | DEL | SH3D19;FAM160A1 | 51.68% | 37.30% | 6 | IntronicEQTL |
| 6 | 170,038,129 | 170,038,446 | DEL | WDR27 | 23.78% | 26.38% | 7 | IntronicEQTL |
| 6 | 170,044,824 | 170,045,154 | DEL | WDR27 | 30.41% | - | 3 | IntronicEQTL |
| 14 | 92,619,420 | 92,620,656 | INS | CPSF2 | 14.67% | 20.65% | 15 | IntronicEQTL |
| 17 | 76,052,946 | 76,053,029 | INS | DNAH17 | 9.07% | - | 3 | IntronicEQTL |
| 20 | 1,546,228 | 1,546,508 | INS | SIRPB2 | 32.82% | 40.56% | 5 | IntronicEQTL |
| 20 | 34,458,566 | 34,458,843 | INS | FER1L4;CPNE1 | 8.30% | 4.21% | 6 | IntronicEQTL |
| 5 | 167,619 | 167,899 | INS | CCDC127;LRRC14B | 9.65% | 4.23% | 3 | IntronicEQTL |
| 6 | 110,102,982 | 110,103,230 | INS | MICAL1;AK9 | 61.00% | 26.96% | 4 | IntronicEQTL |
| 6 | 116,898,686 | 116,898,965 | INS | RSPH4A;KPNA5 | 17.95% | 17.27% | 13 | IntronicEQTL |
| 8 | 61,526,797 | 61,527,076 | INS | RAB2A | 25.68% | 20.85% | 6 | IntronicEQTL |
| 4 | 120,244,573 | 120,244,904 | DEL | C4orf3 | 24.06% | - | 4 | IntergenicEQTL |
| 10 | 124,736,004 | 124,736,285 | INS | PSTK | 63.71% | 42.99% | 3 | IntergenicEQTL |
| 4 | 152,732,823 | 152,738,842 | INS | FAM160A1 | 10.81% | 2.90% | 3 | IntergenicEQTL |
| 9 | 95,680,508 | 95,680,786 | INS | ZNF484;BICD2;CENPP | 14.67% | 9.60% | 4 | IntergenicEQTL |
| Phenotype | CNV | SV/MEI | Total (Unique) | |||
|---|---|---|---|---|---|---|
| Genes | Families | Genes | Families | Genes | Families | |
| ASD | 212 | 50 | 49 | 51 | 258 | 77 |
| LI* | 203 | 54 | 50 | 41 | 252 | 76 |
| RI* | 161 | 43 | 57 | 48 | 217 | 73 |
| Total | 274 | 62 | 75 | 58 | 344 | 86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alibutud, R.; Hansali, S.; Cao, X.; Zhou, A.; Mahaganapathy, V.; Azaro, M.; Gwin, C.; Wilson, S.; Buyske, S.; Bartlett, C.W.; et al. Structural Variations Contribute to the Genetic Etiology of Autism Spectrum Disorder and Language Impairments. Int. J. Mol. Sci. 2023, 24, 13248. https://doi.org/10.3390/ijms241713248
Alibutud R, Hansali S, Cao X, Zhou A, Mahaganapathy V, Azaro M, Gwin C, Wilson S, Buyske S, Bartlett CW, et al. Structural Variations Contribute to the Genetic Etiology of Autism Spectrum Disorder and Language Impairments. International Journal of Molecular Sciences. 2023; 24(17):13248. https://doi.org/10.3390/ijms241713248
Chicago/Turabian StyleAlibutud, Rohan, Sammy Hansali, Xiaolong Cao, Anbo Zhou, Vaidhyanathan Mahaganapathy, Marco Azaro, Christine Gwin, Sherri Wilson, Steven Buyske, Christopher W. Bartlett, and et al. 2023. "Structural Variations Contribute to the Genetic Etiology of Autism Spectrum Disorder and Language Impairments" International Journal of Molecular Sciences 24, no. 17: 13248. https://doi.org/10.3390/ijms241713248
APA StyleAlibutud, R., Hansali, S., Cao, X., Zhou, A., Mahaganapathy, V., Azaro, M., Gwin, C., Wilson, S., Buyske, S., Bartlett, C. W., Flax, J. F., Brzustowicz, L. M., & Xing, J. (2023). Structural Variations Contribute to the Genetic Etiology of Autism Spectrum Disorder and Language Impairments. International Journal of Molecular Sciences, 24(17), 13248. https://doi.org/10.3390/ijms241713248