
Integrating Single-Cell RNA-Seq and Bulk RNA-Seq Data to Explore the Key Role of Fatty Acid Metabolism in Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
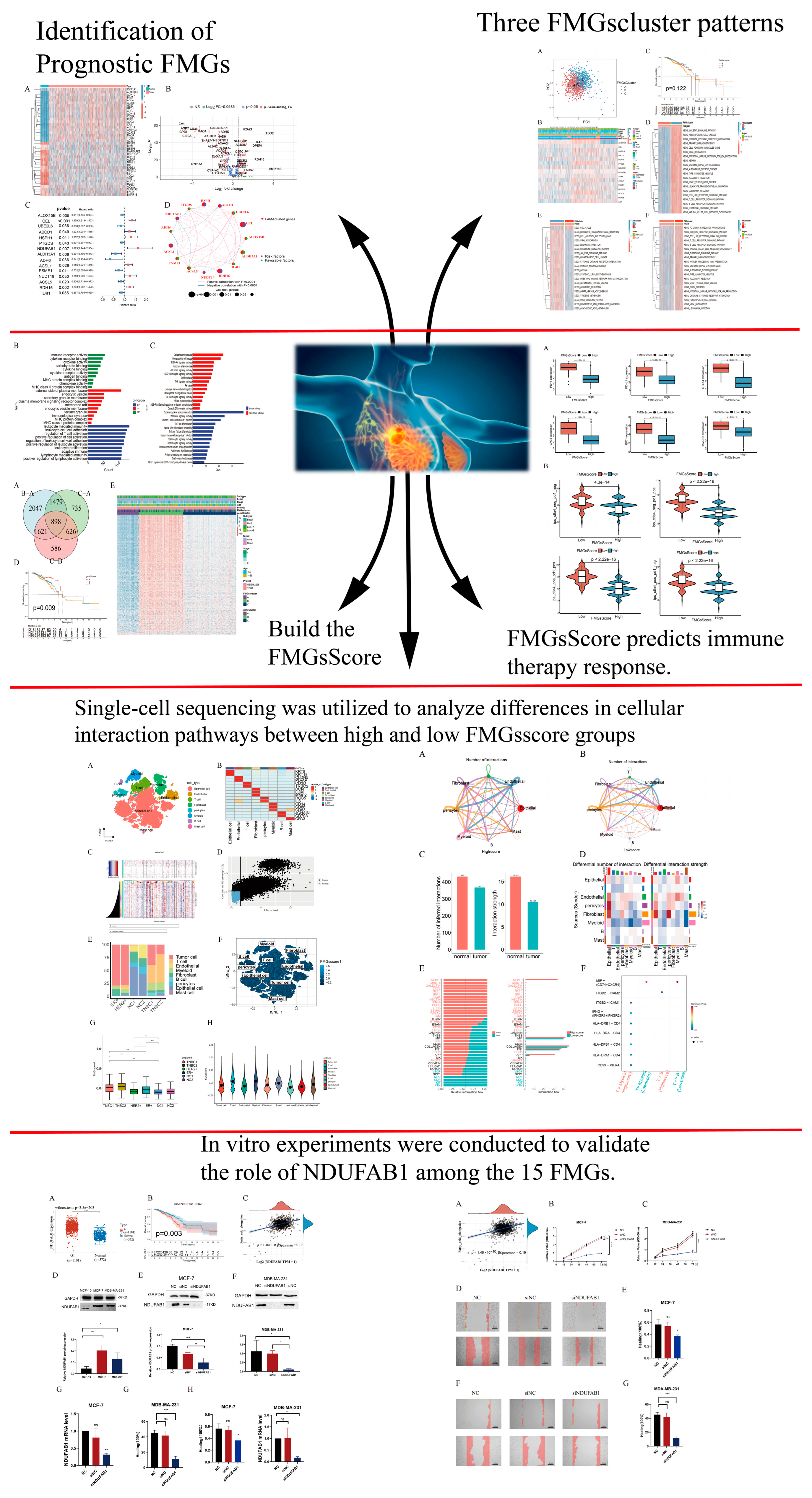
2.1. Identification of Prognostic Fatty Acid Metabolism-Related Genes and Genetic Variation in Breast Cancer
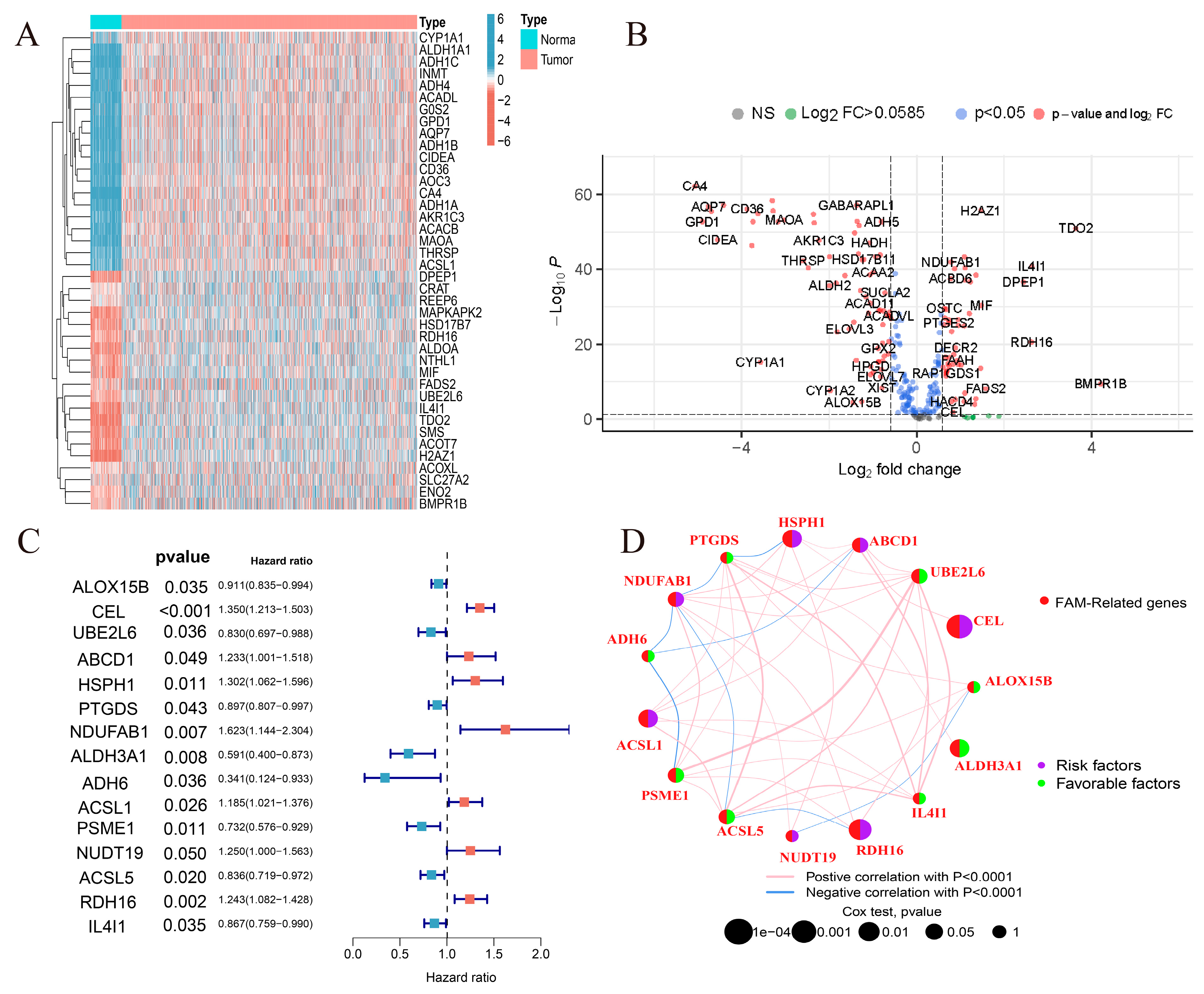
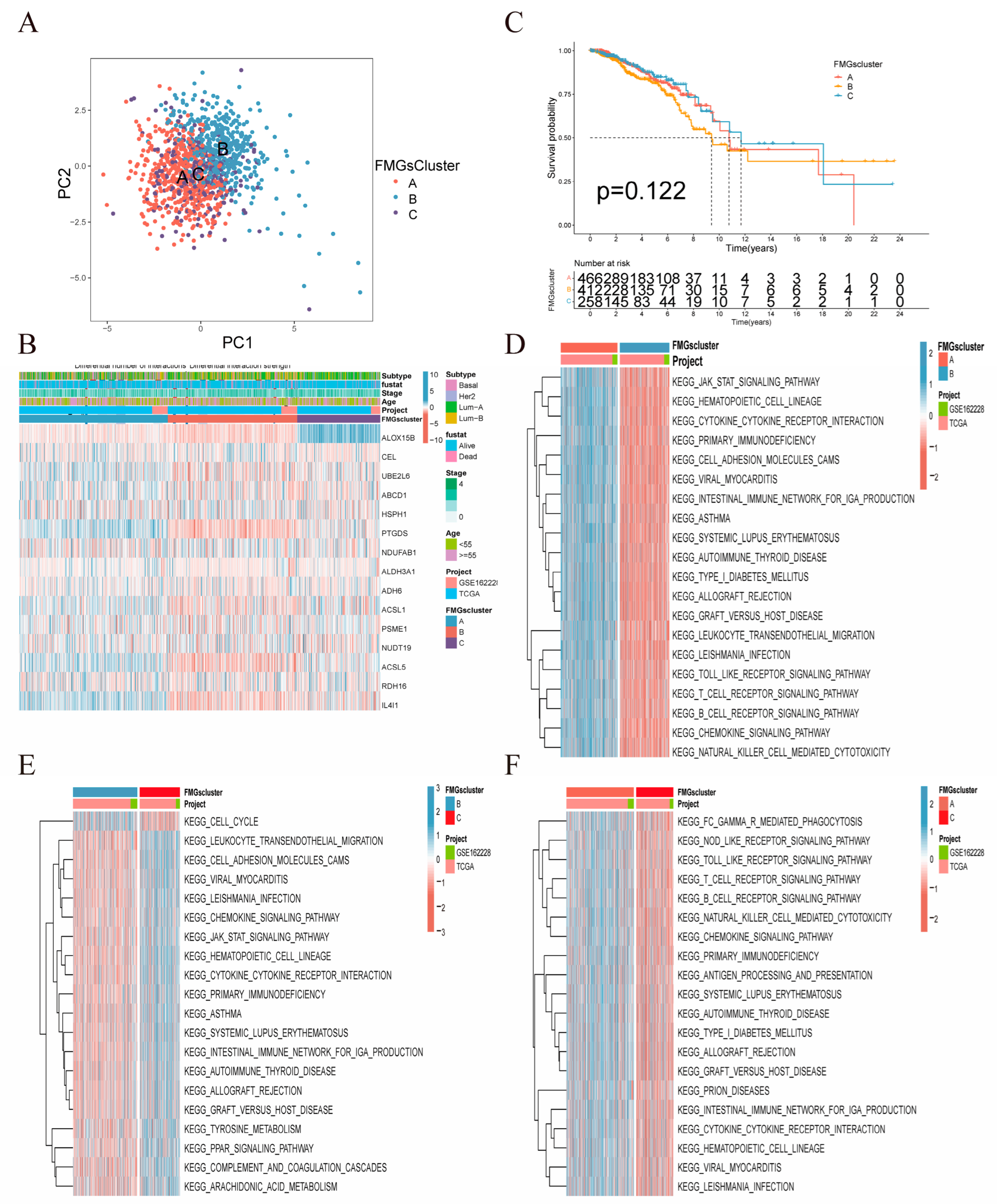
2.2. Immune Infiltration and Biological Functions Associated with FMG Modification Patterns
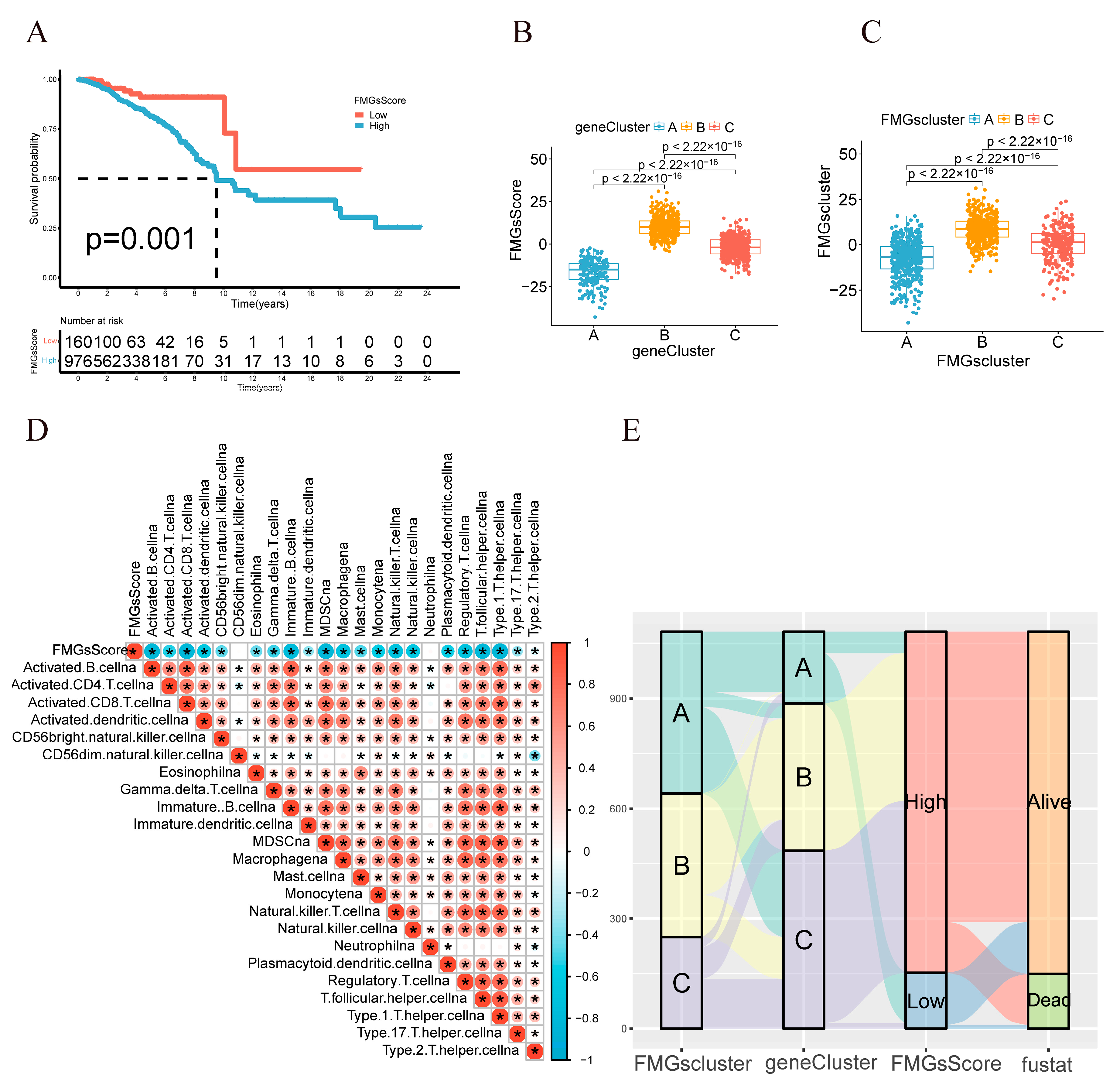
2.3. Construction of Gene Signatures Based on Differential Genes of FMGsCluster
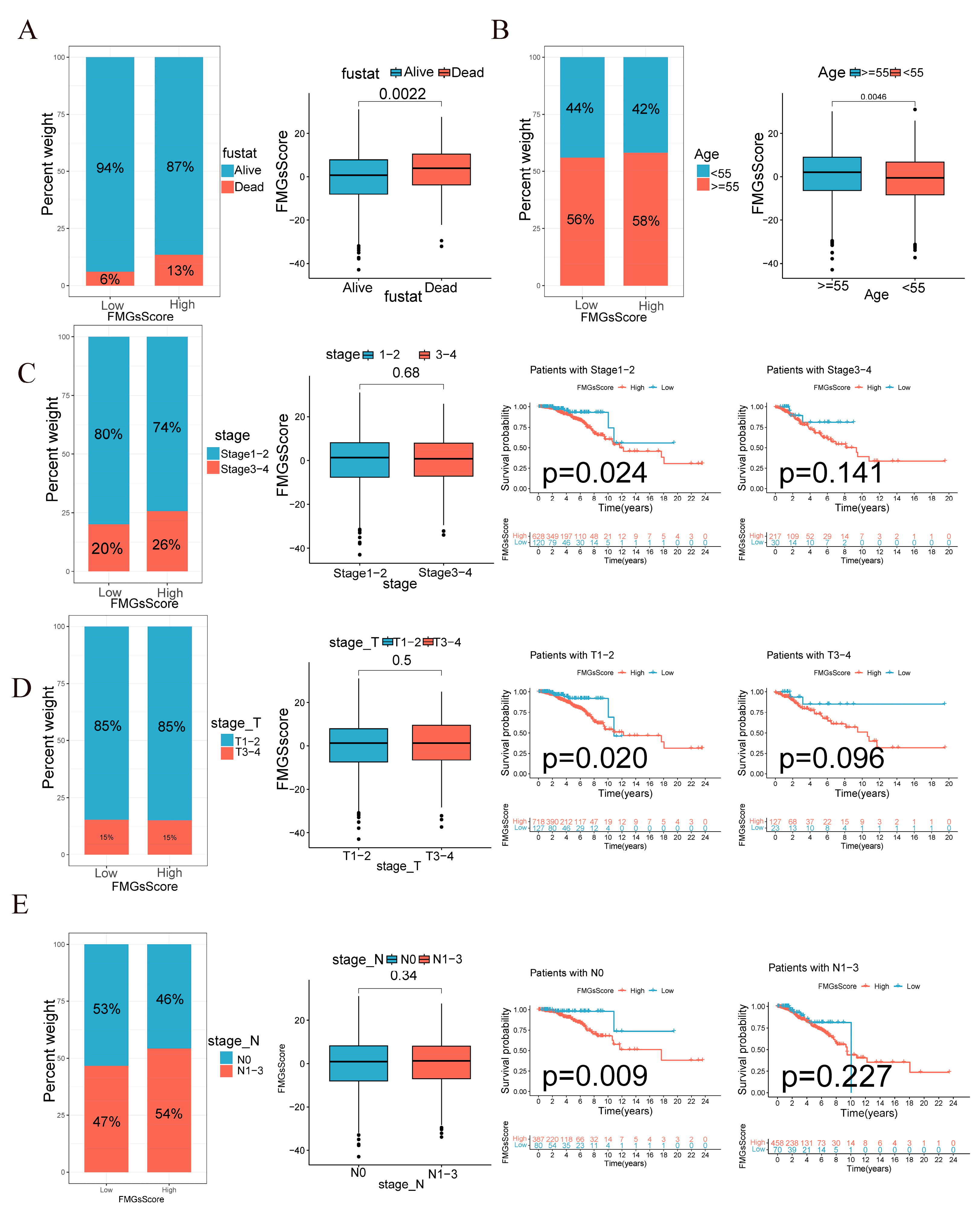
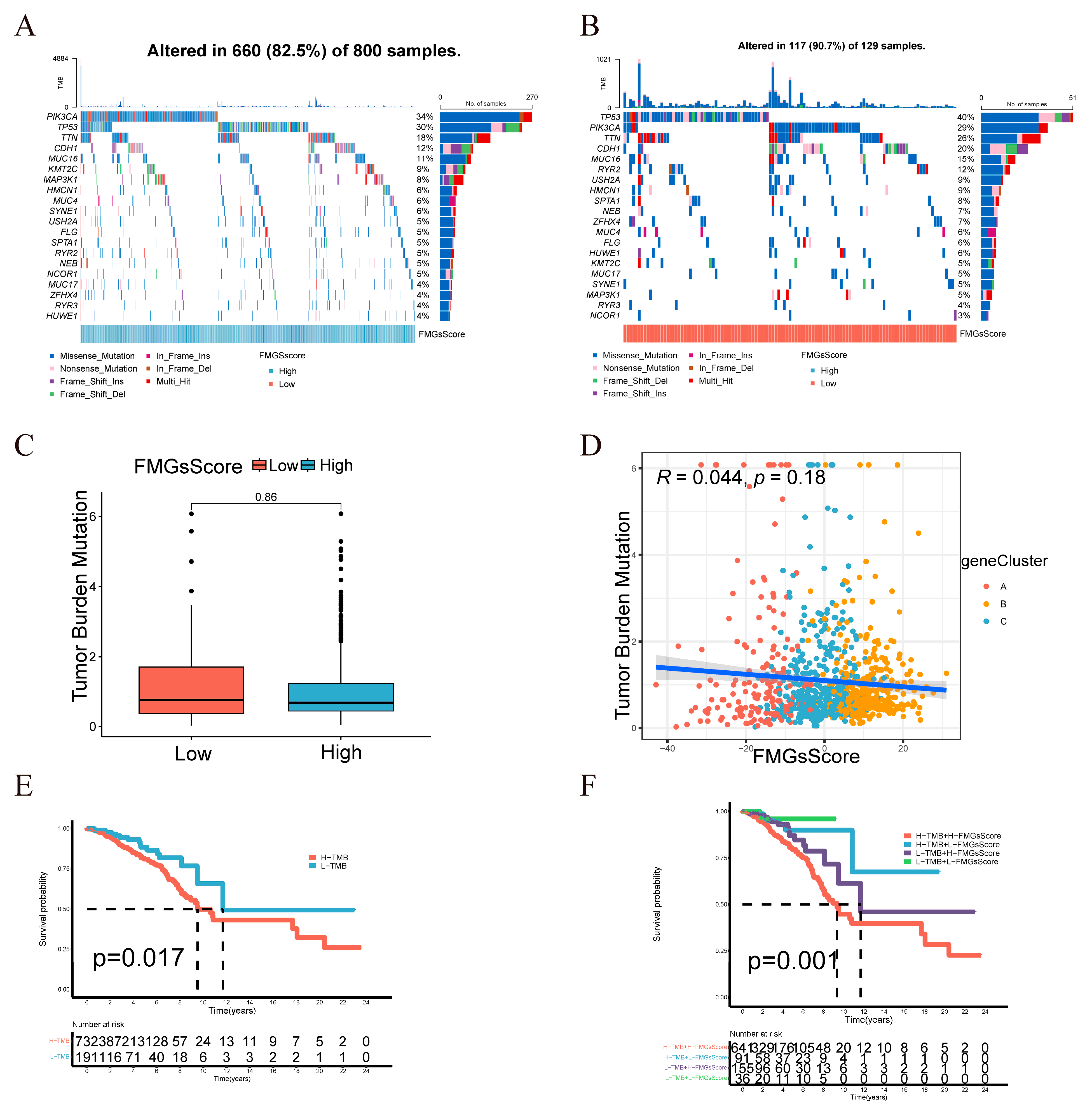
2.4. Clinical and Tumor Somatic Cell Mutation Characteristics between Patient High and Low FMGsScore Groups
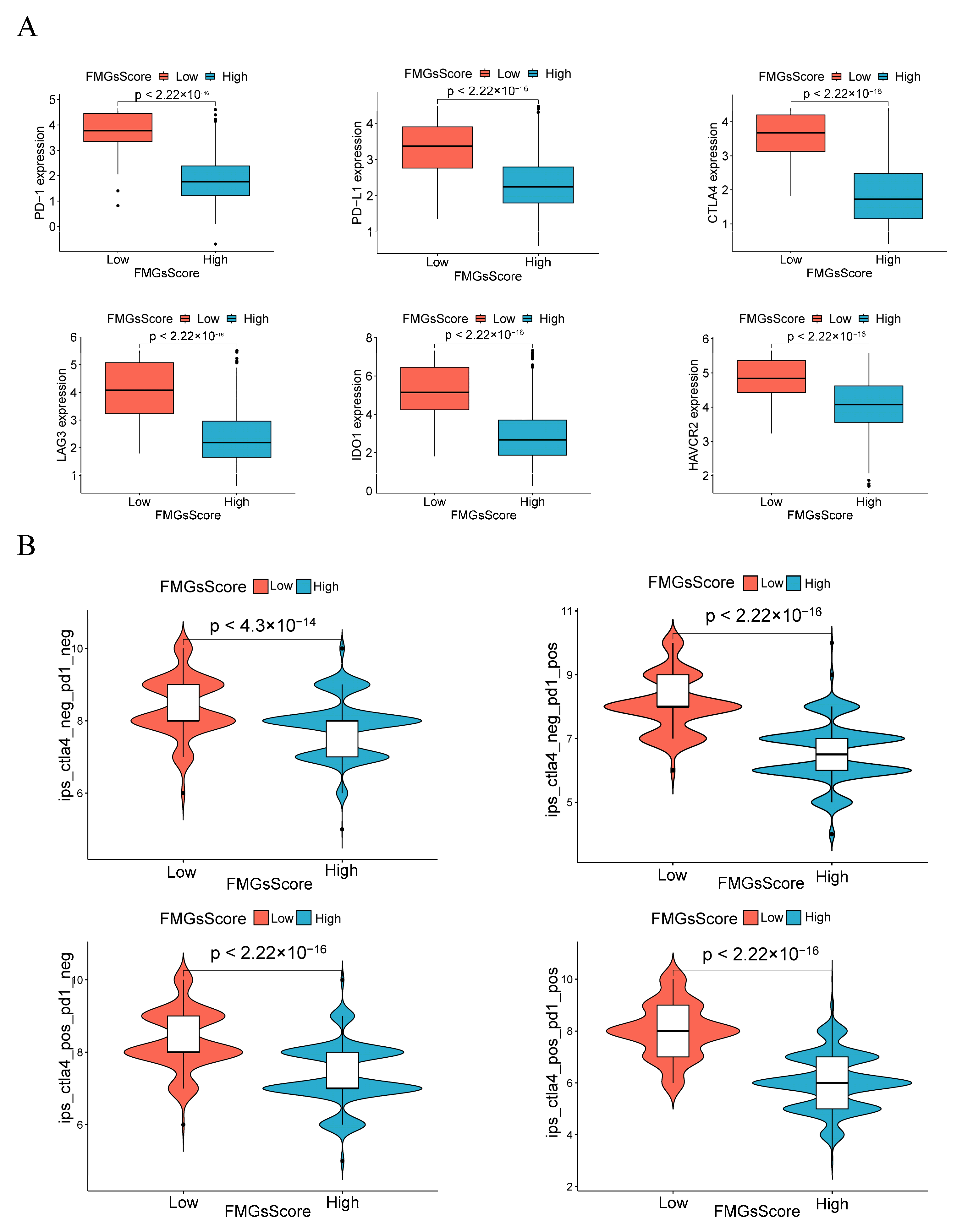
2.5. Effect of FMGsScore on Immunotherapy
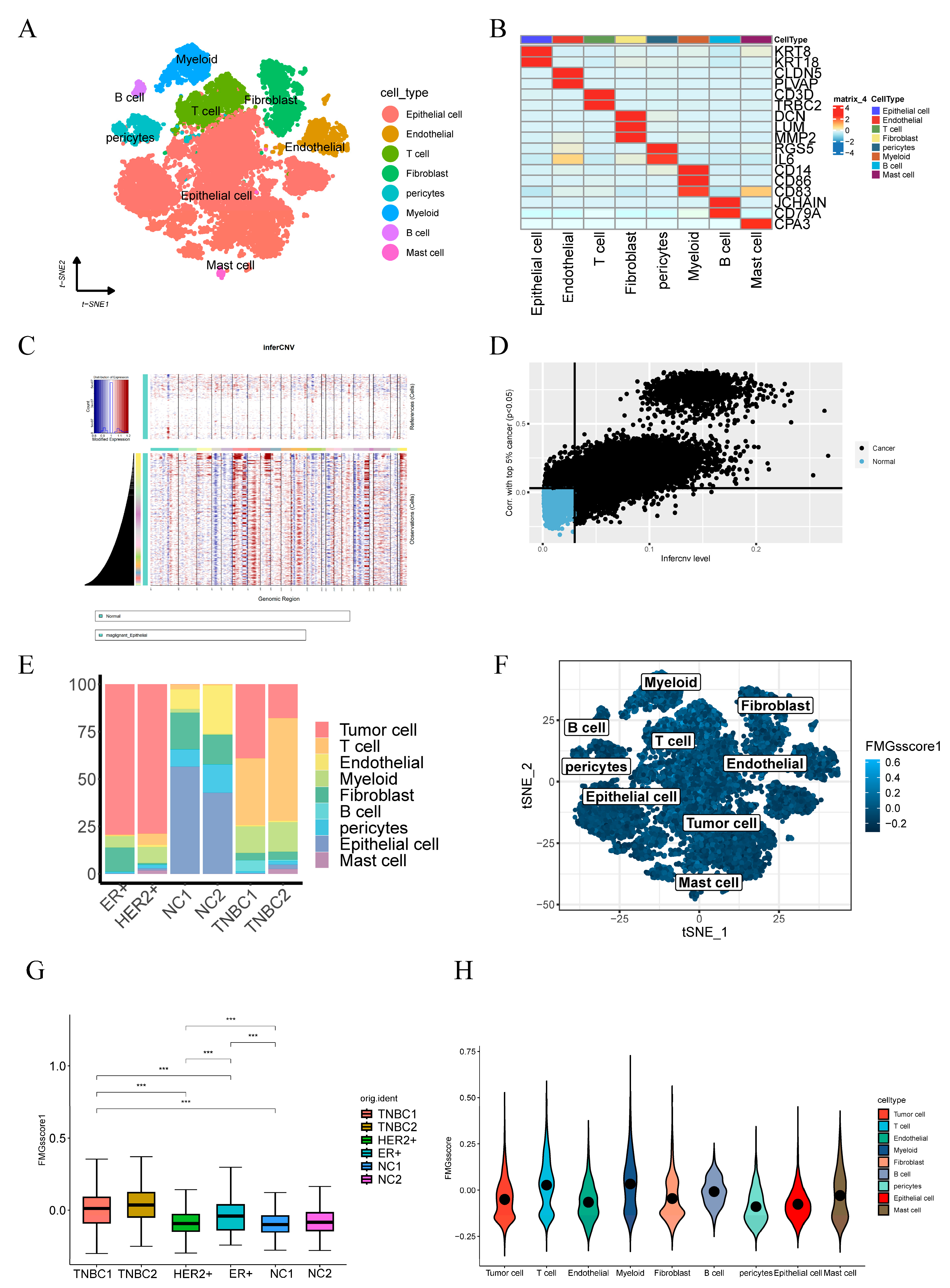
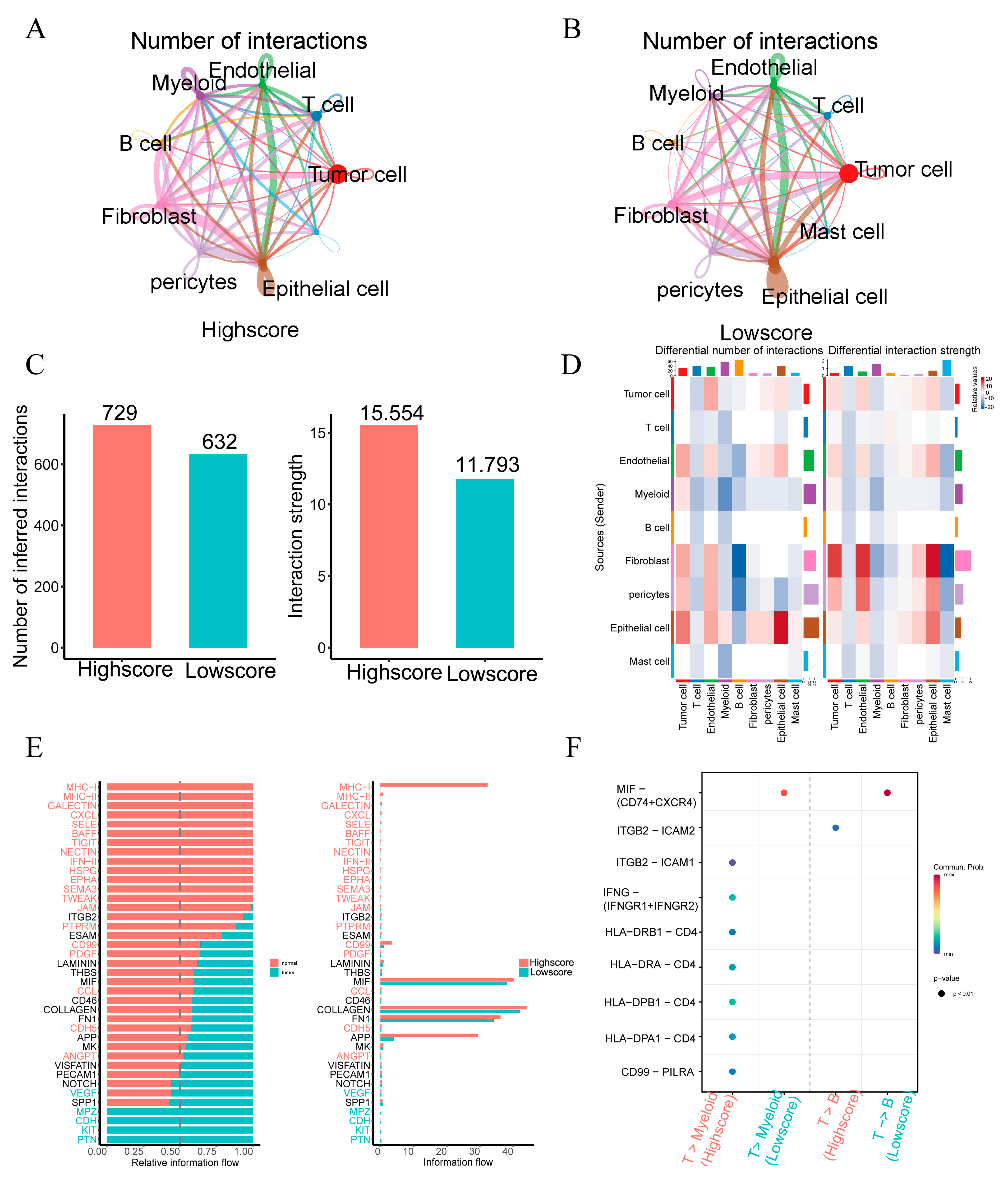
2.6. Single-Cell Profiling Data Reveal the Relationship between 15 Fatty Acid Metabolism Genes and Tumor Immunity
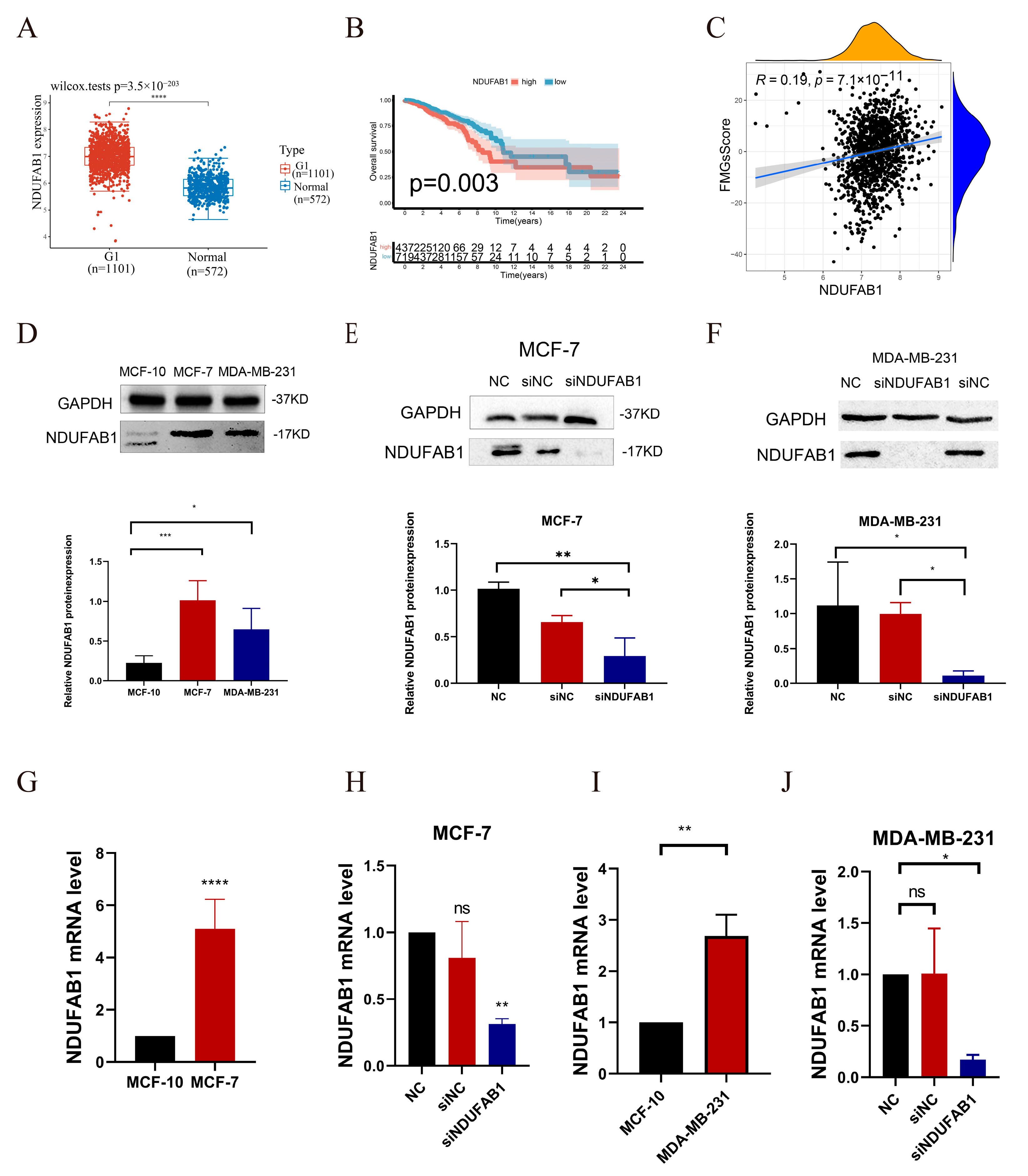
2.7. Critical Role of NDUFAB1 Gene in Migration and Proliferation in Breast Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Breast Cancer Dataset Source
4.2. Produces Fatty Acid Metabolism-Related Genes Associated with Prognosis
4.3. Consensus Clustering of FMG Regulators
4.4. Gene Set Variation Analysis
4.5. Estimation of TME Cell Infiltration
4.6. Construction of the FMGs’ Gene Signature
4.7. Comprehensive Analysis of the FMGsScore Signature with Genomic Mutations, Clinical Information, and Immunity Correlation
4.8. Analysis of Single-Cell Sequencing Data
4.9. Cell Lines Culture and Transfection
4.10. Western Blotting
4.11. qRT-PCR
4.12. CCK8 Assay to Detect Cell Proliferation
4.13. Healing Assay
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glodzik, D.; Bosch, A.; Hartman, J.; Aine, M.; Vallon-Christersson, J.; Reuterswärd, C.; Karlsson, A.; Mitra, S.; Niméus, E.; Holm, K.; et al. Comprehensive Molecular Comparison of BRCA1 Hypermethylated and BRCA1 Mutated Triple Negative Breast Cancers. Nat. Commun. 2020, 11, 3747. [Google Scholar] [CrossRef] [PubMed]
- Britt, K.L.; Cuzick, J.; Phillips, K.-A. Key Steps for Effective Breast Cancer Prevention. Nat. Rev. Cancer 2020, 20, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous Lipids Promote the Growth of Breast Cancer Cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour Fatty Acid Metabolism in the Context of Therapy Resistance and Obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; García-Cañaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory Breast Cancer Biology: The Tumour Microenvironment Is Key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiol. J. Immunopathol. Mol. Cell Biol. 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Yang, Y. Cancer Immunotherapy: Harnessing the Immune System to Battle Cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Diamond, J.R.; Hamilton, E.; Pohlmann, P.R.; Tolaney, S.M.; Chang, C.-W.; Zhang, W.; Iizuka, K.; Foster, P.G.; Molinero, L.; et al. Atezolizumab Plus Nab-Paclitaxel in the Treatment of Metastatic Triple-Negative Breast Cancer with 2-Year Survival Follow-up: A Phase 1b Clinical Trial. JAMA Oncol. 2019, 5, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, H.; Liu, B.; Wei, J. Fatty Acid Metabolism and Cancer Immunotherapy. Curr. Oncol. Rep. 2022, 24, 659–670. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef]
- Chu, Q.; Liu, P.; Song, Y.; Yang, R.; An, J.; Zhai, X.; Niu, J.; Yang, C.; Li, B. Stearate-Derived Very Long-Chain Fatty Acids Are Indispensable to Tumor Growth. EMBO J. 2023, 42, e111268. [Google Scholar] [CrossRef]
- Manzo, T.; Prentice, B.M.; Anderson, K.G.; Raman, A.; Schalck, A.; Codreanu, G.S.; Nava Lauson, C.B.; Tiberti, S.; Raimondi, A.; Jones, M.A.; et al. Accumulation of Long-Chain Fatty Acids in the Tumor Microenvironment Drives Dysfunction in Intrapancreatic CD8+ T Cells. J. Exp. Med. 2020, 217, e20191920. [Google Scholar] [CrossRef]
- Tang, Y.; Tian, W.; Xie, J.; Zou, Y.; Wang, Z.; Li, N.; Zeng, Y.; Wu, L.; Zhang, Y.; Wu, S.; et al. Prognosis and Dissection of Immunosuppressive Microenvironment in Breast Cancer Based on Fatty Acid Metabolism-Related Signature. Front. Immunol. 2022, 13, 843515. [Google Scholar] [CrossRef]
- Weigert, A.; Strack, E.; Snodgrass, R.G.; Brüne, B. MPGES-1 and ALOX5/-15 in Tumor-Associated Macrophages. Cancer Metastasis Rev. 2018, 37, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-H.; Tang, Y.; Yu, H.; Li, H.-D. The Role of Ferroptosis in Breast Cancer Patients: A Comprehensive Analysis. Cell Death Discov. 2021, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The Human ATP-Binding Cassette (ABC) Transporter Superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Hlaváč, V.; Souček, P. Role of Family D ATP-Binding Cassette Transporters (ABCD) in Cancer. Biochem. Soc. Trans. 2015, 43, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, Z.; Zhang, C.; Ouyang, J.; Zhang, G.; Wu, C. A Four-Gene Signature in the Tumor Microenvironment That Significantly Associates with the Prognosis of Patients with Breast Cancer. Gene 2020, 761, 145049. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J. Tumor Microenvironment, Metabolism, and Immunotherapy. N. Engl. J. Med. 2020, 382, 869–871. [Google Scholar] [CrossRef]
- Ecker, C.; Guo, L.; Voicu, S.; Gil-de-Gómez, L.; Medvec, A.; Cortina, L.; Pajda, J.; Andolina, M.; Torres-Castillo, M.; Donato, J.L.; et al. Differential Reliance on Lipid Metabolism as a Salvage Pathway Underlies Functional Differences of T Cell Subsets in Poor Nutrient Environments. Cell Rep. 2018, 23, 741–755. [Google Scholar] [CrossRef]
- Gao, A.; Liu, X.; Lin, W.; Wang, J.; Wang, S.; Si, F.; Huang, L.; Zhao, Y.; Sun, Y.; Peng, G. Tumor-Derived ILT4 Induces T Cell Senescence and Suppresses Tumor Immunity. J. Immunother. Cancer 2021, 9, e001536. [Google Scholar] [CrossRef]
- Bellmunt, J.; Powles, T.; Vogelzang, N.J. A Review on the Evolution of PD-1/PD-L1 Immunotherapy for Bladder Cancer: The Future Is Now. Cancer Treat. Rev. 2017, 54, 58–67. [Google Scholar] [CrossRef]
- Adams, S.; Gatti-Mays, M.E.; Kalinsky, K.; Korde, L.A.; Sharon, E.; Amiri-Kordestani, L.; Bear, H.; McArthur, H.L.; Frank, E.; Perlmutter, J.; et al. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol. 2019, 5, 1205–1214. [Google Scholar] [CrossRef]
- Polk, A.; Svane, I.-M.; Andersson, M.; Nielsen, D. Checkpoint Inhibitors in Breast Cancer-Current Status. Cancer Treat. Rev. 2018, 63, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chikina, M.; Deshpande, R.; Menk, A.V.; Wang, T.; Tabib, T.; Brunazzi, E.A.; Vignali, K.M.; Sun, M.; Stolz, D.B.; et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8 T Cell-Derived Interferon-γ. Immunity 2019, 51, 381–397.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Thennavan, A.; Garay, J.P.; Marron, J.S.; Perou, C.M. MultiK: An Automated Tool to Determine Optimal Cluster Numbers in Single-Cell RNA Sequencing Data. Genome Biol. 2021, 22, 232. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Zhu, X.; Jin, L.; Chen, M.; Zhang, W.; Su, J. SMGR: A Joint Statistical Method for Integrative Analysis of Single-Cell Multi-Omics Data. NAR Genom. Bioinform. 2022, 4, lqac056. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.-J.; Ma, D.; Liu, Y.-Y.; Xiao, Y.; Gong, Y.; Jiang, Y.-Z.; Shao, Z.-M.; Hu, X.; Di, G.-H. Bulk and Single-Cell Transcriptome Profiling Reveal the Metabolic Heterogeneity in Human Breast Cancers. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 2350–2365. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid Bodies Containing Oxidatively Truncated Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 Interacts with PD-1 and TIM-3 to Regulate T Cell Death and Is a Target for Cancer Immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef]
- Gieseke, F.; Kruchen, A.; Tzaribachev, N.; Bentzien, F.; Dominici, M.; Müller, I. Proinflammatory Stimuli Induce Galectin-9 in Human Mesenchymal Stromal Cells to Suppress T-Cell Proliferation. Eur. J. Immunol. 2013, 43, 2741–2749. [Google Scholar] [CrossRef]
- Hou, T.; Zhang, R.; Jian, C.; Ding, W.; Wang, Y.; Ling, S.; Ma, Q.; Hu, X.; Cheng, H.; Wang, X. NDUFAB1 Confers Cardio-Protection by Enhancing Mitochondrial Bioenergetics through Coordination of Respiratory Complex and Supercomplex Assembly. Cell Res. 2019, 29, 754–766. [Google Scholar] [CrossRef]
- Nitta, S.; Kandori, S.; Tanaka, K.; Sakka, S.; Siga, M.; Nagumo, Y.; Negoro, H.; Kojima, T.; Mathis, B.J.; Shimazui, T.; et al. ELOVL5-Mediated Fatty Acid Elongation Promotes Cellular Proliferation and Invasion in Renal Cell Carcinoma. Cancer Sci. 2022, 113, 2738–2752. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.-J.; et al. Polyunsaturated Fatty Acid Biosynthesis Pathway Determines Ferroptosis Sensitivity in Gastric Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 32433–32442. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Huang, C.-S.; Phan, N.-N.; Lu, T.-P.; Liu, C.-Y.; Huang, C.-J.; Chiu, J.-H.; Tseng, L.-M.; Huang, C.-C. Molecular Subtyping of Breast Cancer Intrinsic Taxonomy with Oligonucleotide Microarray and NanoString NCounter. Biosci. Rep. 2021, 41, BSR20211428. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Kao, K.-J.; Chang, K.-M.; Hsu, H.-C.; Huang, A.T. Correlation of Microarray-Based Breast Cancer Molecular Subtypes and Clinical Outcomes: Implications for Treatment Optimization. BMC Cancer 2011, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Chen, Y.; Vaillant, F.; Capaldo, B.D.; Joyce, R.; Song, X.; Bryant, V.L.; Penington, J.S.; Di Stefano, L.; Tubau Ribera, N.; et al. A Single-Cell RNA Expression Atlas of Normal, Preneoplastic and Tumorigenic States in the Human Breast. EMBO J. 2021, 40, e107333. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative Molecular and Clinical Modeling of Clinical Outcomes to PD1 Blockade in Patients with Metastatic Melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinform. Oxf. Engl. 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Wu, W.; Jin, C.; Cui, J.; Diao, Y.; Wang, R.; Xu, R.; Yao, Z.; Li, X. Integrating Single-Cell RNA-Seq and Bulk RNA-Seq Data to Explore the Key Role of Fatty Acid Metabolism in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 13209. https://doi.org/10.3390/ijms241713209
Chen Y, Wu W, Jin C, Cui J, Diao Y, Wang R, Xu R, Yao Z, Li X. Integrating Single-Cell RNA-Seq and Bulk RNA-Seq Data to Explore the Key Role of Fatty Acid Metabolism in Breast Cancer. International Journal of Molecular Sciences. 2023; 24(17):13209. https://doi.org/10.3390/ijms241713209
Chicago/Turabian StyleChen, Yongxing, Wei Wu, Chenxin Jin, Jiaxue Cui, Yizhuo Diao, Ruiqi Wang, Rongxuan Xu, Zhihan Yao, and Xiaofeng Li. 2023. "Integrating Single-Cell RNA-Seq and Bulk RNA-Seq Data to Explore the Key Role of Fatty Acid Metabolism in Breast Cancer" International Journal of Molecular Sciences 24, no. 17: 13209. https://doi.org/10.3390/ijms241713209
APA StyleChen, Y., Wu, W., Jin, C., Cui, J., Diao, Y., Wang, R., Xu, R., Yao, Z., & Li, X. (2023). Integrating Single-Cell RNA-Seq and Bulk RNA-Seq Data to Explore the Key Role of Fatty Acid Metabolism in Breast Cancer. International Journal of Molecular Sciences, 24(17), 13209. https://doi.org/10.3390/ijms241713209