
The Stabilization of S100A9 Structure by Calcium Inhibits the Formation of Amyloid Fibrils

 , , ,
, , ,  , , ,
, , ,  and
and
Abstract
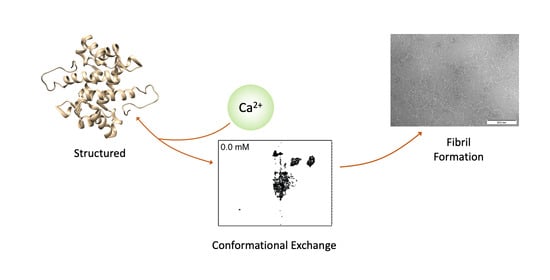
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
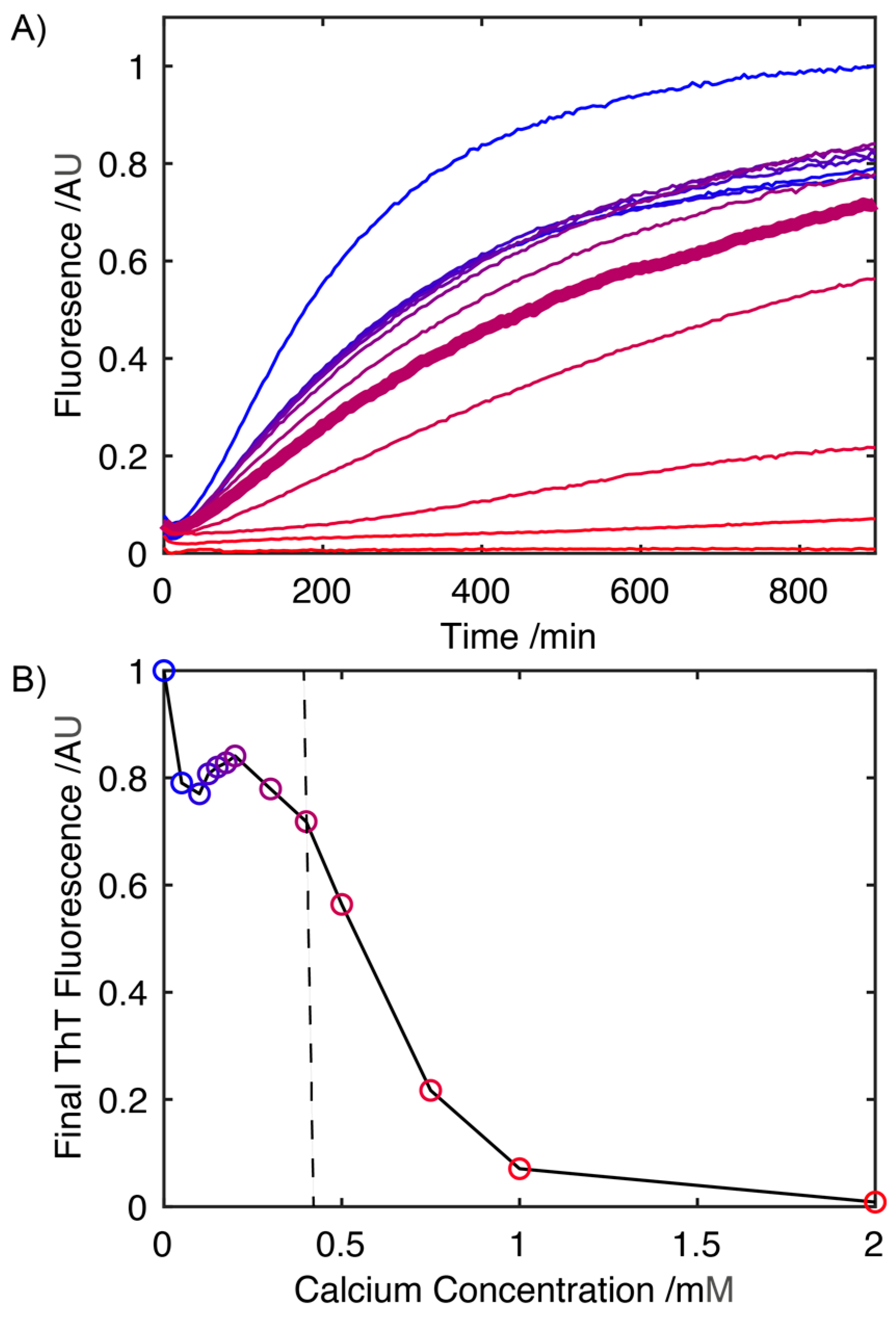
2.1. Influence of Ca2+ on the Rate of S100A9 Fibril Formation
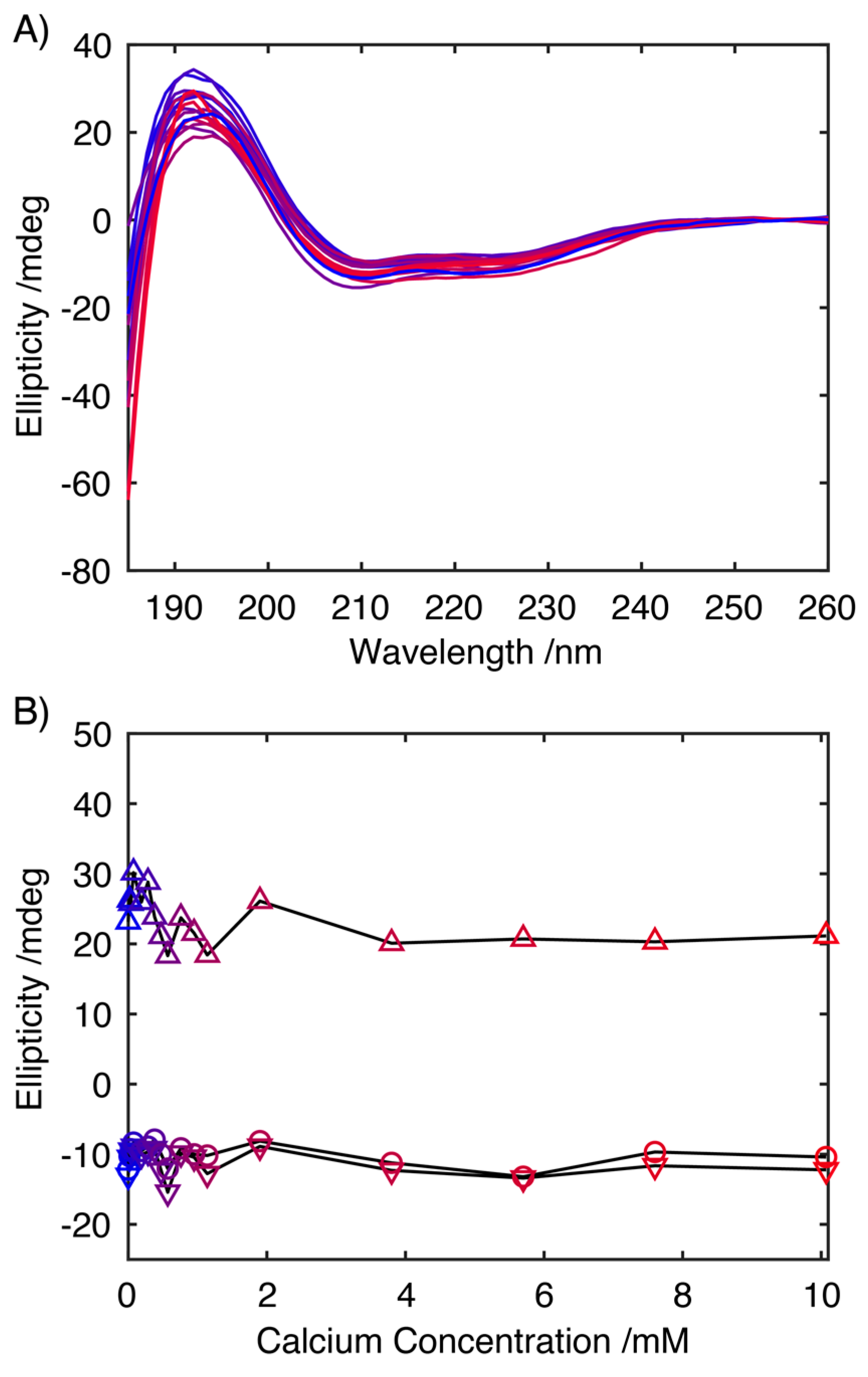
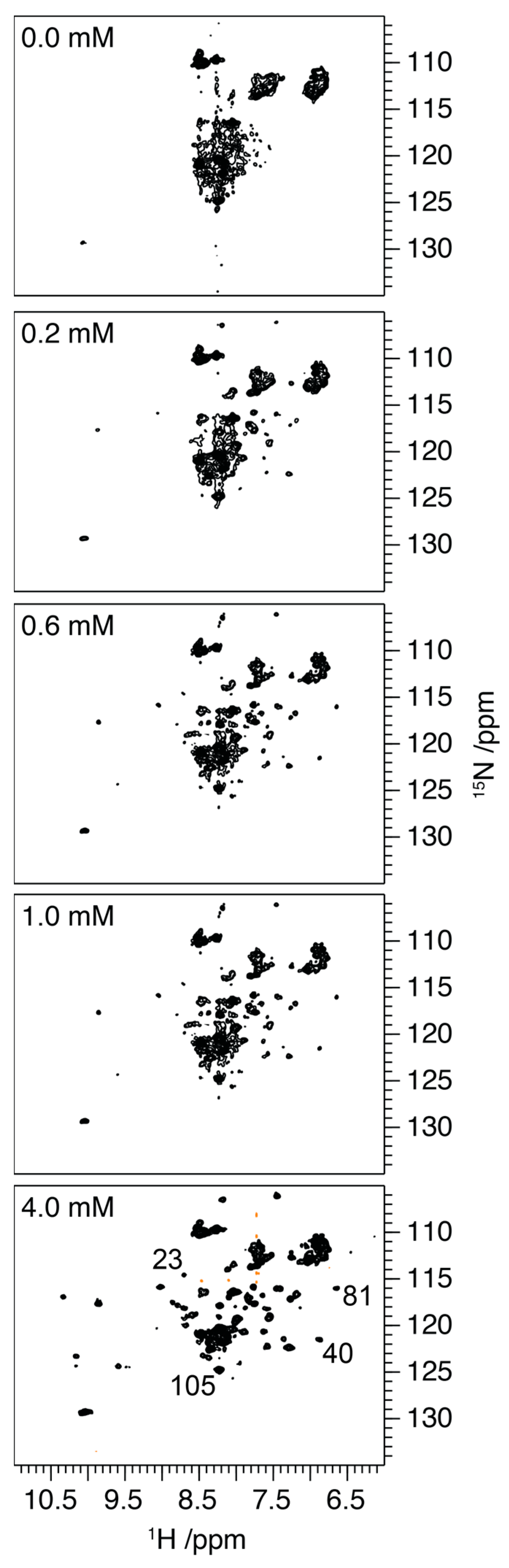
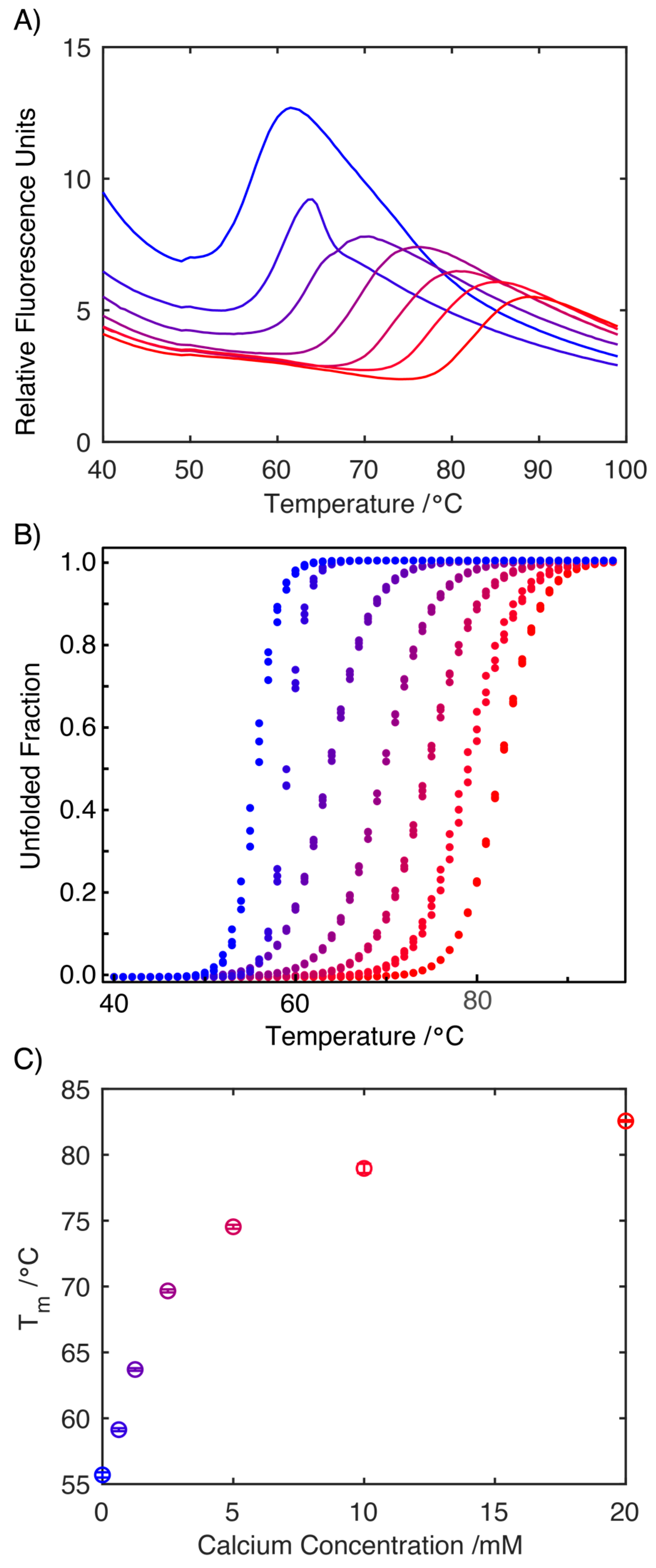
2.2. Influence of Ca2+ on the Structure of S100A9
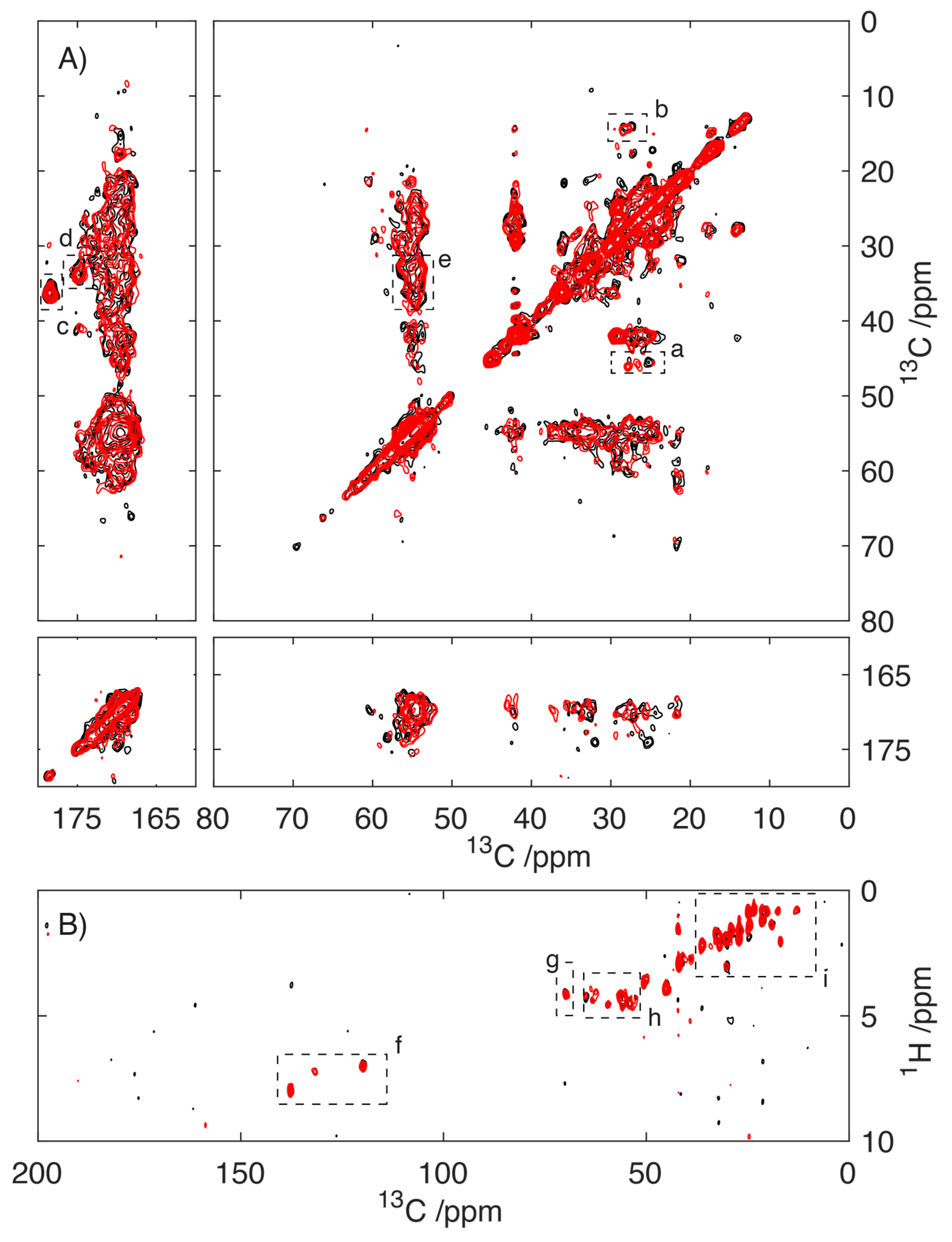
2.3. Does the Presence of Ca2+ Influence the Structure of Fibrillar S100A9?
3. Materials and Methods
3.1. Expression and Purification of S100A9
3.2. Thioflavin-T Fluorescence Fibrilization Assays
3.3. Circular Dichroism
3.4. Differential Scanning Fluorimetry (DSF)
3.5. NMR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Jia, X.; Johansson, P.; Wang, C.; Moskalenko, R.; Steinau, A.; Forsgren, L.; Wagberg, T.; Svensson, J.; Zetterberg, H.; et al. Pro-inflammatory S100A9 Protein as a Robust Biomarker Differentiating Early Stages of Cognitive Impairment in Alzheimer’s Disease. ACS Chem. Neurosci. 2016, 7, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef]
- Akiyama, H.; Ikeda, K.; Katoh, M.; McGeer, E.G.; McGeer, P.L. Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J. Neuroimmunol. 1994, 50, 195–201. [Google Scholar] [CrossRef]
- Kim, H.J.; Chang, K.A.; Ha, T.Y.; Kim, J.; Ha, S.; Shin, K.Y.; Moon, C.; Nacken, W.; Kim, H.S.; Suh, Y.H. S100A9 Knockout Decreases the Memory Impairment and Neuropathology in Crossbreed Mice of Tg2576 and S100A9 Knockout Mice Model. PLoS ONE 2014, 9, e889249. [Google Scholar] [CrossRef]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Warmlander, S.K.; Jarvet, J.; Zhao, L.; Jia, X.; Narayana, V.K.; Shankar, S.K.; Olofsson, A.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef]
- Wang, C.; Iashchishyn, I.A.; Pansieri, J.; Nystrom, S.; Klementieva, O.; Kara, J.; Horvath, I.; Moskalenko, R.; Rofougaran, R.; Gouras, G.; et al. S100A9-Driven Amyloid-Neuroinflammatory Cascade in Traumatic Brain Injury as a Precursor State for Alzheimer’s Disease. Sci. Rep. 2018, 8, 12836. [Google Scholar] [CrossRef]
- Horvath, I.; Iashchishyn, I.A.; Moskalenko, R.A.; Wang, C.; Warmlander, S.; Wallin, C.; Graslund, A.; Kovacs, G.G.; Morozova-Roche, L.A. Co-aggregation of pro-inflammatory S100A9 with alpha-synuclein in Parkinson’s disease: Ex vivo and in vitro studies. J. Neuroinflamm. 2018, 15, 172. [Google Scholar] [CrossRef]
- Zhao, L.N.; Zhang, T.; Zhang, C.; Wang, C.; Morozova-Roche, L.A.; Chew, L.Y.; Mu, Y.G. S100A9 induces aggregation-prone conformation in Abeta peptides: A combined experimental and simulation study. RSC Adv. 2013, 3, 24081–24089. [Google Scholar] [CrossRef]
- Pansieri, J.; Iashchishyn, I.A.; Fakhouri, H.; Ostojic, L.; Malisauskas, M.; Musteikyte, G.; Smirnovas, V.; Schneider, M.M.; Scheidt, T.; Xu, C.K.; et al. Templating S100A9 amyloids on Abeta fibrillar surfaces revealed by charge detection mass spectrometry, microscopy, kinetic and microfluidic analyses. Chem. Sci. 2020, 11, 7031–7039. [Google Scholar] [CrossRef] [PubMed]
- Haruki, K.; Iguchi, Y.; Sahashi, K.; Katsuno, M. Significance of Oligomeric and Fibrillar Species in Amyloidosis: Insights into Pathophysiology and Treatment. Molecules 2021, 26, 5091. [Google Scholar]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef]
- Baracaldo-Santamaría, D.; Avendaño-Lopez, S.S.; Ariza-Salamanca, D.F.; Rodriguez-Giraldo, M.; Calderon-Ospina, C.A.; González-Reyes, R.E.; Nava-Mesa, M.O. Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 9067. [Google Scholar] [CrossRef]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 Proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef] [PubMed]
- Leukert, N.; Vogl, T.; Strupat, K.; Reichelt, R.; Sorg, C.; Roth, J. Calcium-dependent tetramer formation of S100A8 and S100A9 is essential for biological activity. J. Mol. Biol. 2006, 359, 961–972. [Google Scholar] [CrossRef]
- Vogl, T.; Leukert, N.; Barczyk, K.; Strupat, K.; Roth, J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 1298–1306. [Google Scholar] [CrossRef]
- Källberg, E.; Tahvili, S.; Ivars, F.; Leanderson, T. Induction of S100A9 homodimer formation in vivo. Biochem. Biophys. Res. Commun. 2018, 500, 564–568. [Google Scholar] [CrossRef]
- Polakowska, M.; Steczkiewicz, K.; Szczepanowski, R.H.; Wysłouch-Cieszyńska, A. Toward an understanding of the conformational plasticity of S100A8 and S100A9 Ca2+-binding proteins. J. Biol. Chem. 2023, 299, 102952. [Google Scholar] [CrossRef] [PubMed]
- Damo, S.M.; Kehl-Fie, T.E.; Sugitani, N.; Holt, M.E.; Rathi, S.; Murphy, W.J.; Zhang, Y.F.; Betz, C.; Hench, L.; Fritz, G.; et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 3841–3846. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Botelho, H.M.; Morozova-Roche, L.A.; Gomes, C.M. Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 2010, 277, 4578–4590. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.A.; Janes, R.W. Synchrotron radiation circular dichroism (SRCD) spectroscopy: An enhanced method for examining protein conformations and protein interactions. Biochem. Soc. Trans. 2010, 38, 861–873. [Google Scholar] [CrossRef]
- Chang, C.C.; Khan, I.; Tsai, K.L.; Li, H.C.; Yang, L.W.; Chou, R.H.; Yu, C. Blocking the interaction between S100A9 and RAGE V domain using CHAPS molecule: A novel route to drug development against cell proliferation. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2016, 1864, 1558–1569. [Google Scholar] [CrossRef]
- Peti, W.; Smith, L.J.; Redfield, C.; Schwalbe, H. Chemical shifts in denatured proteins: Resonance assignments for denatured ubiquitin and comparisons with other denatured proteins. J. Biomol. NMR 2001, 19, 153–165. [Google Scholar] [CrossRef]
- Schulman, B.A.; Kim, P.S.; Dobson, C.M.; Redfield, C. A residue-specific NMR view of the non-cooperative unfolding of a molten globule. Nat. Struct. Biol. 1997, 4, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.; Carson, W.E. Review of S100A9 biology and its role in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2013, 1835, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Gharibyan, A.L.; Morozova-Roche, L.A. Pro-Inflammatory S100A8 and S100A9 Proteins: Self-Assembly into Multifunctional Native and Amyloid Complexes. Int. J. Mol. Sci. 2012, 13, 2893–2917. [Google Scholar] [CrossRef]
- Lopes, J.P.; Cunha, R.A. What is the extracellular calcium concentration within brain synapses? J. Neurochem. 2019, 149, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Takegoshi, K.; Nakamura, S.; Terao, T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001, 344, 631–637. [Google Scholar] [CrossRef]
- Takegoshi, K.; Nakamura, S.; Terao, T. C-13-H-1 dipolar-driven C-13-C-13 recoupling without C-13 rf irradiation in nuclear magnetic resonance of rotating solids. J. Chem. Phys. 2003, 118, 2325–2341. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef]
- Fritzsching, K.J.; Yang, Y.; Schmidt-Rohr, K.; Hong, M. Practical use of chemical shift databases for protein solid-state NMR: 2D chemical shift maps and amino-acid assignment with secondary-structure information. J. Biomol. NMR 2013, 56, 155–167. [Google Scholar] [CrossRef]
- Miles, A.J.; Ramalli, S.G.; Wallace, B.A. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2022, 31, 37–46. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 1993, 209, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Vadim, K.; Mlynek, G.; Vesper, O.; Pletzer, M.; Wald, J.; Teixeira-Duarte, C.M.; Celia, H.; Garcia-Alai, M.; Nussberger, S.; Buchanan, S.K.; et al. In-depth interrogation of protein thermal unfolding data with MoltenProt. Protein Sci. 2021, 30, 201–217. [Google Scholar]
- Hediger, S.; Meier, B.H.; Kurur, N.D.; Bodenhausen, G.; Ernst, R.R. NMR Cross-Polarization by Adiabatic Passage through the Hartmann-Hahn Condition (Aphh). Chem. Phys. Lett. 1994, 223, 283–288. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Morcombe, C.R.; Gaponenko, V.; Byrd, R.A.; Zilm, K.W. Diluting abundant spins by isotope edited radio frequency field assisted diffusion. J. Am. Chem. Soc. 2004, 126, 7196–7197. [Google Scholar] [CrossRef]
- Marion, D.; Ikura, M.; Tschudin, R.; Bax, A. Rapid Recording of 2D NMR-Spectra without Phase Cycling–Application to the Study of Hydrogen-Exchange in Proteins. J. Magn. Reson. 1989, 85, 393–399. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins Struct. Funct. Bioinform. 2005, 59, 687–696. [Google Scholar] [CrossRef]
- van Beek, J.D. matNMR: A flexible toolbox for processing, analyzing and visualizing magnetic resonance data in Matlab®. J. Magn. Reson. 2007, 187, 19–26. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanders, E.; Csondor, R.; Šulskis, D.; Baronaitė, I.; Smirnovas, V.; Maheswaran, L.; Horrocks, J.; Munro, R.; Georgiadou, C.; Horvath, I.; et al. The Stabilization of S100A9 Structure by Calcium Inhibits the Formation of Amyloid Fibrils. Int. J. Mol. Sci. 2023, 24, 13200. https://doi.org/10.3390/ijms241713200
Sanders E, Csondor R, Šulskis D, Baronaitė I, Smirnovas V, Maheswaran L, Horrocks J, Munro R, Georgiadou C, Horvath I, et al. The Stabilization of S100A9 Structure by Calcium Inhibits the Formation of Amyloid Fibrils. International Journal of Molecular Sciences. 2023; 24(17):13200. https://doi.org/10.3390/ijms241713200
Chicago/Turabian StyleSanders, Ella, Rebecca Csondor, Darius Šulskis, Ieva Baronaitė, Vytautas Smirnovas, Luckshi Maheswaran, Jack Horrocks, Rory Munro, Christina Georgiadou, Istvan Horvath, and et al. 2023. "The Stabilization of S100A9 Structure by Calcium Inhibits the Formation of Amyloid Fibrils" International Journal of Molecular Sciences 24, no. 17: 13200. https://doi.org/10.3390/ijms241713200
APA StyleSanders, E., Csondor, R., Šulskis, D., Baronaitė, I., Smirnovas, V., Maheswaran, L., Horrocks, J., Munro, R., Georgiadou, C., Horvath, I., Morozova-Roche, L. A., & Williamson, P. T. F. (2023). The Stabilization of S100A9 Structure by Calcium Inhibits the Formation of Amyloid Fibrils. International Journal of Molecular Sciences, 24(17), 13200. https://doi.org/10.3390/ijms241713200