
Construction of a Matrix Cancer-Associated Fibroblast Signature Gene-Based Risk Prognostic Signature for Directing Immunotherapy in Patients with Breast Cancer Using Single-Cell Analysis and Machine Learning

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Analysis of BRCA Single-Cell Datasets Revealed the Heterogeneity of CAFs
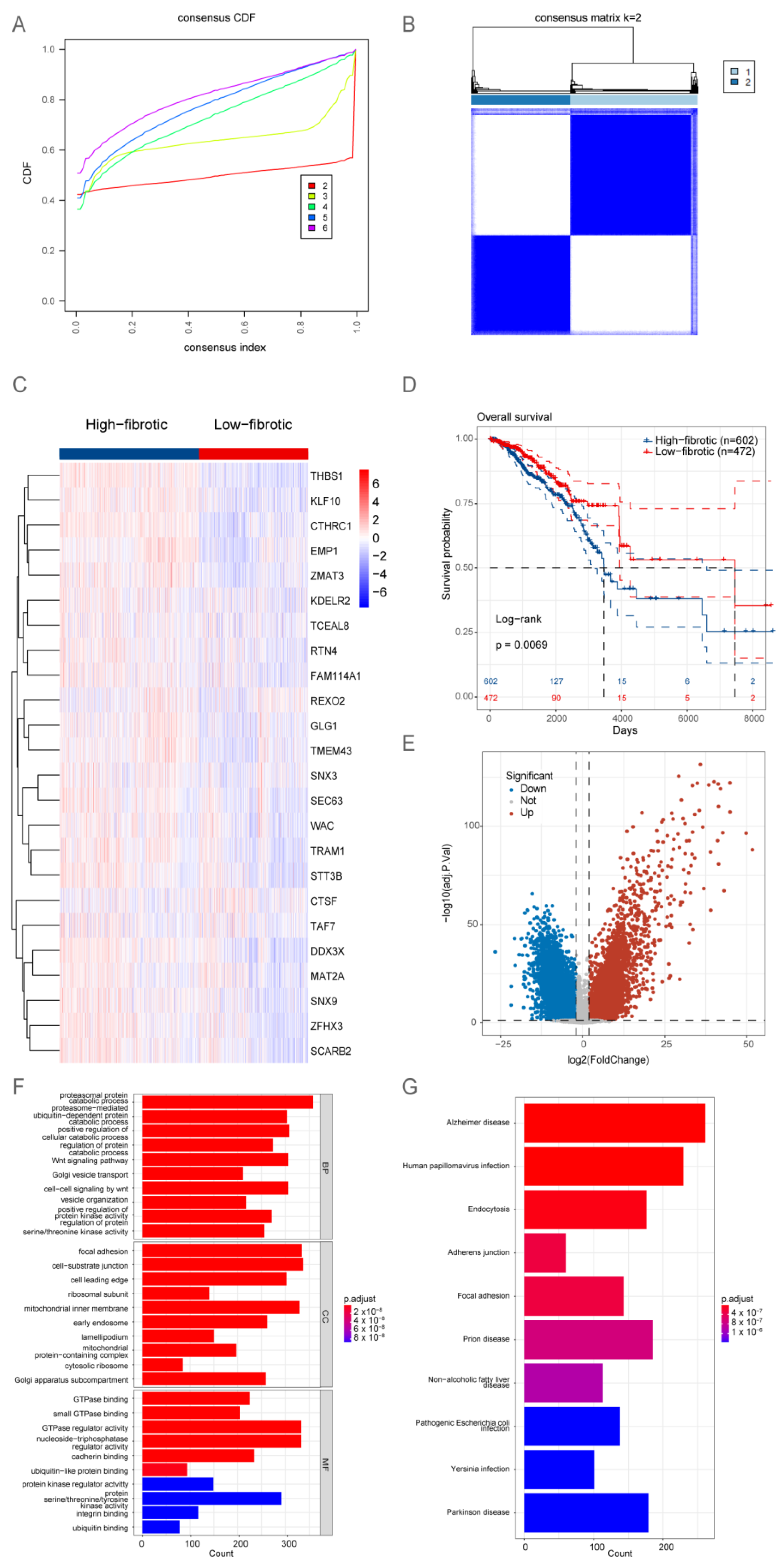
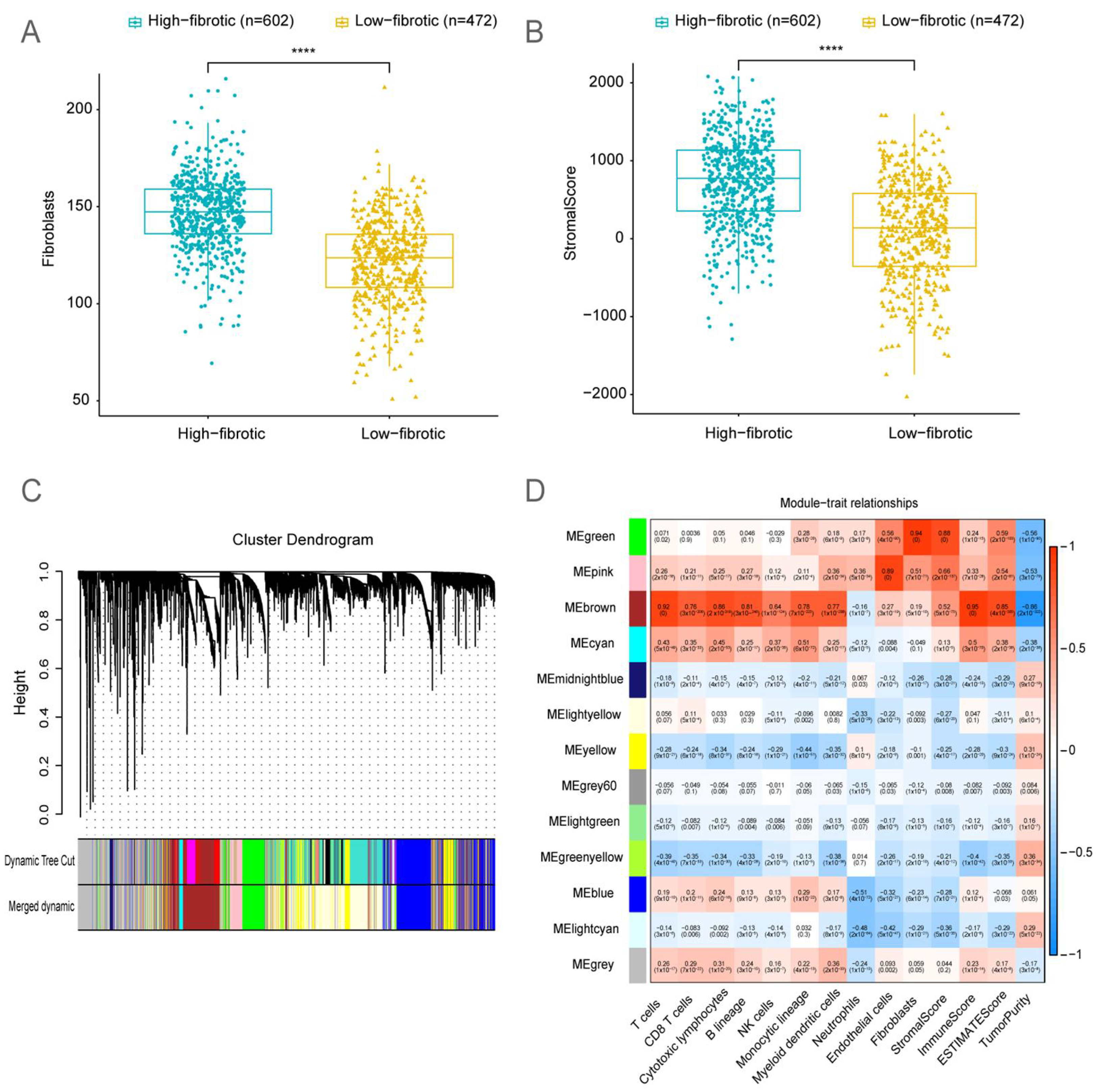
2.2. Bulk RNA-Seq Analysis Revealed That the Accumulation of mCAFs Determines the Poor Prognosis of Patients
2.3. Identification of Fibroblast-Related Module Genes Using Weighted Gene Co-Expression Network Analysis (WGCNA)
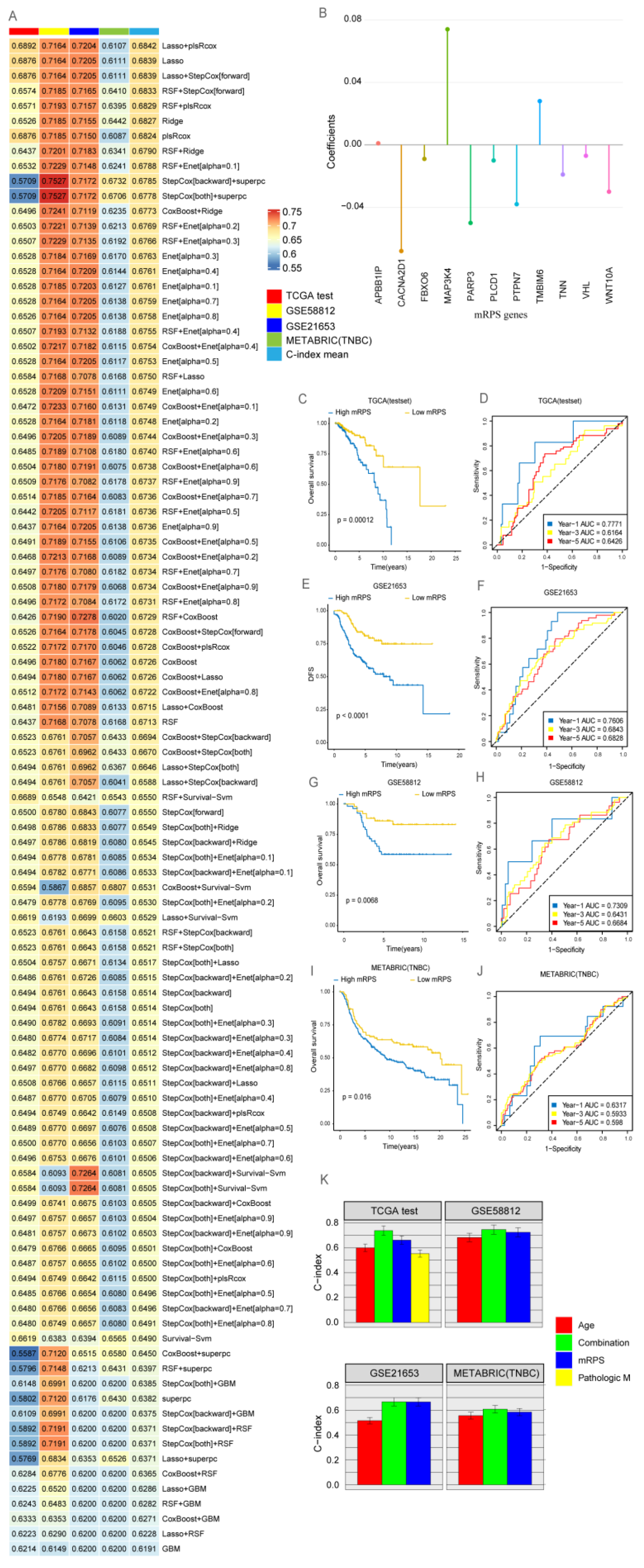
2.4. Development of a Consensus Signature for Predicting the Prognosis of BRCA
2.5. The Clinical Value of mRPS
3. Discussion
4. Materials and Methods
4.1. Transcriptome Analysis Data and Clinical Annotations
4.2. Biological Variation Analysis and the Enrichment Analysis
4.3. BRCA Immune Landscape
4.4. WGCNA
4.5. Machine Learning-Based Construction of an mRPS Risk Prognostic Signature for BRCA
4.6. Application of mRPS in Clinical Treatment
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Dong, G.; Chen, P.; Xu, Y.; Liu, T.; Yin, R. Cancer-associated fibroblasts: Key criminals of tumor pre-metastatic niche. Cancer Lett. 2023, 566, 216234. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Ang, X.; Xu, Z.; Li, S.; Zhang, J.; Pei, C.; Zhou, F. A pan-cancer study of PD-1 and CTLA-4 as therapeutic targets. Transl. Cancer Res. 2021, 10, 3993–4001. [Google Scholar] [CrossRef] [PubMed]
- Metropulos, A.E.; Munshi, H.G.; Principe, D.R. The difficulty in translating the preclinical success of combined TGFβ and immune checkpoint inhibition to clinical trial. EBioMedicine 2022, 86, 104380. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Conroy, M.; Naidoo, J. Immune-related adverse events and the balancing act of immunotherapy. Nat. Commun. 2022, 13, 392. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Kendall, F.; Khozin, S.; Goosen, R.; Hu, J.; Laramie, J.; Ringel, M.; Schork, N. Artificial intelligence and machine learning in clinical development: A translational perspective. NPJ Digit. Med. 2019, 2, 69. [Google Scholar] [CrossRef]
- Zhang, B.; Shi, H.; Wang, H. Machine Learning and AI in Cancer Prognosis, Prediction, and Treatment Selection: A Critical Approach. J. Multidiscip. Healthc. 2023, 16, 1779–1791. [Google Scholar] [CrossRef]
- Galbo, P.M.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14, Erratum in Immunity 2019, 51, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Kim, H.-K.; Bhattarai, K.R.; Junjappa, R.P.; Ahn, J.H.; Pagire, S.H.; Yoo, H.J.; Han, J.; Lee, D.; Kim, K.-W.; Kim, H.-R.; et al. TMBIM6/BI-1 contributes to cancer progression through assembly with mTORC2 and AKT activation. Nat. Commun. 2020, 11, 4012. [Google Scholar] [CrossRef]
- Liang, J.; Deng, Y.; Zhang, Y.; Wu, B.; Zhou, J. PRLR and CACNA2D1 Impact the Prognosis of Breast Cancer by Regulating Tumor Immunity. J. Pers. Med. 2022, 12, 2086. [Google Scholar] [CrossRef]
- Sollome, J.J.; Thavathiru, E.; Camenisch, T.D.; Vaillancourt, R.R. HER2/HER3 regulates extracellular acidification and cell migration through MTK1 (MEKK4). Cell. Signal. 2014, 26, 70–82. [Google Scholar] [CrossRef][Green Version]
- Mu, H.; Wang, N.; Zhao, L.; Li, S.; Li, Q.; Chen, L.; Luo, X.; Qiu, Z.; Li, L.; Ren, G.; et al. Methylation of PLCD1 and adenovirus-mediated PLCD1 overexpression elicits a gene therapy effect on human breast cancer. Exp. Cell Res. 2015, 332, 179–189, Erratum in Exp. Cell Res. 2022, 413, 113047. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Liu, L.; Deng, S.; Ji, W.; Liu, Y.; Wang, X.; Wang, R.; Zhao, X.; Gao, E. Comprehensive analysis of PTPN gene family revealing PTPN7 as a novel biomarker for immuno-hot tumors in breast cancer. Front. Genet. 2022, 13, 981603. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; He, L.; Richards, E.J.; Challa, S.; Xu, C.-X.; Permuth-Wey, J.; Lancaster, J.M.; Coppola, D.; Sellers, T.A.; Djeu, J.Y.; et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene 2013, 33, 679–689. [Google Scholar] [CrossRef]
- Karicheva, O.; Rodriguez-Vargas, J.M.; Wadier, N.; Martin-Hernandez, K.; Vauchelles, R.; Magroun, N.; Tissier, A.; Schreiber, V.; Dantzer, F. PARP3 controls TGFβ and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 2016, 7, 64109–64123. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, B.; Qu, W.; Cao, Y.; Li, J.; Zhao, H. Systematic analysis of the expression and prognosis relevance of FBXO family reveals the significance of FBXO1 in human breast cancer. Cancer Cell Int. 2021, 21, 130. [Google Scholar] [CrossRef]
- Kirikoshi, H.; Sekihara, H.; Katoh, M. Up-regulation of WNT10A by tumor necrosis factor α and Helicobacter pylori in gastric cancer. Int. J. Oncol. 2001, 19, 533–536. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Buchlis, G.; Fridlender, Z.G.; Sun, J.; Kapoor, V.; Cheng, G.; Haas, A.; Cheung, H.K.; Zhang, X.; Corbley, M.; et al. Systemic Blockade of Transforming Growth Factor-β Signaling Augments the Efficacy of Immunogene Therapy. Cancer Res 2008, 68, 10247–10256. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Yi, M.; Jiao, Y.; Chu, Q.; Wu, K. Blocking TGF-β Signaling to Enhance The Efficacy Of Immune Checkpoint Inhibitor. Onco Targets Ther. 2019, 12, 9527–9538. [Google Scholar] [CrossRef]
- Wu, Z.-H.; Li, N.; Gao, Z.-Z.; Chen, G.; Nie, L.; Zhou, Y.-Q.; Jiang, M.-Z.; Chen, Y.; Chen, J.; Mei, X.-F.; et al. Development of the Novel Bifunctional Fusion Protein BR102 That Simultaneously Targets PD-L1 and TGF-β for Anticancer Immunotherapy. Cancers 2022, 14, 4964. [Google Scholar] [CrossRef]
- Gao, Z.-Z.; Li, C.; Chen, G.; Yuan, J.-J.; Zhou, Y.-Q.; Jiao, J.-Y.; Nie, L.; Qi, J.; Yang, Y.; Chen, S.-Q.; et al. Optimization strategies for expression of a novel bifunctional anti-PD-L1/TGFBR2-ECD fusion protein. Protein Expr. Purif. 2022, 189, 105973. [Google Scholar] [CrossRef] [PubMed]
- Pereira, W.J.; Almeida, F.M.; Conde, D.; Balmant, K.M.; Triozzi, P.M.; Schmidt, H.W.; Dervinis, C.; Pappas, G.J.; Kirst, M. Asc-Seurat: Analytical single-cell Seurat-based web application. BMC Bioinform. 2021, 22, 556. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three Differential Expression Analysis Methods for RNA Sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 175, e62528. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218, Erratum in Genome Biol. 2016, 17, 249. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- McEligot, A.J.; Poynor, V.; Sharma, R.; Panangadan, A. Logistic LASSO Regression for Dietary Intakes and Breast Cancer. Nutrients 2020, 12, 2652. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, T.; Xu, F.; Li, S.; Zheng, S.; Lyu, J.; Yin, H. Prediction of prognosis in elderly patients with sepsis based on machine learning (random survival forest). BMC Emerg. Med. 2022, 22, 26. [Google Scholar] [CrossRef]
- Deist, T.M.; Dankers, F.J.W.M.; Valdes, G.; Wijsman, R.; Hsu, I.; Oberije, C.; Lustberg, T.; Soest, J.; Hoebers, F.; Jochems, A.; et al. Machine learning algorithms for outcome prediction in (chemo)radiotherapy: An empirical comparison of classifiers. Med. Phys. 2018, 45, 3449–3459, Erratum in Med. Phys. 2019, 46, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Varma, S.; Sunshine, M.; Elloumi, F.; Ofori-Atta, K.; Lee, S.; Trepel, J.B.; Meltzer, P.S.; Doroshow, J.H.; Pommier, Y. RNA Sequencing of the NCI-60: Integration into CellMiner and CellMiner CDB. Cancer Res 2019, 79, 3514–3524. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, B.; Chen, Q.; Ye, Z.; Zeng, L.; Huang, C.; Xie, Y.; Zhang, R.; Shen, H. Construction of a Matrix Cancer-Associated Fibroblast Signature Gene-Based Risk Prognostic Signature for Directing Immunotherapy in Patients with Breast Cancer Using Single-Cell Analysis and Machine Learning. Int. J. Mol. Sci. 2023, 24, 13175. https://doi.org/10.3390/ijms241713175
Huang B, Chen Q, Ye Z, Zeng L, Huang C, Xie Y, Zhang R, Shen H. Construction of a Matrix Cancer-Associated Fibroblast Signature Gene-Based Risk Prognostic Signature for Directing Immunotherapy in Patients with Breast Cancer Using Single-Cell Analysis and Machine Learning. International Journal of Molecular Sciences. 2023; 24(17):13175. https://doi.org/10.3390/ijms241713175
Chicago/Turabian StyleHuang, Biaojie, Qiurui Chen, Zhiyun Ye, Lin Zeng, Cuibing Huang, Yuting Xie, Rongxin Zhang, and Han Shen. 2023. "Construction of a Matrix Cancer-Associated Fibroblast Signature Gene-Based Risk Prognostic Signature for Directing Immunotherapy in Patients with Breast Cancer Using Single-Cell Analysis and Machine Learning" International Journal of Molecular Sciences 24, no. 17: 13175. https://doi.org/10.3390/ijms241713175
APA StyleHuang, B., Chen, Q., Ye, Z., Zeng, L., Huang, C., Xie, Y., Zhang, R., & Shen, H. (2023). Construction of a Matrix Cancer-Associated Fibroblast Signature Gene-Based Risk Prognostic Signature for Directing Immunotherapy in Patients with Breast Cancer Using Single-Cell Analysis and Machine Learning. International Journal of Molecular Sciences, 24(17), 13175. https://doi.org/10.3390/ijms241713175