
PON-Fold: Prediction of Substitutions Affecting Protein Folding Rate


Abstract
1. Introduction
2. Results and Discussion
2.1. Selection of Data Sets
2.2. Three-Class Classifier
2.3. Regression Predictor
2.4. Blind Test Performance
2.5. PON-Fold Application to Domain-Wide Analysis of Folding Effects
2.6. PON-Fold Web Application
3. Conclusions
4. Materials and Methods
4.1. Data Sets
4.2. Features
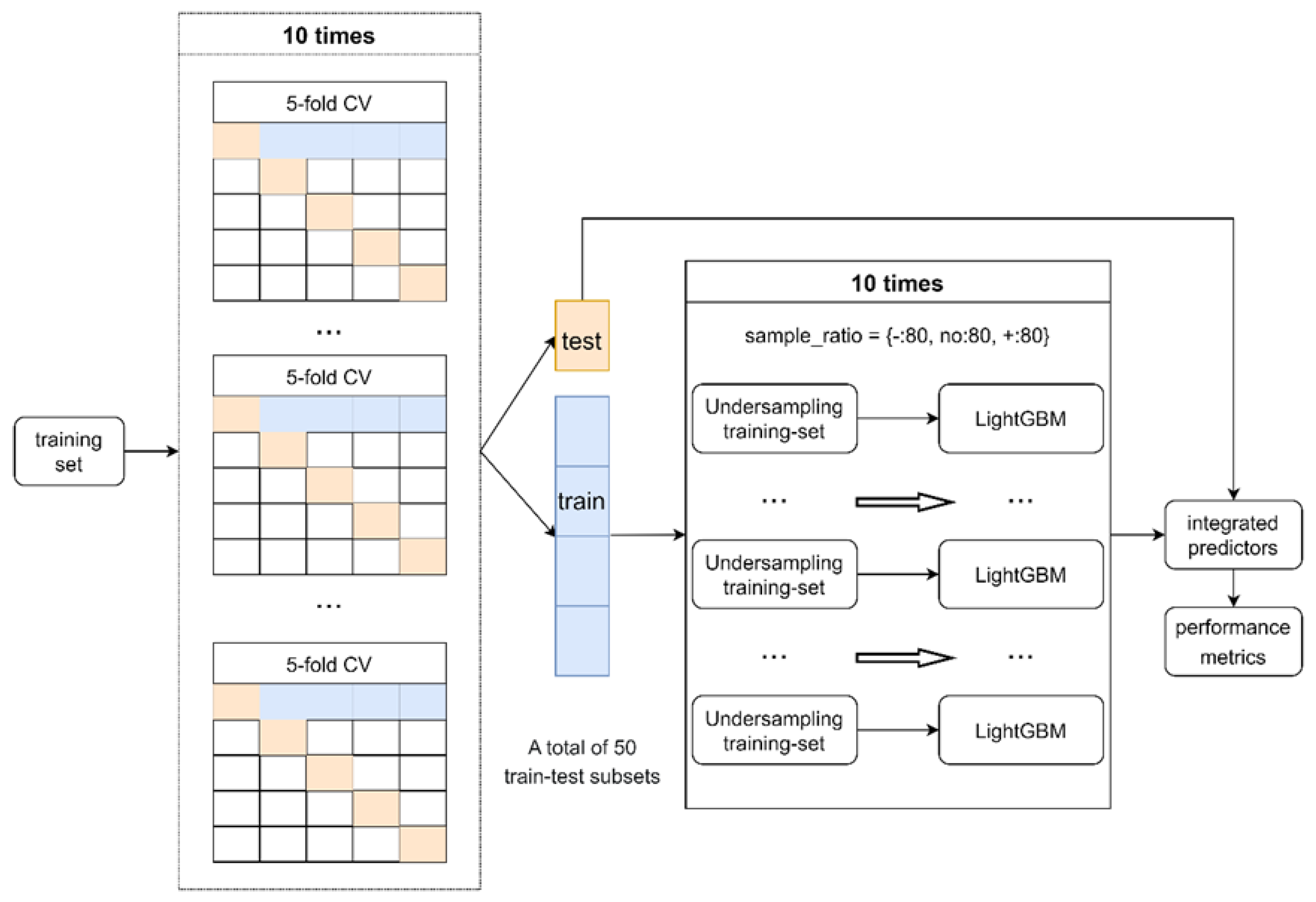
4.3. Training Machine Learning Predictor
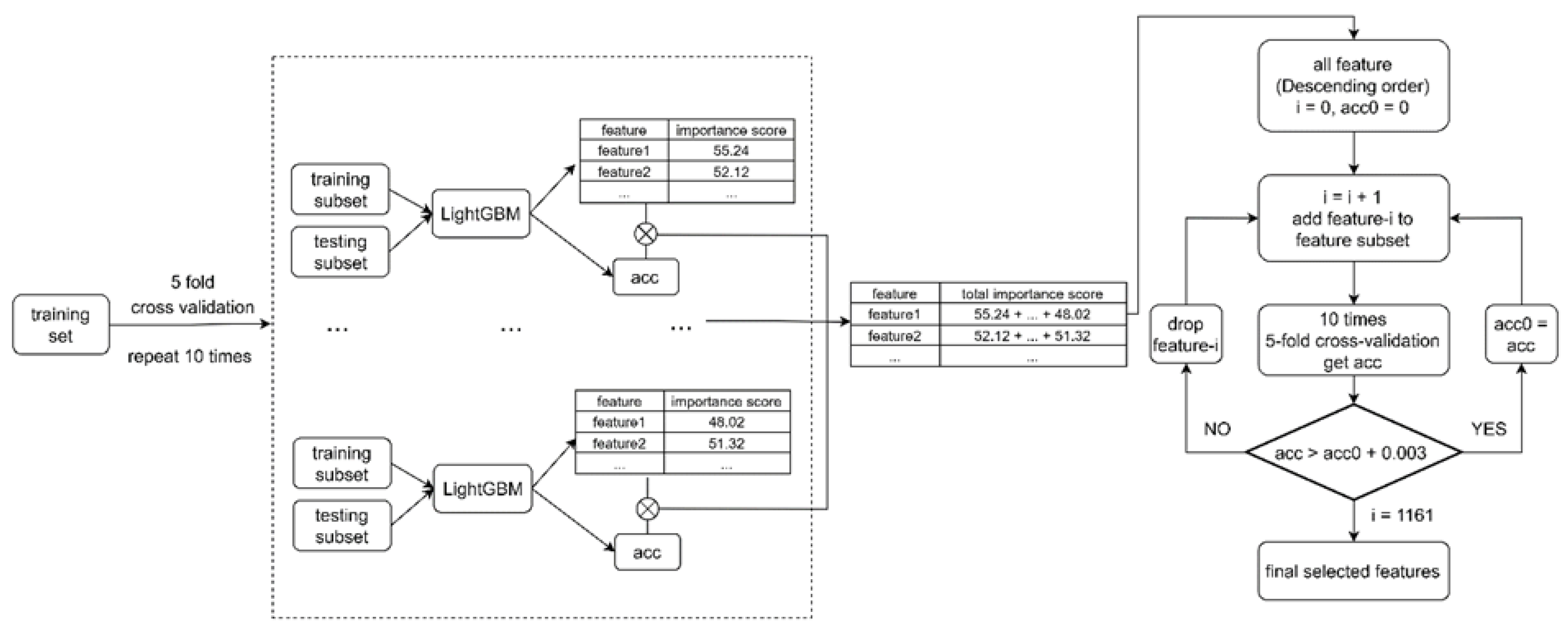
4.4. Feature Selection
4.5. Performance Assessment
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bogatyreva, N.S.; Osypov, A.A.; Ivankov, D.N. KineticDB: A database of protein folding kinetics. Nucleic Acids Res. 2009, 37, D342–D346. [Google Scholar] [CrossRef][Green Version]
- Chaudhary, P.; Naganathan, A.N.; Gromiha, M.M. Prediction of change in protein unfolding rates upon point mutations in two state proteins. Biochim. Biophys. Acta 2016, 1864, 1104–1109. [Google Scholar] [CrossRef]
- Manavalan, B.; Kuwajima, K.; Lee, J. PFDB: A standardized protein folding database with temperature correction. Sci. Rep. 2019, 9, 1588. [Google Scholar] [CrossRef]
- Wagaman, A.S.; Coburn, A.; Brand-Thomas, I.; Dash, B.; Jaswal, S.S. A comprehensive database of verified experimental data on protein folding kinetics. Protein Sci. 2014, 23, 1808–1812. [Google Scholar] [CrossRef]
- Chang, C.C.; Tey, B.T.; Song, J.; Ramanan, R.N. Towards more accurate prediction of protein folding rates: A review of the existing Web-based bioinformatics approaches. Brief. Bioinform. 2015, 16, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Taddei, N.; White, P.M.; Bucciantini, M.; Magherini, F.; Stefani, M.; Dobson, C.M. Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struct. Biol. 1999, 6, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Naganathan, A.N.; Muñoz, V. Insights into protein folding mechanisms from large scale analysis of mutational effects. Proc. Natl. Acad. Sci. USA 2010, 107, 8611–8616. [Google Scholar] [CrossRef]
- Munson, M.; Anderson, K.S.; Regan, L. Speeding up protein folding: Mutations that increase the rate at which Rop folds and unfolds by over four orders of magnitude. Fold. Des. 1997, 2, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Gromiha, M.M. Real value prediction of protein folding rate change upon point mutation. J. Comput. Aided Mol. Des. 2012, 26, 339–347. [Google Scholar] [CrossRef]
- Huang, L.T.; Gromiha, M.M. First insight into the prediction of protein folding rate change upon point mutation. Bioinformatics 2010, 26, 2121–2127. [Google Scholar] [CrossRef]
- Huang, L.T. Finding simple rules for discriminating folding rate change upon single mutation by statistical and learning methods. Protein Pept. Lett. 2014, 21, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Naganathan, A.N.; Gromiha, M.M. Folding RaCe: A robust method for predicting changes in protein folding rates upon point mutations. Bioinformatics 2015, 31, 2091–2097. [Google Scholar] [CrossRef] [PubMed]
- Mallik, S.; Das, S.; Kundu, S. Predicting protein folding rate change upon point mutation using residue-level coevolutionary information. Proteins 2016, 84, 3–8. [Google Scholar] [CrossRef]
- Zhang, Z.; Witham, S.; Petukh, M.; Moroy, G.; Miteva, M.; Ikeguchi, Y.; Alexov, E. A rational free energy-based approach to understanding and targeting disease-causing missense mutations. J. Am. Med. Inform. Assoc. 2013, 20, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, M. Solubility of proteins. ADMET DMPK 2020, 8, 391–399. [Google Scholar] [CrossRef]
- Yang, Y.; Niroula, A.; Shen, B.; Vihinen, M. PON-Sol: Prediction of effects of amino acid substitutions on protein solubility. Bioinformatics 2016, 32, 2032–2034. [Google Scholar] [CrossRef]
- Yang, Y.; Zeng, L.; Vihinen, M. PON-Sol2: Prediction of effects of variants on protein solubility. Int. J. Mol. Sci. 2021, 22, 8027. [Google Scholar] [CrossRef]
- Yang, Y.; Shao, A.; Vihinen, M. PON-All, amino acid substitution tolerance predictor for all organisms. Front. Mol. Biosci. 2022, 9, 867572. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, X.; Zhu, G.; Niroula, A.; Lv, Q.; Vihinen, M. ProTstab—Predictor for cellular protein stability. BMC Genom. 2019, 20, 804. [Google Scholar] [CrossRef]
- Vihinen, M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genom. 2012, 13 (Suppl. S4). [Google Scholar] [CrossRef]
- Vihinen, M. Guidelines for reporting and using prediction tools for genetic variation analysis. Hum. Mutat. 2013, 34, 275–282. [Google Scholar] [CrossRef]
- Schaafsma, G.C.; Vihinen, M. Representativeness of variation benchmark datasets. BMC Bioinform. 2018, 19, 461. [Google Scholar] [CrossRef]
- Väliaho, J.; Smith, C.I.E.; Vihinen, M. BTKbase: The mutation database for X-linked agammaglobulinemia. Hum. Mutat. 2006, 27, 1209–1217. [Google Scholar] [CrossRef]
- Väliaho, J.; Faisal, I.; Ortutay, C.; Smith, C.I.E.; Vihinen, M. Characterization of all possible single nucleotide change –caused amino acid substitutions in the kinase domain of Bruton tyrosine kinase. Hum. Mutat. 2015, 36, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, G.C.; Vihinen, M. Genetic variation in Bruton tyrosine kinase. In Agammaglobulinemia; Plebani, A., Lougaris, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 75–85. [Google Scholar]
- Schaafsma, G.C.P.; Väliaho, J.; Wang, Q.; Berglöf, A.; Zain, R.; Smith, C.I.E.; Vihinen, M. BTKbase, Bruton tyrosin kinase variant database in X-linked agammaglolubinemia: Looking back and ahead. Hum. Mutat. 2023, 2023, 5797541. [Google Scholar] [CrossRef]
- Niroula, A.; Urolagin, S.; Vihinen, M. PON-P2: Prediction method for fast and reliable identification of harmful variants. PLoS ONE 2015, 10, e0117380. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Joseph, R.E.; Wales, T.E.; Fulton, D.B.; Engen, J.R.; Andreotti, A.H. Achieving a Graded Immune Response: BTK Adopts a Range of Active/Inactive Conformations Dictated by Multiple Interdomain Contacts. Structure 2017, 25, 1481–1494.e1484. [Google Scholar] [CrossRef]
- Schaafsma, G.C.P.; Vihinen, M. Large differences in proportions of harmful and benign amino acid substitutions between proteins and diseases. Hum. Mutat. 2017, 38, 839–848. [Google Scholar] [CrossRef]
- Marcotte, D.J.; Liu, Y.T.; Arduini, R.M.; Hession, C.A.; Miatkowski, K.; Wildes, C.P.; Cullen, P.F.; Hong, V.; Hopkins, B.T.; Mertsching, E.; et al. Structures of human Bruton’s tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 2010, 19, 429–439. [Google Scholar] [CrossRef]
- Bone, R.; Springer, J.P.; Atack, J.R. Structure of inositol monophosphatase, the putative target of lithium therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 10031–10035. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zhou, T.; Lafleur, K.; Nevado, C.; Caflisch, A. Kinase selectivity potential for inhibitors targeting the ATP binding site: A network analysis. Bioinformatics 2010, 26, 198–204. [Google Scholar] [CrossRef]
- Nair, P.S.; Vihinen, M. VariBench: A benchmark database for variations. Hum. Mutat. 2013, 34, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Ben Chorin, A.; Masrati, G.; Kessel, A.; Narunsky, A.; Sprinzak, J.; Lahav, S.; Ashkenazy, H.; Ben-Tal, N. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. A Publ. Protein Soc. 2020, 29, 258–267. [Google Scholar] [CrossRef]
- Morcos, F.; Hwa, T.; Onuchic, J.N.; Weigt, M. Direct coupling analysis for protein contact prediction. Methods Mol. Biol. 2014, 1137, 55–70. [Google Scholar] [CrossRef]
- Shen, B.; Vihinen, M. Conservation and covariance in PH domain sequences: Physicochemical profile and information theoretical analysis of XLA-causing mutations in the Btk PH domain. Protein Eng. Des. Sel. 2004, 17, 267–276. [Google Scholar] [CrossRef]
- Lockwood, S.; Krishnamoorthy, B.; Ye, P. Neighborhood properties are important determinants of temperature sensitive mutations. PLoS ONE 2011, 6, e28507. [Google Scholar] [CrossRef]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef]
- Tien, M.Z.; Meyer, A.G.; Sydykova, D.K.; Spielman, S.J.; Wilke, C.O. Maximum allowed solvent accessibilites of residues in proteins. PLoS ONE 2013, 8, e80635. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Aarsand, A.K.; Røraas, T.; Fernandez-Calle, P.; Ricos, C.; Díaz-Garzón, J.; Jonker, N.; Perich, C.; González-Lao, E.; Carobene, A.; Minchinela, J.; et al. The biological variation data critical appraisal checklist: A standard for evaluating studies on biological variation. Clin. Chem. 2018, 64, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.A.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Folding Rate Decreasing (−) | No Effect (No) | Folding Rate Increasing (+) | Total | |
|---|---|---|---|---|
| Training set | 520 | 136 | 106 | 762 |
| Blind test set | 133 | 39 | 18 | 190 |
| Total | 653 | 175 | 124 | 952 |
| Rank | Feature Name | Score | Description |
|---|---|---|---|
| 1 | C-score | 55.23 | Conservation score |
| 2 | rp | 36.11 | Relative position of variation in sequence |
| 3 | rsa | 34.95 | Relative solvent accessibility |
| 4 | TOBD000102 | 30.58 | Optimization-derived potential obtained for large set of decoys |
| 5 | SIMK990102 | 26.75 | Distance-dependent statistical potential (contacts within 5–7.5 Å) |
| 6 | THOP960101 | 24.22 | Mixed quasichemical and optimization-based protein contact potential |
| 7 | VENM980101 | 23.08 | Statistical potential derived by the maximization of the perceptron criterion |
| 8 | BONM030106 | 17.72 | Distances between centers of interacting side chains in the parallel orientation |
| 9 | window_Y | 16.35 | The proportion of Y within a neighborhood window of 25 positions |
| 10 | MOOG990101 | 15.31 | Quasichemical potential derived from interfacial regions of protein–protein complexes |
| 11 | BONM030105 | 14.42 | Distances between centers of interacting side chains in the intermediate orientation |
| 12 | window_NonPolarAA | 10.77 | The proportion of nonpolar residues within a neighborhood window of 25 positions |
| 13 | BETM990101 | 10.65 | Modified version of the Miyazawa–Jernigan transfer energy |
| 14 | MIYS850103 | 8.38 | Quasichemical energy of interactions in an average buried environment |
| 15 | window_ChargedAA | 8.35 | Proportion of charged residues within a neighborhood window of 25 positions |
| 16 | window_PosAA | 8.10 | Proportion of positively charged residues within a neighborhood window of 25 positions |
| 17 | BONM030101 | 7.63 | Quasichemical statistical potential for the antiparallel orientation of interacting side groups |
| 18 | window_F | 7.62 | Proportion of F within a neighborhood window of 25 positions |
| 19 | SKOJ970101 | 7.01 | Statistical potential derived by the quasichemical approximation |
| 20 | window_R | 4.77 | Proportion of R within a neighborhood window of 25 positions |
| 21 | window_D | 4.22 | Proportion of D within a neighborhood window of 25 positions |
| 22 | AURR980118 | 2.90 | Normalized positional residue frequency at helix termini C” |
| 23 | LIWA970101 | 2.85 | Modified version of the Miyazawa-Jernigan transfer energy |
| 24 | GARJ730101 | 1.43 | Partition coefficient |
| 25 | BULH740101 | 1.38 | Transfer free energy to surface |
| 26 | OVEJ920103 | 0.98 | Environment-specific amino acid substitution matrix for beta residues |
| 27 | QIAN880128 | 0.98 | Weights for coil at the window position of −5 |
| 28 | AURR980102 | 0.99 | Normalized positional residue frequency at helix termini N‴ |
| 29 | NADH010101 | 0.18 | Hydropathy scale based on self-information values in the two-state model (5% accessibility) |
| 30 | g2_g6 | 0.001 | Negatively charged amino acid (D, E) substitution by residues in group other amino acids (A, T) |
| 31 | V_A | 0.001 | Valine substitution by alanine |
| 10-Time 5-Fold CV | Blind Test | ||||
|---|---|---|---|---|---|
| Performance Metrics | With All Features a | With 31 Selected Features | With All Features | With 31 Selected Features | |
| TP | − | 55.4/27.4 | 55.9/27.6 | 65.0/31.0 | 60.0/28.6 |
| No | 13.3/24.4 | 14.9/27.4 | 11.0/17.9 | 10.0/16.2 | |
| + | 9.9/22.8 | 11.6/26.9 | 8.0/28.1 | 7.0/24.6 | |
| TN | − | 34.7/70.9 | 35.6/73.2 | 33.0/74.4 | 32.0/67.1 |
| No | 92.6/76.1 | 93.8/78.5 | 118.0/101.8 | 110.0/104.1 | |
| + | 104.7/81.0 | 106.3/83.5 | 123.0/90.7 | 125.0/88.2 | |
| FP | − | 15.2/31.4 | 14.2/29.0 | 24.0/52.2 | 25.0/59.5 |
| No | 32.9/26.1 | 31.7/23.7 | 33.0/24.8 | 41.0/22.6 | |
| + | 26.6/21.3 | 25.0/18.7 | 49.0/36.0 | 47.0/38.4 | |
| FN | − | 48.1/23.8 | 47.6/23.5 | 68.0/32.4 | 73.0/34.8 |
| No | 14.6/26.7 | 12.9/23.8 | 28.0/45.5 | 29.0/47.1 | |
| + | 12.2/28.3 | 10.4/24.2 | 10.0/35.2 | 11.0/38.7 | |
| PRE | − | 0.785/0.472 | 0.797/0.493 | 0.730/0.372 | 0.706/0.324 |
| No | 0.289/0.484 | 0.322/0.535 | 0.250/0.418 | 0.196/0.419 | |
| + | 0.271/0.519 | 0.317/0.591 | 0.140/0.439 | 0.130/0.391 | |
| REC | − | 0.535/0.535 | 0.540/0.540 | 0.489/0.489 | 0.451/0.451 |
| No | 0.479/0.479 | 0.536/0.536 | 0.282/0.282 | 0.256/0.256 | |
| + | 0.445/0.445 | 0.527/0.527 | 0.444/0.444 | 0.389/0.389 | |
| F1 | − | 0.636/0.499 | 0.643/0.514 | 0.586/0.423 | 0.550/0.377 |
| No | 0.357/0.479 | 0.399/0.534 | 0.265/0.337 | 0.222/0.318 | |
| + | 0.333/0.475 | 0.393/0.553 | 0.213/0.442 | 0.194/0.390 | |
| Macro-F1 | All | 0.442/0.485 | 0.478/0.533 | 0.355/0.400 | 0.322/0.362 |
| ACC | All | 0.512/0.486 | 0.538/0.534 | 0.442/0.405 | 0.405/0.366 |
| GC2 | All | 0.050/0.070 | 0.078/0.114 | 0.007/0.015 | 0.017/0.039 |
| Rank | Feature Name | Score | Description |
|---|---|---|---|
| 1 | rsa | 36.000 | Relative solvent accessibility |
| 2 | C-score | 29.361 | Conservation score |
| 3 | rp | 26.525 | Relative position of variation in sequence |
| 4 | ZHAC000105 | 21.893 | Environment-dependent residue contact energies (rows = strand, cols = coil) |
| 5 | MOOG990101 | 18.082 | Quasichemical potential derived from interfacial regions of protein–protein complexes |
| 6 | ZHAC000102 | 16.647 | Environment-dependent residue contact energies (rows = helix, cols = strand) |
| 7 | BASU010101 | 16.462 | Optimization-based potential derived by the modified perceptron criterion |
| 8 | window_T | 15.404 | Proportion of T within a neighborhood window of 25 positions |
| 9 | SIMK990105 | 15.355 | Distance-dependent statistical potential (contacts longer than 12 Å) |
| 10 | window_PolarAA | 15.248 | Proportion of polar residues within a neighborhood window of 25 positions |
| 11 | KESO980102 | 14.054 | Quasichemical energy in an average protein environment derived from interfacial regions of protein–protein complexes |
| 12 | SIMK990102 | 12.777 | Distance-dependent statistical potential (contacts within 5–7.5 Å) |
| 13 | window_G | 11.407 | Proportion of G within a neighborhood window of 25 positions |
| 14 | window_Y | 9.909 | Proportion of Y within a neighborhood window of 25 positions |
| 15 | window_F | 9.617 | Proportion of F within a neighborhood window of 25 positions |
| 16 | window_PosAA | 9.447 | Proportion of positively charged residues within a neighborhood window of 25 positions |
| 17 | window_V | 8.810 | Proportion of V within a neighborhood window of 25 positions |
| 18 | window_D | 7.500 | Proportion of D within a neighborhood window of 25 positions |
| 19 | SNEP660101 | 1.844 | Principal component I |
| 20 | BULH740101 | 1.159 | Transfer free energy to surface |
| 21 | LAWE840101 | 1.155 | Transfer free energy, CHP/water |
| With All Features | With 21 Selected Features | |
|---|---|---|
| PCC | 0.449 | 0.525 |
| MAE | 0.609 | 0.581 |
| MSE | 0.674 | 0.603 |
| R2 | 0.167 | 0.255 |
| PON-Fold | Folding RaCe | |
|---|---|---|
| PCC | 0.330 | 0.170 |
| MAE | 0.672 | 0.952 |
| MSE | 0.817 | 1.632 |
| R2 | −0.021 | −1.040 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Chong, Z.; Vihinen, M. PON-Fold: Prediction of Substitutions Affecting Protein Folding Rate. Int. J. Mol. Sci. 2023, 24, 13023. https://doi.org/10.3390/ijms241613023
Yang Y, Chong Z, Vihinen M. PON-Fold: Prediction of Substitutions Affecting Protein Folding Rate. International Journal of Molecular Sciences. 2023; 24(16):13023. https://doi.org/10.3390/ijms241613023
Chicago/Turabian StyleYang, Yang, Zhang Chong, and Mauno Vihinen. 2023. "PON-Fold: Prediction of Substitutions Affecting Protein Folding Rate" International Journal of Molecular Sciences 24, no. 16: 13023. https://doi.org/10.3390/ijms241613023
APA StyleYang, Y., Chong, Z., & Vihinen, M. (2023). PON-Fold: Prediction of Substitutions Affecting Protein Folding Rate. International Journal of Molecular Sciences, 24(16), 13023. https://doi.org/10.3390/ijms241613023