

From Pathogenesis to Therapeutics: A Review of 150 Years of Huntington’s Disease Research

Abstract
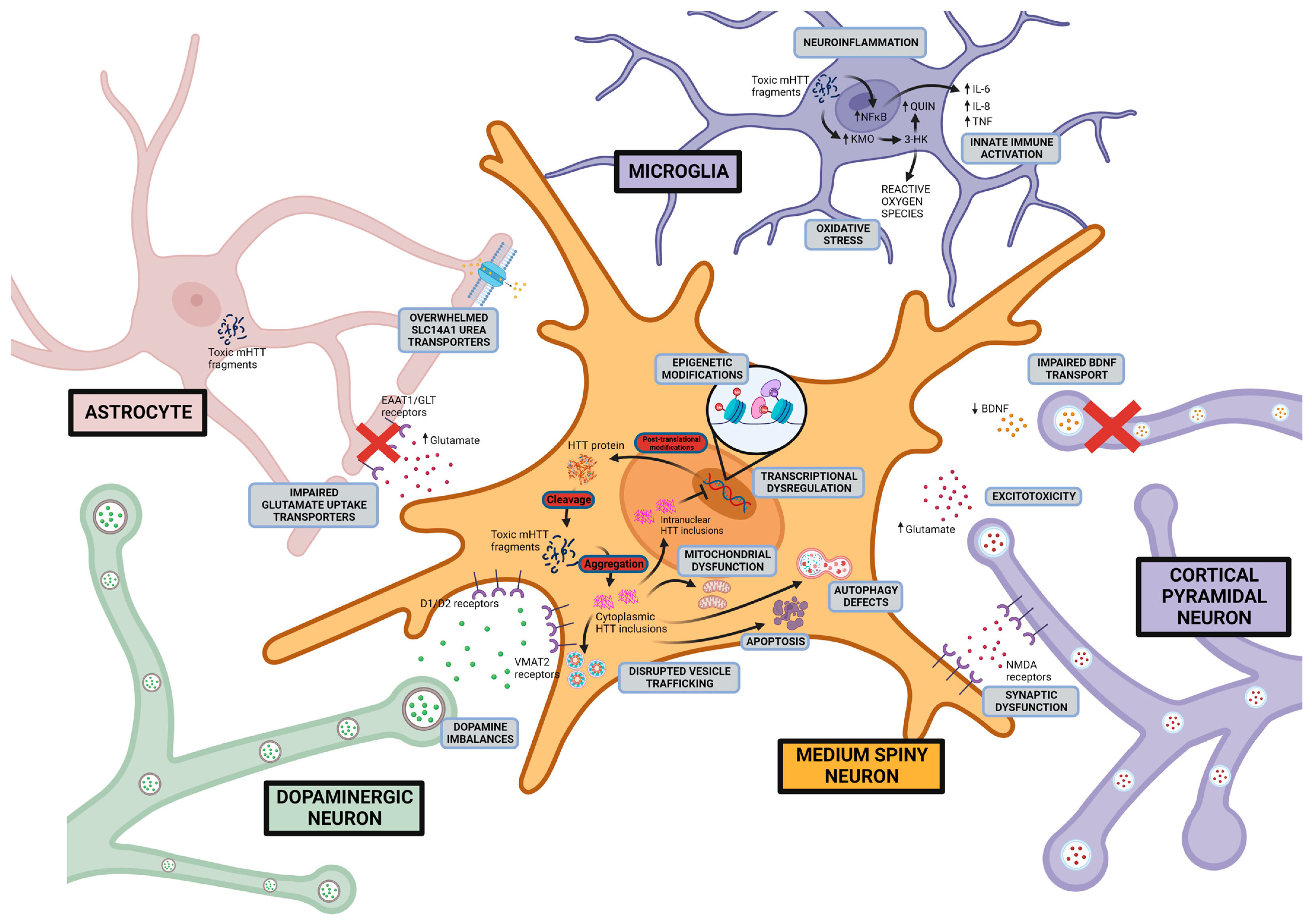
1. Introduction
2. Clinical Implications
2.1. Prevalence and Diagnosis
2.2. Clinical Presentation
2.3. Huntington’s Disease—A Disease of the Basal Ganglia
3. Genetic Aetiology
3.1. Genetic Disease Modifiers
3.2. Huntingtin Structure and Function
4. Hallmarks of Disease
4.1. Neurodegeneration of Striatal GABAergic Medium Spiny Neurons
4.2. Neurodegeneration in Other Brain Regions
4.3. Toxic or Neuroprotective mHTT Inclusions?
4.4. HD Biomarkers
4.5. HD as a Peripheral Disease?
5. Animal Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Models | ||||||||
|---|---|---|---|---|---|---|---|---|
| Toxic Compound | Species | Type of Administration | Mechanism of Action | HD-Related Phenotypes | Reference | |||
| Motor | Neuronal Loss | mHTT Aggreg. | Early Death 1 | |||||
| Quinolinic acid | Rattus norvegicus, Mus musculus, non-human primates | Intrastriatal injections | Glutamate excitotoxicity | ✓ | ✓ | X | ✓ | [194,195,196,197,198,199,200,201,202,203,204] |
| 3-Nitropropionic | Rattus norvegicus, Mus musculus, non-human primates | Systemic, intrastriatal injections | Mitochondrial dysfunction via succinate dehydrogenase inhibition | ✓ | ✓ | X | ✓ | [118,119,195,205,206,207,208,209,210,211,212,213] |
| Mouse Models | ||||||||
| Animal Model | Species | Construct + Promoter | Polyglutamine Repeat Size | HD-Related Phenotypes | Reference | |||
| Motor | Neuronal Loss | mHTT Aggreg. | Early Death 1 | |||||
| (a) Random integration transgenic models | ||||||||
| R6/2 | Mus musculus | 1 Kb 5′UTR + exon 1 of human HTT + 262 bp of intron 1 Human HTT promoter | 144 | ✓ | ✓ | ✓ | ✓ | [214,215,216,217,218] |
| R6/1 | Mus musculus | 1 Kb 5′UTR + exon 1 of human HTT + 262 bp of intron 1 Human HTT promoter | 116 | ✓ | ✓ | ✓ | ✓ | [216] |
| N171-82Q | Mus musculus | First 171 amino acids of human HTT cDNA Mouse Prp promoter | 82 | ✓ | ✓ | ✓ | ✓ | [219,220] |
| HD94 | Mus musculus | 100 bp UTR + human/mouse exon 1 (chimeric) + 600 bp intron 1. CamKIIa promoter | 94 | ✓ | ✓ | ✓ | ✓ | [221] |
| Shortstop | Mus musculus | First 171 amino acids of human HTT Human HTT promoter | 128 | X | X | ✓ | X | [156] |
| N118-82Q | Mus musculus | First 118 amino acids of human HTT cDNA Mouse Prp promoter | 82 | X | X | ✓ | ✓ | [222] |
| N586-82Q | Mus musculus | First 586 amino acids of human HTT cDNA Mouse Prp promoter | 82 | ✓ | ✓ | ✓ | ✓ | [222] |
| HTT-160Q-31 | Mus musculus | First 208 amino acids of human HTT cDNA GFAP promoter | 160 | ✓ | X | ✓ | ✓ | [117] |
| Ubi-G-HTT84Q | Mus musculus | First 153 amino acids of human HTT cDNA fused to EGFP Ubiquitin promoter | 84 | ✓ | X | ✓ | ✓ | [223] |
| HD150QG, HD190QG | Mus musculus | First 67 amino acids of human HTT cDNA fused to EGFP Human HTT promoter | 150, 190 | ✓ | X | ✓ | ✓ | [224] |
| BACHD | Mus musculus | Full length human HTT Human HTT promoter | 97 | ✓ | ✓ | ✓ | X | [225] |
| YAC72 | Mus musculus | Full length human HTT Human HTT promoter | 72 | X | ✓ | ✓ | X | [157] |
| YAC128 | Mus musculus | Full length human HTT Human HTT promoter | 128 | ✓ | ✓ | ✓ | X | [226,227] |
| Hu97/18 | Mus musculus | Full length human HTT, two copies of the full length human HTT alleles Human HTT promoter | 97 | ✓ | ✓ | X | X | [228] |
| iFL148Q | Mus musculus | Inducible full length human HTT cDNA Mouse Prp promoter | 148 | ✓ | X | ✓ | ✓ | [229] |
| BACHD rats | Rattus norvegicus | Full length human HTT Human HTT promoter | 97 | ✓ | ✓ | ✓ | X | [230,231] |
| HD51 | Rattus norvegicus | First 727 amino acids of rat HTT cDNA Rat HTT promoter | 51 | ✓ | ✓ | ✓ | X | [232,233,234] |
| (b) Knock-in models | ||||||||
| HdhQ50 | Mus musculus | Replacement of mouse Hdh exon 1 with human HTT with 50 CAG repeats Mouse HTT promoter | 50 | X | X | X | X | [160] |
| HdhQ92, HdhQ111, HdhQ140, HdhQ150 | Mus musculus | Replacement of mouse Hdh exon 1 with human HTT with 92, 111, 140, 150 CAG repeats. Mouse HTT promoter | 92, 111, 140, 150 | ✓ | ✓ | ✓ | X | [154,158,159,235,236,237] |
| zQ175 | Mus musculus | Replacement of mouse Hdh exon 1 with human HTT with 188 CAG repeats Mouse HTT promoter | 188 | ✓ | ✓ | ✓ | ✓ | [238] |
| (c) Knock-out models | ||||||||
| Animal Model | Species | HD-Related Phenotypes | Reference | |||||
| Hdhex5 (mutation at mouse HTT exon 5) | Mus musculus | -Homozygous knockouts are embryonically lethal at day 8.5 -Heterozygous animals show motor defects and neuronal loss | [239] | |||||
| Deletion of exon 4 and 5 in mouse HTT | Mus musculus | -Homozygous knockouts are embryonically lethal at day 8.5 -Heterozygous animals showcase no overt phenotype and are phenotypically normal | [240] | |||||
| Deletion of a 2.8 kb of 5′ flanking sequence, entire exon 1 and 0.6 kb of 3′ intron of mouse HTT | Mus musculus | -Homozygous knockouts are embryonically lethal at day 8.5–10.5 -Heterozygous animals showcase no overt phenotype and are phenotypically normal | [241] | |||||
| Large Mammalian Models | ||||||||
| Animal Model | Species | Construct + Promoter | Polyglutamine Repeat Size | HD-Related Phenotypes | Reference | |||
| Motor | Neuronal Loss | mHTT Aggreg. | Early Death 1 | |||||
| Transgenic sheep (OVT73) | Ovis aries | Full length human HTT cDNA + 1.1 kb human HTT genomic DNA upstream of exon 1 + 3′ bovine growth hormone UTR Human HTT promoter | 73 | X | X | ✓ | X | [59,178,186,242,243] |
| Transgenic pig #1 | Sus domesticus | First 1100 amino acids of Sus domesticus HTT cDNA Rat neuron-specific enolase (NSE) promoter | 75 | No reports of these animals | [187] | |||
| Transgenic pig #2 | Sus domesticus | First 208 amino acids of human HTT cDNA Cytomegalovirus enhancer/chicken beta-actin promoter | 105 | ✓ | ✓ | ✓ | ✓ | [188] |
| Transgenic pig #3 | Sus domesticus | First 548 amino acids of human HTT cDNA Human HTT promoter | 124 | ✓ | ✓ | ✓ | X | [189,244] |
| Knock-in pig | Sus domesticus | Replacement of pig HTT exon 1 with human HTT exon 1 with 150 CAG repeats Human HTT promoter | 150 | ✓ | ✓ | ✓ | ✓ | [190] |
| Transgenic monkeys #1 | Macaca fascicularis | Lentiviral injection of the first 171 amino acids of human HTT cDNA into rhesus macaque’s striatum PGK-1 lentiviral promoter | 82 | ✓ | X | ✓ | X | [191] |
| Transgenic monkeys #2 | Macaca mulatta | Human HTT exon 1 + GFP gene Human Ubiquitin promoter | 84 | ✓ | ✓ | ✓ | ✓ | [192,245] |
| Transgenic monkeys #3 (rHD6, rHD7, rHD8) | Macaca mulatta | First 508 amino acids of human HTT cDNA Human HTT promoter | 67, 70, 72 | ✓ | ✓ | ✓ | X | [193] |
6. Molecular Mechanisms and Therapeutic Development
6.1. Excitotoxicity-Induced Neurodegeneration
Treatments Targeting Excitotoxicity
6.2. Dopaminergic Imbalance
Treatments Targeting the Dopaminergic Pathway
6.3. Mitochondrial Dysfunction
Treatments Targeting Mitochondrial Dysfunction
6.4. Neuroinflammation
Treatments Targeting Neuroinflammation
6.5. mHTT Protein Misfolding and Endoplasmic Reticulum Stress
Treatments Directly Targeting mHTT Protein
7. Invasive Therapeutics
8. Genetic Mechanisms and Therapeutics
8.1. BDNF
8.2. CREB/CBP
8.3. REST
8.4. PGC1α
8.5. Epigenetic Modifications
8.6. Treatments Targeting Transcriptional Dysregulation
8.7. DNA Targeting Approaches
8.8. RNA-Targeting Approaches
| Drug Name | Mechanism of Action | Current Status (Highest Phase) | Clinical Trial Number | Supporting Reference | |||
|---|---|---|---|---|---|---|---|
| I | II | III | IV | ||||
| Excitotoxicity | |||||||
| Amantadine | NMDA inhibitor | ? | NCT00001930 | [263,264] | |||
| Dimebon (laterepirdine) | NMDA inhibitor | X | NCT00497159, NCT00920946, NCT00387270, NCT01085266, NCT00931073 | [463] | |||
| BN82451 | Na+ channel inhibitor | X | NCT02231580 | [267] | |||
| Memantine | NMDA inhibitor | X | NCT01458470, NCT00652457 | [464] | |||
| Riluzole | Na+ channel inhibitor | X | NCT00277602 | [266] | |||
| Dopaminergic pathway | |||||||
| Bupropion | Dopamine receptor antagonist | X | NCT01914965 | [465] | |||
| Deutetrabenzine | VMAT2 inhibitor | ✓ | FDA-approved | [286] | |||
| Olanzapine, Tiapride | Dopamine receptor antagonist | / | NCT00632645 | ||||
| OMS643762 | Phosphodiesterase inhibitor | X | NCT02074410 | ||||
| PF-02545920 | Phosphodiesterase inhibitor | X | NCT02342548, NCT02197130, NCT01806896 | ||||
| Risperidone | Dopamine receptor antagonist | / | NCT04201834, NCT04071639 | ||||
| Rolipram | Phosphodiesterase inhibitor | ✓ | NCT01602900 | [291] | |||
| SOM3355 | VMAT2 inhibitor | / | NCT03575676, NCT05475483 | ||||
| Tetrabenazine | VMAT2 inhibitor | ✓ | FDA-approved | [284,285,466] | |||
| Valbenazine | VMAT2 inhibitor | ✓ | NCT04102579, NCT04400331 | [288] | |||
| Mitochondrial dysfunction | |||||||
| Creatine | Antioxidant | X | NCT04400331, NCT04102579, NCT01412151, NCT01411150, NCT01411163, NCT00712426, NCT00592995 | [311,312] | |||
| Coenzyme Q10 | Cofactor in electron transport chain | X | NCT00608881, NCT00920699 | [308] | |||
| Eicosapentaenoic acid | Omega-3 fatty acid | X | NCT00146211 | [305,306] | |||
| Fenofibrate | PPARα agonist | / | NCT03515213 | ||||
| Resveratrol | Anti-fungal agent | / | NCT02336633 | ||||
| Triheptanoin | Substrates for Krebs cycle | / | NCT02453061 | [315] | |||
| Neuroinflammation | |||||||
| Laquinimod | Reduces expression of Bax | X | NCT02215616 | [346,349] | |||
| Neflamapimod | p38α MAPK inhibitor | X | NCT03980938 | ||||
| VX15/2503 (pepinemab) | SEMA4D inhibitor | ? | NCT02481674 | [345] | |||
| Minocycline | Antibiotic | X | NCT00277355, NCT00029874, IND 60943 | [352] | |||
| mHTT aggregation and autophagy | |||||||
| Cysteamine | Transglutaminase inhibitor | X | NCT02101957 | [377] | |||
| Epigallocatechin Gallate | Antioxidant | / | NCT01357681 | [374,467] | |||
| Metformin | Inhibitor of mTOR | ? | NCT04826692 | [366] | |||
| Nilotinib | Selective Bcr-Abl tyrosine kinase inhibitor | / | NCT03764215 | ||||
| PBT2 | Metal chelator | ✓ | NCT01590888 | [378] | |||
| Pridopidine | Sigma1-receptor agonist | X | NCT03019289, NCT02494778, NCT01306929, NCT04556656, NCT02006472, NCT00724048, NCT00665223 | [370,371,373] | |||
| Selisistat | SirT1 inhibitor | X | NCT01485952, NCT01485965, NCT01521585, NCT01521832 | [468,469] | |||
| Transcriptional dysregulation | |||||||
| Lithium + Valproate | HDAC inhibitor | ? | NCT00095355 | [438] | |||
| Phenylbutyrate | HDAC inhibitor | ? | NCT00212316 | [470] | |||
| DNA/RNA targeting approaches | |||||||
| AMT-130 | Nonselective miRNA | / | NCT04120493 | ||||
| Branaplam | Splicing modifier introducing a pseudoexon | / | NCT05111249 | [471] | |||
| PTC518 | Splicing modifier introducing a premature STOP codon | / | NCT05358717 | ||||
| Tominersen (IONIS-HTTRX, RG6042) | mHTT allele non-selective antisense oligonucleotide | X | NCT03761849, NCT02519036, NCT03342053, NCT03842969 | [461,472] | |||
| WVE-120101, WVE-120102 | mHTT allele selective antisense oligonucleotide | X | NCT04617860, NCT03225833, NCT03225846 | [473] | |||
| Other | |||||||
| ANX005 | Targets C1q of the complement system | / | NCT04514367 | ||||
| Dextromethorphan/quinidine | Morphinan/class I antiarrhythmic agent | / | NCT03854019 | ||||
| SRX246 | Vasopressin 1a antagonist | / | NCT02507284 | [474] | |||
| Ursodiol | Bile acid | / | NCT00514774 | ||||
9. 150 Years of HD Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huntington, G. On chorea. 1872. Med. Surg. Rep. 1872, 26, 317–321. [Google Scholar]
- Nance, M.A. Huntington Disease: Clinical, Genetic, and Social Aspects. J. Geriatr. Psychiatry Neurol. 1998, 11, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 2001, 14, 219–226. [Google Scholar]
- Evans, S.J.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef]
- Morrison, P.; Harding-Lester, S.; Bradley, A. Uptake of Huntington disease predictive testing in a complete population. Clin. Genet. 2011, 80, 281–286. [Google Scholar] [CrossRef]
- Adachi, Y.; Nakashima, K. Population genetic study of Huntington’s disease--prevalence and founder’s effect in the San-in area, western Japan. Nihon Rinsho. Jpn. J. Clin. Med. 1999, 57, 900–904. [Google Scholar]
- Chang, C.M.; Yu, Y.L.; Fong, K.Y.; Wong, M.T.; Chan, Y.W.; Ng, T.H.; Leung, C.M.; Chan, V. Huntington’s disease in Hong Kong Chinese: Epidemiology and clinical picture. Clin. Exp. Neurol. 1994, 31, 43–51. [Google Scholar]
- Leung, C.M.; Chan, Y.W.; Chang, C.M.; Yu, Y.L.; Chen, C.N. Huntington’s disease in Chinese: A hypothesis of its origin. J. Neurol. Neurosurg. Psychiatry 1992, 55, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; MacGregor, J.M.; Beighton, P.H. The prevalence of Huntington’s chorea in South Africa. S. Afr. Med. J. 1980, 58, 193–196. [Google Scholar] [PubMed]
- Kay, C.; Fisher, E.R.; Hayden, M.R. Huntington’s Disease, 4th ed.; Bates, G.P., Tabrizi, S.J., Jones, L., Eds.; Oxford University Press: Oxford, UK, 2014; Chapter 7. [Google Scholar]
- Fisher, E.R.; Hayden, M.R. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 2014, 29, 105–114. [Google Scholar] [CrossRef]
- Hoppitt, T.; Pall, H.; Calvert, M.; Gill, P.; Yao, G.; Ramsay, J.; James, G.; Conduit, J.; Sackley, C. Systematic Review of the Incidence and Prevalence of Long-Term Neurological Conditions in the UK. Neuroepidemiology 2011, 36, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Morrison, P.J. Accurate prevalence and uptake of testing for Huntington’s disease. Lancet Neurol. 2010, 9, 1147. [Google Scholar] [CrossRef] [PubMed]
- Wexler, A. Stigma, history, and Huntington’s disease. Lancet 2010, 376, 18–19. [Google Scholar] [CrossRef]
- Wexler, N.S. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar]
- Pridmore, S.A. The prevalence of Huntington’s disease in Tasmania. Med. J. Aust. 1990, 153, 133–134. [Google Scholar] [CrossRef]
- Kirilenko, N.B.; Fedotov, V.P.; Baryshnikova, N.V.; Dadali, E.L.; Poliakov, A.V. Nozological spectrum of hereditary diseases of the nervous system in the cities of Volgograd and Volzhsky. Genetika 2004, 40, 1262–1267. [Google Scholar] [CrossRef]
- Sveinsson, O.; Halldórsson, S.; Olafsson, E. An Unusually Low Prevalence of Huntington’s Disease in Iceland. Eur. Neurol. 2012, 68, 48–51. [Google Scholar] [CrossRef]
- Sipilä, J.O.T.; Hietala, M.; Siitonen, A.; Päivärinta, M.; Majamaa, K. Epidemiology of Huntington’s disease in Finland. Park. Relat. Disord. 2015, 21, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.; Karadima, G.; Vassos, E.; Kalfakis, N.; Kladi, A.; Christodoulou, K.; Vassilopoulos, D. Huntington’s disease in Greece: The experience of 14 years. Clin. Genet. 2011, 80, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Sepčić, J.; Antonelli, L.; Šepić-Grahovac, D.; Materljari, E. Epidemiology of Huntington’s Disease in Rijeka District, Yugoslavia. Neuroepidemiology 1989, 8, 105–108. [Google Scholar] [CrossRef]
- Przuntek, H.; Steigerwald, A. Epidemiologic study of Huntington disease in the catchment area of the Würzburg University Neurologic Clinic with special reference to the Lower Franconia district. Nervenarzt 1987, 58, 424–427. [Google Scholar] [PubMed]
- Leger, J.; Ronauil, R.; Vallat, J. Huntington’s chorea in Limousin: Statistical and clinical study. Rev. Med. Limoges 1974, 5, 147–153. [Google Scholar]
- Petit, H.; Salomez, J.L. Huntington’s disease. Contribution of clinical and epidemiological data to genetic counseling. J. Genet. Hum. 1985, 33, 91–102. [Google Scholar]
- Peterlin, B.; Kobal, J.; Teran, N.; Flisar, D.; Lovrečić, L. Epidemiology of Huntington’s disease in Slovenia. Acta Neurol. Scand. 2009, 119, 371–375. [Google Scholar] [CrossRef]
- Mattsson, B. Huntington’s choera in sweden. Acta Psychiatr. Scand. 1974, 50, 221–235. [Google Scholar] [CrossRef]
- Al-Jader, L.N.; Harper, P.S.; Krawczak, M.; Palmer, S.R. The Frequency of Inherited Disorders Database: Prevalence of Huntington Disease. Public Health Genom. 2001, 4, 148–157. [Google Scholar] [CrossRef]
- Maat-Kievit, A.; Helderman-van den Enden, P.; Losekoot, M.; de Knijff, P.; Belfroid, R.; Vegter-van der Vlis, M.; Roos, R.; Breuning, M. Using a roster and haplotyping is useful in risk assessment for persons with intermediate and reduced penetrance alleles in Huntington disease. Am. J. Med. Genet. 2001, 105, 737–744. [Google Scholar] [CrossRef]
- Saugstad, L.; Ødegård, Ø. Huntington’s chorea in Norway. Psychol. Med. 2009, 16, 39–48. [Google Scholar] [CrossRef]
- Burguera, J.A.; Solís, P.; Salazar, A. Estimate of the prevalence of Huntington disease in the Valencia region using the capture-recapture method. Rev. Neurol. 1997, 25, 1845–1847. [Google Scholar]
- Ruiz, J.; Ortin, A.; Cacho, J.; Alburquerque, J. Corea de Huntington: Estudio epidemiológico en la provincia de Salamanca. Arch. Neurobiol. 1985, 48, 302–303. [Google Scholar]
- Squitieri, F.; Griguoli, A.; Capelli, G.; Porcellini, A.; D’Alessio, B. K12 Epidemiology Of Huntington Disease: First Post-htt Gene Analysis Of Prevalence In Italy. J. Neurol. Neurosurg. Psychiatry 2016, 85, A82–A83. [Google Scholar] [CrossRef]
- Gassivaro Gallo, P.; Buhagiar, M.; Cuschieri, A.; Viviani, F. Huntington’s chorea (HD) in Malta: Epidemiology and origins. Int. J. Anthropol. 1999, 14, 115–125. [Google Scholar] [CrossRef]
- Morrison, P.; Johnston, W.; Nevin, N. The epidemiology of Huntington’s disease in Northern Ireland. J. Med. Genet. 1995, 32, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, I.; Hernández, A.; Arias, S. Huntington disease mutation in Venezuela: Age of onset, haplotype analyses and geographic aggregation. J. Hum. Genet. 2007, 53, 127. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.E.; Ochoa, A.; Boll, M.-C.; Sosa, A.L.; Yescas, P.; López, M.; Macias, R.; Familiar, I.; Rasmussen, A. Clinical and genetic characteristics of Mexican Huntington’s disease patients. Mov. Disord. 2009, 24, 2012–2015. [Google Scholar] [CrossRef]
- Reed, T.E.; Chandler, J.H. Huntington’s chorea in Michigan. I. Demography and genetics. Am. J. Hum. Genet. 1958, 10, 201–225. [Google Scholar]
- Wright, H.H.; Still, C.N.; Abramson, R.K. Huntington’s Disease in Black Kindreds in South Carolina. JAMA Neurol. 1981, 38, 412–414. [Google Scholar] [CrossRef]
- Folstein, S.E.; Chase, G.A.; Wahl, W.; McDonnell, A.M.; Folstein, M.F. Huntington disease in Maryland: Clinical aspects of racial variation. Am. J. Hum. Genet. 1987, 41, 168–179. [Google Scholar]
- Kokmen, E.; Özekmekçi, F.S.; Beard, C.M.; O’Brien, P.C.; Kurland, L.T. Incidence and Prevalence of Huntington’s Disease in Olmsted County, Minnesota (1950 Through 1989). JAMA Neurol. 1994, 51, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, A.; Coiteux, C.; Trudeau, J.; Fullum, G. Huntington’s chorea in French Canadians. Preliminary Study. l’Union Med. Can. 1964, 93, 1178. [Google Scholar]
- Shokeir, M.H.K. Investigations on Huntington’s disease in the Canadian Prairies. Clin. Genet. 1975, 7, 345–348. [Google Scholar] [CrossRef]
- Mccusker, E.A.; Casse, R.F.; Graham, S.J.; Wllllams, D.B.; Lazarus, R. Prevalence of Huntington disease in New South Wales in 1996. Med. J. Aust. 2000, 173, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Scrimgeour, E.M.; Pfumojena, J.W. Huntington disease in black Zimbabwean families living near the Mozambique border. Am. J. Med Genet. 1992, 44, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Scrimgeour, E.M. Huntington’s disease in Tanzania. J. Med. Genet. 1981, 18, 200–203. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Lai, C.-H. Nationwide Population-Based Epidemiologic Study of Huntington’s Disease in Taiwan. Neuroepidemiology 2010, 35, 250–254. [Google Scholar] [CrossRef]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet. J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- van der Burg, J.M.; Bjorkqvist, M.; Brundin, P. Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.B.; Bates, G.P.; Steffan, J.; Saft, C.; Tabrizi, S.J. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015, 14, 1135–1142. [Google Scholar] [CrossRef]
- Sassone, J.; Colciago, C.; Cislaghi, G.; Silani, V.; Ciammola, A. Huntington’s disease: The current state of research with peripheral tissues. Exp. Neurol. 2009, 219, 385–397. [Google Scholar] [CrossRef]
- Aziz, N.A.; van der Burg, J.M.; Landwehrmeyer, G.B.; Brundin, P.; Stijnen, T.; Group, E.S.; Roos, R.A. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008, 71, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Djousse, L.; Knowlton, B.; Cupples, L.A.; Marder, K.; Shoulson, I.; Myers, R.H. Weight loss in early stage of Huntington’s disease. Neurology 2002, 59, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.O.; Ho, A.K.; Barker, R.A. Weight changes in Huntington’s disease. Eur. J. Neurol. 2006, 13, e7. [Google Scholar] [CrossRef]
- Arnulf, I.; Nielsen, J.; Lohmann, E.; Schiefer, J.; Wild, E.; Jennum, P.; Konofal, E.; Walker, M.; Oudiette, D.; Tabrizi, S.; et al. Rapid Eye Movement Sleep Disturbances in Huntington Disease. Arch. Neurol. 2008, 65, 482–488. [Google Scholar] [CrossRef]
- Morton, A.J. Circadian and sleep disorder in Huntington’s disease. Exp. Neurol. 2013, 243, 34–44. [Google Scholar] [CrossRef]
- Keum, J.W.; Shin, A.; Gillis, T.; Mysore, J.S.; Abu Elneel, K.; Lucente, D.; Hadzi, T.; Holmans, P.; Jones, L.; Orth, M.; et al. The HTT CAG-Expansion Mutation Determines Age at Death but Not Disease Duration in Huntington Disease. Am. J. Hum. Genet. 2016, 98, 287–298. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Thu, D.C.; Tippett, L.J.; Faull, R.L. New Perspectives on the Neuropathology in Huntington’s Disease in the Human Brain and its Relation to Symptom Variation. J. Huntingt. Dis. 2012, 1, 143–153. [Google Scholar] [CrossRef]
- Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and consistency. Mov. Disord. 1996, 11, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Reilmann, R. E1 Diagnostic criteria for huntington’s disease based on natural history. J. Neurol. Neurosurg. Psychiatry 2014, 87, A45.1–A45. [Google Scholar] [CrossRef]
- Lanciego, J.L.; Luquin, N.; Obeso, J.A. Functional Neuroanatomy of the Basal Ganglia. Cold Spring Harb. Perspect. Med. 2012, 2, a009621. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.B.; Kreitzer, A.C. Reassessing Models of Basal Ganglia Function and Dysfunction. Annu. Rev. Neurosci. 2014, 37, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Snell, R.G.; MacMillan, J.C.; Cheadle, J.P.; Fenton, I.; Lazarou, L.P.; Davies, P.; MacDonald, M.E.; Gusella, J.F.; Harper, P.S.; Shaw, D.J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet. 1993, 4, 393–397. [Google Scholar] [CrossRef]
- Foroud, T.; Gray, J.; Ivashina, J.; Conneally, P.M. Differences in duration of Huntington’s disease based on age at onset. J. Neurol. Neurosurg. Psychiatry 1999, 66, 52–56. [Google Scholar] [CrossRef]
- Lee, J.-M.; Ramos, E.M.; Lee, J.-H.; Gillis, T.; Mysore, J.S.; Hayden, M.R.; Warby, S.C.; Morrison, P.; Nance, M.; Ross, C.A.; et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 2012, 78, 690–695. [Google Scholar] [CrossRef]
- Wexler, N.S.; Young, A.B.; Tanzi, R.E.; Travers, H.; Starosta-Rubinstein, S.; Penney, J.B.; Snodgrass, S.R.; Shoulson, I.; Gomez, F.; Ramos Arroyo, M.A.; et al. Homozygotes for Huntington’s disease. Nature 1987, 326, 194–197. [Google Scholar] [CrossRef]
- Cubo, E.; Martinez-Horta, S.I.; Santalo, F.S.; Descalls, A.M.; Calvo, S.; Gil-Polo, C.; Munoz, I.; Llano, K.; Mariscal, N.; Diaz, D.; et al. Clinical manifestations of homozygote allele carriers in Huntington disease. Neurology 2019, 92, e2101–e2108. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Takahashi, T.; Katada, S.; Onodera, O. Polyglutamine Diseases: Where does Toxicity Come from? What is Toxicity? Where are We Going? J. Mol. Cell Biol. 2010, 2, 180–191. [Google Scholar] [CrossRef]
- Trottier, Y.; Biancalana, V.; Mandel, J.L. Instability of CAG repeats in Huntington’s disease: Relation to parental transmission and age of onset. J. Med. Genet. 1994, 31, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; Persichetti, F.; McNeil, S.M.; Mysore, J.S.; Mysore, S.S.; MacDonald, M.E.; Myers, R.H.; Gusella, J.F.; Wexler, N.S.; US-Venezuela Collaborative Research Group. Factors associated with HD CAG repeat instability in Huntington disease. J. Med. Genet. 2007, 44, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Quarrell, O.; O’Donovan, K.L.; Bandmann, O.; Strong, M. The Prevalence of Juvenile Huntington’s Disease: A Review of the Literature and Meta-Analysis. PLoS Curr. 2012, 4, e4f8606b742ef3. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, J.G.; van der Velde, E.A.; Roos, R.A.; Bruyn, G.W. Juvenile Huntington disease. Hum. Genet. 1986, 73, 235–239. [Google Scholar] [CrossRef]
- Squitieri, F.; Frati, L.; Ciarmiello, A.; Lastoria, S.; Quarrell, O. Juvenile Huntington’s disease: Does a dosage-effect pathogenic mechanism differ from the classical adult disease? Mech. Ageing Dev. 2006, 127, 208–212. [Google Scholar] [CrossRef]
- Telenius, H.; Kremer, B.; Goldberg, Y.P.; Theilmann, J.; Andrew, S.E.; Zeisler, J.; Adam, S.; Greenberg, C.; Ives, E.J.; Clarke, L.A.; et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet. 1994, 6, 409–414. [Google Scholar] [CrossRef]
- De Rooij, K.E.; De Koning Gans, P.A.; Roos, R.A.; Van Ommen, G.J.; Den Dunnen, J.T. Somatic expansion of the (CAG) n repeat in Huntington disease brains. Hum. Genet. 1995, 95, 270–274. [Google Scholar] [CrossRef]
- Kennedy, L.; Evans, E.; Chen, C.M.; Craven, L.; Detloff, P.J.; Ennis, M.; Shelbourne, P.F. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 2003, 12, 3359–3367. [Google Scholar] [CrossRef]
- Pinto, R.M.; Arning, L.; Giordano, J.V.; Razghandi, P.; Andrew, M.A.; Gillis, T.; Correia, K.; Mysore, J.S.; Grote Urtubey, D.-M.; Parwez, C.R.; et al. Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2020, 29, 2551–2567. [Google Scholar] [CrossRef]
- Gonitel, R.; Moffitt, H.; Sathasivam, K.; Woodman, B.; Detloff, P.J.; Faull, R.L.; Bates, G.P. DNA instability in postmitotic neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 3467–3472. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Tam, M.; Wang, Y.H.; Montgomery, S.E.; Dar, A.C.; Cleary, J.D.; Nichol, K. Slipped-strand DNAs formed by long (CAG)*(CTG) repeats: Slipped-out repeats and slip-out junctions. Nucleic Acids Res. 2002, 30, 4534–4547. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wilson, J.H. Nucleotide Excision Repair, Mismatch Repair, and R-Loops Modulate Convergent Transcription-Induced Cell Death and Repeat Instability. PLoS ONE 2012, 7, e46807. [Google Scholar] [CrossRef]
- Pinto, R.M.; Dragileva, E.; Kirby, A.; Lloret, A.; Lopez, E.; St Claire, J.; Panigrahi, G.B.; Hou, C.; Holloway, K.; Gillis, T.; et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLoS Genet. 2013, 9, e1003930. [Google Scholar] [CrossRef] [PubMed]
- Dragileva, E.; Hendricks, A.; Teed, A.; Gillis, T.; Lopez, E.T.; Friedberg, E.C.; Kucherlapati, R.; Edelmann, W.; Lunetta, K.L.; MacDonald, M.E.; et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 2009, 33, 37–47. [Google Scholar] [CrossRef]
- Tome, S.; Manley, K.; Simard, J.P.; Clark, G.W.; Slean, M.M.; Swami, M.; Shelbourne, P.F.; Tillier, E.R.; Monckton, D.G.; Messer, A.; et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. 2013, 9, e1003280. [Google Scholar] [CrossRef] [PubMed]
- Loupe, J.M.; Pinto, R.M.; Kim, K.H.; Gillis, T.; Mysore, J.S.; Andrew, M.A.; Kovalenko, M.; Murtha, R.; Seong, I.; Gusella, J.F.; et al. Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum. Mol. Genet. 2020, 29, 3044–3053. [Google Scholar] [CrossRef]
- Goold, R.; Flower, M.; Moss, D.H.; Medway, C.; Wood-Kaczmar, A.; Andre, R.; Farshim, P.; Bates, G.P.; Holmans, P.; Jones, L.; et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet. 2019, 28, 650–661. [Google Scholar] [CrossRef]
- Gusella, J.F.; MacDonald, M.E.; Lee, J.M. Genetic modifiers of Huntington’s disease. Mov. Disord. 2014, 29, 1359–1365. [Google Scholar] [CrossRef]
- Huntington Study Group, P.I. At risk for Huntington disease: The PHAROS (Prospective Huntington At Risk Observational Study) cohort enrolled. Arch. Neurol. 2006, 63, 991–996. [Google Scholar]
- Huntington Study Group COHORT Investigators; Dorsey, E. R. Characterization of a large group of individuals with huntington disease and their relatives enrolled in the COHORT study. PLoS ONE 2012, 7, e29522. [Google Scholar]
- Huntington Study Group TREND-HD Investigators. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch. Neurol. 2008, 65, 1582–1589. [Google Scholar]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington’s disease decades before diagnosis: The Predict-HD study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Orth, M. Observing Huntington’s disease: The European Huntington’s Disease Network’s REGISTRY. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1409. [Google Scholar] [CrossRef] [PubMed]
- Landwehrmeyer, G.B.; Fitzer-Attas, C.J.; Giuliano, J.D.; Gonçalves, N.; Anderson, K.E.; Cardoso, F.; Ferreira, J.J.; Mestre, T.A.; Stout, J.C.; Sampaio, C. Data Analytics from Enroll-HD, a Global Clinical Research Platform for Huntington’s Disease. Mov. Disord. Clin. Pract. 2017, 4, 212–224. [Google Scholar] [CrossRef]
- Li, J.L.; Hayden, M.R.; Almqvist, E.W.; Brinkman, R.R.; Durr, A.; Dode, C.; Morrison, P.J.; Suchowersky, O.; Ross, C.A.; Margolis, R.L.; et al. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. Am. J. Hum. Genet. 2003, 73, 682–687. [Google Scholar] [CrossRef]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e14. [Google Scholar] [CrossRef]
- Lobanov, S.V.; McAllister, B.; McDade-Kumar, M.; Landwehrmeyer, G.B.; Orth, M.; Rosser, A.E.; Paulsen, J.S.; Lee, J.-M.; MacDonald, M.E.; Gusella, J.F.; et al. Huntington’s disease age at motor onset is modified by the tandem hexamer repeat in TCERG1. Genom. Med. 2022, 7, 53. [Google Scholar] [CrossRef]
- Nakamori, M.; Panigrahi, G.B.; Lanni, S.; Gall-Duncan, T.; Hayakawa, H.; Tanaka, H.; Luo, J.; Otabe, T.; Li, J.; Sakata, A.; et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet. 2020, 52, 146–159. [Google Scholar] [CrossRef]
- Lin, B.; Rommens, J.M.; Graham, R.K.; Kalchman, M.; MacDonald, H.; Nasir, J.; Delaney, A.; Goldberg, Y.P.; Hayden, M.R. Differential 3′ polyadenylation of the Huntington disease gene results in two mRNA species with variable tissue expression. Hum. Mol. Genet. 1993, 2, 1541–1545. [Google Scholar] [CrossRef]
- Hughes, A.C.; Mort, M.; Elliston, L.; Thomas, R.M.; Brooks, S.P.; Dunnett, S.B.; Jones, L. Identification of Novel Alternative Splicing Events in the Huntingtin Gene and Assessment of the Functional Consequences Using Structural Protein Homology Modelling. J. Mol. Biol. 2014, 426, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Ruzo, A.; Ismailoglu, I.; Popowski, M.; Haremaki, T.; Croft, G.F.; Deglincerti, A.; Brivanlou, A.H. Discovery of Novel Isoforms of Huntingtin Reveals a New Hominid-Specific Exon. PLoS ONE 2015, 10, e0127687. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998, 95, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the Caspase-6 Site Is Required for Neuronal Dysfunction and Degeneration Due to Mutant Huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef]
- Palidwor, G.A.; Shcherbinin, S.; Huska, M.R.; Rasko, T.; Stelzl, U.; Arumughan, A.; Foulle, R.; Porras, P.; Sanchez-Pulido, L.; Wanker, E.E.; et al. Detection of Alpha-Rod Protein Repeats Using a Neural Network and Application to Huntingtin. PLoS Comput. Biol. 2009, 5, e1000304. [Google Scholar] [CrossRef]
- Schaefer, M.H.; Fontaine, J.-F.; Vinayagam, A.; Porras, P.; Wanker, E.E.; Andrade-Navarro, M.A. HIPPIE: Integrating Protein Interaction Networks with Experiment Based Quality Scores. PLoS ONE 2012, 7, e31826. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Bin, H.; Cheng, J.; Seefelder, M.; Engler, T.; Pfeifer, G.; Oeckl, P.; Otto, M.; Moser, F.; Maurer, M.; et al. The cryo-electron microscopy structure of huntingtin. Nature 2018, 555, 117–120. [Google Scholar] [CrossRef]
- Seong, I.S.; Woda, J.M.; Song, J.-J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.-M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2009, 19, 573–583. [Google Scholar] [CrossRef]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Plotkin, J.L.; Surmeier, D.J. Corticostriatal synaptic adaptations in Huntington’s disease. Curr. Opin. Neurobiol. 2015, 33, 53–62. [Google Scholar]
- Rauskolb, S.; Zagrebelsky, M.; Dreznjak, A.; Deogracias, R.; Matsumoto, T.; Wiese, S.; Erne, B.; Sendtner, M.; Schaeren-Wiemers, N.; Korte, M.; et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 2010, 30, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Strand, A.D.; Baquet, Z.C.; Aragaki, A.K.; Holmans, P.; Yang, L.; Cleren, C.; Beal, M.F.; Jones, L.; Kooperberg, C.; Olson, J.M.; et al. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 2007, 27, 11758–11768. [Google Scholar] [CrossRef] [PubMed]
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010, 19, 3053–3067. [Google Scholar] [CrossRef]
- Bradford, J.; Shin, J.Y.; Roberts, M.; Wang, C.E.; Li, X.J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. USA 2009, 106, 22480–22485. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Myers, R.H.; Vonsattel, J.P.; Paskevich, P.A.; Kiely, D.K.; Stevens, T.J.; Cupples, L.A.; Richardson, E.P., Jr.; Bird, E.D. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J. Neuropathol. Exp. Neurol. 1991, 50, 729–742. [Google Scholar] [CrossRef]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef]
- Estrada-Sanchez, A.M.; Rebec, G.V. Role of cerebral cortex in the neuropathology of Huntington’s disease. Front. Neural. Circuits 2013, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Rosas, H.D.; Salat, D.H.; Lee, S.Y.; Zaleta, A.K.; Pappu, V.; Fischl, B.; Greve, D.; Hevelone, N.; Hersch, S.M. Cerebral cortex and the clinical expression of Huntington’s disease: Complexity and heterogeneity. Brain 2008, 131, 1057–1068. [Google Scholar] [CrossRef]
- Rosas, H.D.; Hevelone, N.D.; Zaleta, A.K.; Greve, D.N.; Salat, D.H.; Fischl, B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 2005, 65, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Thu, D.C.; Oorschot, D.E.; Tippett, L.J.; Nana, A.L.; Hogg, V.M.; Synek, B.J.; Luthi-Carter, R.; Waldvogel, H.J.; Faull, R.L. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain 2010, 133, 1094–1110. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Thu, D.C.; Tippett, L.J.; Oorschot, D.E.; Hogg, V.M.; Roxburgh, R.; Synek, B.J.; Waldvogel, H.J.; Faull, R.L. Cortical interneuron loss and symptom heterogeneity in Huntington disease. Ann. Neurol. 2014, 75, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Nana, A.L.; Kim, E.H.; Thu, D.C.; Oorschot, D.E.; Tippett, L.J.; Hogg, V.M.; Synek, B.J.; Roxburgh, R.; Waldvogel, H.J.; Faull, R.L. Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington’s disease. J. Huntingt. Dis. 2014, 3, 45–64. [Google Scholar] [CrossRef]
- Mehrabi, N.F.; Waldvogel, H.J.; Tippett, L.J.; Hogg, V.M.; Synek, B.J.; Faull, R.L. Symptom heterogeneity in Huntington’s disease correlates with neuronal degeneration in the cerebral cortex. Neurobiol. Dis. 2016, 96, 67–74. [Google Scholar] [CrossRef]
- Singh-Bains, M.K.; Mehrabi, N.F.; Sehji, T.; Austria, M.D.R.; Tan, A.Y.S.; Tippett, L.J.; Dragunow, M.; Waldvogel, H.J.; Faull, R.L.M. Cerebellar degeneration correlates with motor symptoms in Huntington disease. Ann. Neurol. 2019, 85, 396–405. [Google Scholar] [CrossRef]
- Singh-Bains, M.K.; Tippett, L.J.; Hogg, V.M.; Synek, B.J.; Roxburgh, R.H.; Waldvogel, H.J.; Faull, R.L. Globus pallidus degeneration and clinicopathological features of Huntington disease. Ann. Neurol. 2016, 80, 185–201. [Google Scholar] [CrossRef]
- Albin, R.L.; Young, A.B.; Penney, J.B.; Handelin, B.; Balfour, R.; Anderson, K.D.; Markel, D.S.; Tourtellotte, W.W.; Reiner, A. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington’s disease. N. Engl. J. Med. 1990, 322, 1293–1298. [Google Scholar] [CrossRef]
- Ahveninen, L.M.; Stout, J.C.; Georgiou-Karistianis, N.; Lorenzetti, V.; Glikmann-Johnston, Y. Reduced amygdala volumes are related to motor and cognitive signs in Huntington’s disease: The IMAGE-HD study. NeuroImage Clin. 2018, 18, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Dogan, I.; Eickhoff, S.B.; Schulz, J.B.; Shah, N.J.; Laird, A.R.; Fox, P.T.; Reetz, K. Consistent Neurodegeneration and Its Association with Clinical Progression in Huntington’s Disease: A Coordinate-Based Meta-Analysis. Neurodegener. Dis. 2013, 12, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.L.; Zhang, J.; Begeti, F.; Guzman, N.V.; Lazar, A.S.; Rowe, J.B.; Barker, R.A.; Hampshire, A. The role of the amygdala during emotional processing in Huntington’s disease: From pre-manifest to late stage disease. Neuropsychologia 2015, 70, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Van den Stock, J.; De Winter, F.L.; Ahmad, R.; Sunaert, S.; Van Laere, K.; Vandenberghe, W.; Vandenbulcke, M. Functional brain changes underlying irritability in premanifest Huntington’s disease. Hum. Brain Mapp. 2015, 36, 2681–2690. [Google Scholar] [CrossRef]
- Spargo, E.; Everall, I.P.; Lantos, P.L. Neuronal loss in the hippocampus in Huntington’s disease: A comparison with HIV infection. J. Neurol. Neurosurg. Psychiatry 1993, 56, 487–491. [Google Scholar] [CrossRef]
- Lange, H.; Thorner, G.; Hopf, A.; Schroder, K.F. Morphometric studies of the neuropathological changes in choreatic diseases. J. Neurol. Sci. 1976, 28, 401–425. [Google Scholar] [CrossRef]
- Wakai, M.; Takahashi, A.; Hashizume, Y. A histometrical study on the globus pallidus in Huntington’s disease. J. Neurol. Sci. 1993, 119, 18–27. [Google Scholar] [CrossRef]
- Faure, A.; Höhn, S.; Von Hörsten, S.; Delatour, B.; Raber, K.; Le Blanc, P.; Desvignes, N.; Doyère, V.; El Massioui, N. Altered emotional and motivational processing in the transgenic rat model for Huntington’s disease. Neurobiol. Learn. Mem. 2011, 95, 92–101. [Google Scholar] [CrossRef]
- van den Bogaard, S.J.A.; Dumas, E.M.; Ferrarini, L.; Milles, J.; van Buchem, M.A.; van der Grond, J.; Roos, R.A.C. Shape analysis of subcortical nuclei in Huntington’s disease, global versus local atrophy—Results from the TRACK-HD study. J. Neuro. Sci. 2011, 307, 60–68. [Google Scholar] [CrossRef][Green Version]
- Chen, M.; Wolynes, P.G. Aggregation landscapes of Huntingtin exon 1 protein fragments and the critical repeat length for the onset of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 4406–4411. [Google Scholar]
- McGowan, D.P.; van Roon-Mom, W.; Holloway, H.; Bates, G.P.; Mangiarini, L.; Cooper, G.J.; Faull, R.L.; Snell, R.G. Amyloid-like inclusions in Huntington’s disease. Neuroscience 2000, 100, 677–680. [Google Scholar] [CrossRef]
- Cooper, J.K.; Schilling, G.; Peters, M.F.; Herring, W.J.; Sharp, A.H.; Kaminsky, Z.; Masone, J.; Khan, F.A.; Delanoy, M.; Borchelt, D.R. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet. 1998, 7, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J. Clin. Investig. 2015, 125, 1979–1986. [Google Scholar] [CrossRef]
- Byrne, L.M.; Wild, E.J. Cerebrospinal Fluid Biomarkers for Huntington’s Disease. J. Huntingt. Dis. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Valor, L.M. Transcription, epigenetics and ameliorative strategies in Huntington’s Disease: A genome-wide perspective. Mol. Neurobiol. 2015, 51, 406–423. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar]
- Nagai, Y.; Inui, T.; Popiel, H.A.; Fujikake, N.; Hasegawa, K.; Urade, Y.; Goto, Y.; Naiki, H.; Toda, T. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 2007, 14, 332–340. [Google Scholar] [CrossRef]
- Miller, J.; Arrasate, M.; Brooks, E.; Libeu, C.P.; Legleiter, J.; Hatters, D.; Curtis, J.; Cheung, K.; Krishnan, P.; Mitra, S.; et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 2011, 7, 925–934. [Google Scholar] [CrossRef]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Menalled, L.B.; Sison, J.D.; Dragatsis, I.; Zeitlin, S.; Chesselet, M.F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 2003, 465, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef]
- Slow, E.J.; Graham, R.K.; Osmand, A.P.; Devon, R.S.; Lu, G.; Deng, Y.; Pearson, J.; Vaid, K.; Bissada, N.; Wetzel, R.; et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl. Acad. Sci. USA 2005, 102, 11402–11407. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.A.; Leavitt, B.R.; LePiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; Auerbach, W.; White, J.K.; Srinidhi, J.; Auerbach, A.; Ryan, A.; Duyao, M.P.; Vrbanac, V.; Weaver, M.; Gusella, J.F.; et al. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum. Mol. Genet. 1999, 8, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; White, J.K.; Gutekunst, C.A.; Vrbanac, V.; Weaver, M.; Li, X.J.; Li, S.H.; Yi, H.; Vonsattel, J.P.; Gusella, J.F.; et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 2000, 9, 503–513. [Google Scholar] [CrossRef]
- White, J.K.; Auerbach, W.; Duyao, M.P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; MacDonald, M.E. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat. Genet. 1997, 17, 404–410. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Cox, D.C.; Cooper, T.A. Non-canonical RAN Translation of CGG Repeats Has Canonical Requirements. Mol. Cell. 2016, 62, 155–156. [Google Scholar]
- Banez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef]
- Yang, S.; Yang, H.; Huang, L.; Chen, L.; Qin, Z.; Li, S.; Li, X.J. Lack of RAN-mediated toxicity in Huntington’s disease knock-in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4411–4417. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Langbehn, D.R.; Leavitt, B.R.; Roos, R.A.; Durr, A.; Craufurd, D.; Kennard, C.; Hicks, S.L.; Fox, N.C.; Scahill, R.I. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: Cross-sectional analysis of baseline data. Lancet Neurol. 2009, 8, 791–801. [Google Scholar] [CrossRef]
- Kinnunen, K.M.; Schwarz, A.J.; Turner, E.C.; Pustina, D.; Gantman, E.C.; Gordon, M.F.; Joules, R.; Mullin, A.P.; Scahill, R.I.; Georgiou-Karistianis, N.; et al. Volumetric MRI-Based Biomarkers in Huntington’s Disease: An Evidentiary Review. Front. Neurol. 2021, 12, 712555. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.M.; McColgan, P.; Robertson, N.; Tabrizi, S.J.; Zetterberg, H.; Wild, E.J. Cerebrospinal fluid inflammatory biomarkers reflect clinical severity in Huntington’s disease. PLoS ONE 2016, 11, e0163479. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, R.; Romer, M.; Oakes, D.; Rosengren, L.; Kieburtz, K. Levels of the light subunit of neurofilament triplet protein in cerebrospinal fluid in Huntington’s disease. Park. Relat. Disord. 2009, 15, 245–248. [Google Scholar] [CrossRef]
- Vinther-Jensen, T.; Börnsen, L.; Budtz-Jørgensen, E.; Ammitzbøll, C.; Larsen, I.U.; Hjermind, L.E.; Sellebjerg, F.; Nielsen, J.E. Selected CSF biomarkers indicate no evidence of early neuroinflammation in Huntington disease. Neurol. Neuroimmunol. Neuroinflammation. 2016, 3, e287. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: A retrospective cohort analysis. Lancet Neurol. 2017, 16, 601–609. [Google Scholar] [CrossRef]
- Southwell, A.L.; Smith, S.E.; Davis, T.R.; Caron, N.S.; Villanueva, E.B.; Xie, Y.; Collins, J.A.; Ye, M.L.; Sturrock, A.; Leavitt, B.R.; et al. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Sci. Rep. 2015, 5, 12166. [Google Scholar] [CrossRef]
- Silajdzic, E.; Bjorkqvist, M. A Critical Evaluation of Wet Biomarkers for Huntington’s Disease: Current Status and Ways forward. J. Huntingt. Dis. 2018, 7, 109–135. [Google Scholar] [CrossRef]
- Cybulska, K.; Perk, L.; Booij, J.; Laverman, P.; Rijpkema, M. Huntington’s Disease: A Review of the Known PET Imaging Biomarkers and Targeting Radiotracers. Molecules 2020, 25, 482. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Charles, P.; Seguin, F.; Barritault, J.; Coussieu, C.; Perin, L.; Le Bouc, Y.; Gervais, C.; Carcelain, G.; Vassault, A.; et al. Early energy deficit in Huntington disease: Identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007, 2, e647. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.R.; Broadhurst, D.; Dunn, W.B.; Ellis, D.I.; Michell, A.W.; Vacher, C.; Mosedale, D.E.; Kell, D.B.; Barker, R.A.; Grainger, D.J.; et al. Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 2006, 129, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Patassini, S.; Begley, P.; Reid, S.J.; Xu, J.; Church, S.J.; Curtis, M.; Dragunow, M.; Waldvogel, H.J.; Unwin, R.D.; Snell, R.G.; et al. Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington’s disease. Biochem. Biophys. Res. Commun. 2015, 468, 161–166. [Google Scholar] [CrossRef]
- Chiang, M.C.; Chen, H.M.; Lee, Y.H.; Chang, H.H.; Wu, Y.C.; Soong, B.W.; Chen, C.M.; Wu, Y.R.; Liu, C.S.; Niu, D.M.; et al. Dysregulation of C/EBPalpha by mutant Huntingtin causes the urea cycle deficiency in Huntington’s disease. Hum. Mol. Genet. 2007, 16, 483–498. [Google Scholar] [CrossRef]
- Handley, R.R.; Reid, S.J.; Brauning, R.; Maclean, P.; Mears, E.R.; Fourie, I.; Patassini, S.; Cooper, G.J.S.; Rudiger, S.R.; McLaughlan, C.J.; et al. Brain urea increase is an early Huntington’s disease pathogenic event observed in a prodromal transgenic sheep model and HD cases. Proc. Natl. Acad. Sci. USA 2017, 114, E11293–E11302. [Google Scholar] [CrossRef]
- Mears, E. Investigating the Molecular Mechanisms of Huntington’s Disease; The University of Auckland: Auckland, New Zealand, 2018. [Google Scholar]
- Mahoney, C.A.; Arieff, A.I. Uremic encephalopathies: Clinical, biochemical, and experimental features. Am. J. Kidney Dis. 1982, 2, 324–336. [Google Scholar]
- Gropman, A.L.; Summar, M.; Leonard, J.V. Neurological implications of urea cycle disorders. J. Inherit. Metab. Dis. 2007, 30, 865–879. [Google Scholar] [CrossRef]
- Leonard, J.V.; Morris, A.A. Urea cycle disorders. Semin. Neonatol. 2002, 7, 27–35. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. Lessons from animal models of Huntington’s disease. Trends Genet. 2002, 18, 202–209. [Google Scholar] [CrossRef]
- Pouladi, M.A.; Morton, A.J.; Hayden, M.R. Choosing an animal model for the study of Huntington’s disease. Nat. Rev. Neurosci. 2013, 14, 708–721. [Google Scholar] [CrossRef]
- Menalled, L.; Brunner, D. Animal models of Huntington’s disease for translation to the clinic: Best practices. Mov. Disord. 2014, 29, 1375–1390. [Google Scholar]
- Jacobsen, J.C.; Bawden, C.S.; Rudiger, S.R.; McLaughlan, C.J.; Reid, S.J.; Waldvogel, H.J.; MacDonald, M.E.; Gusella, J.F.; Walker, S.K.; Kelly, J.M.; et al. An ovine transgenic Huntington’s disease model. Hum. Mol. Genet. 2010, 19, 1873–1882. [Google Scholar] [CrossRef]
- Uchida, M.; Shimatsu, Y.; Onoe, K.; Matsuyama, N.; Niki, R.; Ikeda, J.E.; Imai, H. Production of transgenic miniature pigs by pronuclear microinjection. Transgenic Res. 2001, 10, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, C.E.; Zhao, B.; Li, W.; Ouyang, Z.; Liu, Z.; Yang, H.; Fan, P.; O’Neill, A.; Gu, W.; et al. Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum. Mol. Genet. 2010, 19, 3983–3994. [Google Scholar] [CrossRef]
- Baxa, M.; Hruska-Plochan, M.; Juhas, S.; Vodicka, P.; Pavlok, A.; Juhasova, J.; Miyanohara, A.; Nejime, T.; Klima, J.; Macakova, M.; et al. A transgenic minipig model of Huntington’s Disease. J. Huntingt. Dis. 2013, 2, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002.e13. [Google Scholar] [CrossRef]
- Palfi, S.; Brouillet, E.; Jarraya, B.; Bloch, J.; Jan, C.; Shin, M.; Conde, F.; Li, X.J.; Aebischer, P.; Hantraye, P.; et al. Expression of mutated huntingtin fragment in the putamen is sufficient to produce abnormal movement in non-human primates. Mol. Ther. 2007, 15, 1444–1451. [Google Scholar] [CrossRef]
- Yang, S.H.; Cheng, P.H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.J.; Cheng, E.C.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, J.; et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef]
- Chan, A.W.; Jiang, J.; Chen, Y.; Li, C.; Prucha, M.S.; Hu, Y.; Chi, T.; Moran, S.; Rahim, T.; Li, S.; et al. Progressive cognitive deficit, motor impairment and striatal pathology in a transgenic Huntington disease monkey model from infancy to adulthood. PLoS ONE 2015, 10, e0122335. [Google Scholar] [CrossRef]
- Albin, R.L.; Reiner, A.; Anderson, K.D.; Penney, J.B.; Young, A.B. Striatal and nigral neuron subpopulations in rigid Huntington’s disease: Implications for the functional anatomy of chorea and rigidity-akinesia. Ann. Neurol. 1990, 27, 357–365. [Google Scholar] [CrossRef]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991, 11, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 1986, 321, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Bordelon, Y.M.; Chesselet, M.-F.; Nelson, D.; Welsh, F.; Erecińska, M. Energetic Dysfunction in Quinolinic Acid-Lesioned Rat Striatum. J. Neurochem. 1997, 69, 1629–1639. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Randall, T.S.; Cahill, D.W.; Sanberg, P.R. Asymmetrical motor behavior in rats with unilateral striatal excitotoxic lesions as revealed by the elevated body swing test. Brain Res. 1995, 676, 231–234. [Google Scholar] [CrossRef]
- Emerich, D.F.; Thanos, C.G.; Goddard, M.; Skinner, S.J.; Geany, M.S.; Bell, W.J.; Bintz, B.; Schneider, P.; Chu, Y.; Babu, R.S.; et al. Extensive neuroprotection by choroid plexus transplants in excitotoxin lesioned monkeys. Neurobiol. Dis. 2006, 23, 471–480. [Google Scholar] [CrossRef]
- Foster, A.C.; Miller, L.P.; Oldendorf, W.H.; Schwarcz, R. Studies on the disposition of quinolinic acid after intracerebral or systemic administration in the rat. Exp. Neurol. 1984, 84, 428–440. [Google Scholar] [CrossRef]
- Kendall, A.L.; David, F.; Rayment, G.; Torres, E.M.; Annett, L.E.; Dunnett, S.B. The influence of excitotoxic basal ganglia lesions on motor performance in the common marmoset. Brain 2000, 123 Pt 7, 1442–1458. [Google Scholar] [CrossRef]
- McLin, J.P.; Thompson, L.M.; Steward, O. Differential susceptibility to striatal neurodegeneration induced by quinolinic acid and kainate in inbred, outbred and hybrid mouse strains. Eur. J. Neurosci. 2006, 24, 3134–3140. [Google Scholar] [CrossRef]
- Vazey, E.M.; Chen, K.; Hughes, S.M.; Connor, B. Transplanted adult neural progenitor cells survive, differentiate and reduce motor function impairment in a rodent model of Huntington’s disease. Exp. Neurol. 2006, 199, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Alston, T.A.; Mela, L.; Bright, H.J. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. USA 1977, 74, 3767–3771. [Google Scholar] [CrossRef] [PubMed]
- Blum, D.; Gall, D.; Cuvelier, L.; Schiffmann, S.N. Topological analysis of striatal lesions induced by 3-nitropropionic acid in the Lewis rat. Neuroreport 2001, 12, 1769–1772. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Koutouzis, T.K.; Freeman, T.B.; Hauser, R.A.; Cahill, D.W.; Sanberg, P.R. Hyperactivity and hypoactivity in a rat model of Huntington’s disease: The systemic 3-nitropropionic acid model. Brain Res. Brain Res. Protoc. 1997, 1, 253–257. [Google Scholar] [CrossRef]
- Coles, C.J.; Edmondson, D.E.; Singer, T.P. Inactivation of succinate dehydrogenase by 3-nitropropionate. J. Biol. Chem. 1979, 254, 5161–5167. [Google Scholar] [CrossRef]
- Ludolph, A.C.; He, F.; Spencer, P.S.; Hammerstad, J.; Sabri, M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can. J. Neurol. Sci. 1991, 18, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Ouary, S.; Bizat, N.; Altairac, S.; Menetrat, H.; Mittoux, V.; Conde, F.; Hantraye, P.; Brouillet, E. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: Implications for neuroprotection studies. Neuroscience 2000, 97, 521–530. [Google Scholar] [CrossRef]
- Palfi, S.; Ferrante, R.J.; Brouillet, E.; Beal, M.F.; Dolan, R.; Guyot, M.C.; Peschanski, M.; Hantraye, P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J. Neurosci. 1996, 16, 3019–3025. [Google Scholar] [CrossRef]
- Palfi, S.; Leventhal, L.; Goetz, C.G.; Hantraye, T.; Roitberg, B.Z.; Sramek, J.; Emborg, M.; Kordower, J.H. Delayed onset of progressive dystonia following subacute 3-nitropropionic acid treatment in Cebus apella monkeys. Mov. Disord. 2000, 15, 524–530. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Chen, J.; Ley, J.J.; Becker, D.A.; Beal, M.F. A novel azulenyl nitrone antioxidant protects against MPTP and 3-nitropropionic acid neurotoxicities. Exp. Neurol. 2005, 191, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Lione, L.A.; Carter, R.J.; Hunt, M.J.; Bates, G.P.; Morton, A.J.; Dunnett, S.B. Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. J. Neurosci. 1999, 19, 10428–10437. [Google Scholar] [CrossRef]
- Luesse, H.G.; Schiefer, J.; Spruenken, A.; Puls, C.; Block, F.; Kosinski, C.M. Evaluation of R6/2 HD transgenic mice for therapeutic studies in Huntington’s disease: Behavioral testing and impact of diabetes mellitus. Behav. Brain Res. 2001, 126, 185–195. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Kubilus, J.K.; Smith, K.; Cormier, K.; Del Signore, S.J.; Guelin, E.; Ryu, H.; Hersch, S.M.; Ferrante, R.J. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington’s disease transgenic mice. J. Comp. Neurol. 2005, 490, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Del Mar, N.; Meade, C.; Goldowitz, D.; Reiner, A. Differential changes in striatal projection neurons in R6/2 transgenic mice for Huntington’s disease. Neurobiol. Dis. 2002, 11, 369–385. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.L.; Ramaswamy, S.; Gasmi, M.; Bartus, R.T.; Herzog, C.D.; Brandon, E.P.; Zhou, L.; Pitzer, M.R.; Berry-Kravis, E.M.; Kordower, J.H. Viral delivery of glial cell line-derived neurotrophic factor improves behavior and protects striatal neurons in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 9345–9350. [Google Scholar] [CrossRef]
- Schilling, G.; Becher, M.W.; Sharp, A.H.; Jinnah, H.A.; Duan, K.; Kotzuk, J.A.; Slunt, H.H.; Ratovitski, T.; Cooper, J.K.; Jenkins, N.A.; et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999, 8, 397–407. [Google Scholar] [CrossRef]
- Yamamoto, A.; Lucas, J.J.; Hen, R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000, 101, 57–66. [Google Scholar] [CrossRef]
- Tebbenkamp, A.T.; Swing, D.; Tessarollo, L.; Borchelt, D.R. Premature death and neurologic abnormalities in transgenic mice expressing a mutant huntingtin exon-2 fragment. Hum. Mol. Genet. 2011, 20, 1633–1642. [Google Scholar] [CrossRef]
- Cheng, P.H.; Li, C.L.; Her, L.S.; Chang, Y.F.; Chan, A.W.; Chen, C.M.; Yang, S.H. Significantly differential diffusion of neuropathological aggregates in the brain of transgenic mice carrying N-terminal mutant huntingtin fused with green fluorescent protein. Brain Struct. Funct. 2013, 218, 283–294. [Google Scholar] [CrossRef]
- Kotliarova, S.; Jana, N.R.; Sakamoto, N.; Kurosawa, M.; Miyazaki, H.; Nekooki, M.; Doi, H.; Machida, Y.; Wong, H.K.; Suzuki, T.; et al. Decreased expression of hypothalamic neuropeptides in Huntington disease transgenic mice with expanded polyglutamine-EGFP fluorescent aggregates. J. Neurochem. 2005, 93, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; Andre, V.M.; Wilburn, B.; Lu, X.H.; Tao, J.; Yamazaki, I.; Li, S.H.; Sun, Y.E.; et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef] [PubMed]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.Z.; et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Slow, E.J.; Hossain, S.M.; Leavitt, B.R.; Hayden, M.R. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 2005, 25, 4169–4180. [Google Scholar] [CrossRef]
- Southwell, A.L.; Warby, S.C.; Carroll, J.B.; Doty, C.N.; Skotte, N.H.; Zhang, W.; Villanueva, E.B.; Kovalik, V.; Xie, Y.; Pouladi, M.A.; et al. A fully humanized transgenic mouse model of Huntington disease. Hum. Mol. Genet. 2013, 22, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Igarashi, S.; Nakamura, M.; Gafni, J.; Torcassi, C.; Schilling, G.; Crippen, D.; Wood, J.D.; Sawa, A.; Jenkins, N.A.; et al. Progressive phenotype and nuclear accumulation of an amino-terminal cleavage fragment in a transgenic mouse model with inducible expression of full-length mutant huntingtin. Neurobiol. Dis. 2006, 21, 381–391. [Google Scholar] [CrossRef]
- Yu-Taeger, L.; Petrasch-Parwez, E.; Osmand, A.P.; Redensek, A.; Metzger, S.; Clemens, L.E.; Park, L.; Howland, D.; Calaminus, C.; Gu, X.; et al. A novel BACHD transgenic rat exhibits characteristic neuropathological features of Huntington disease. J. Neurosci. 2012, 32, 15426–15438. [Google Scholar] [CrossRef]
- Manfre, G.; Clemensson, E.K.H.; Kyriakou, E.I.; Clemensson, L.E.; van der Harst, J.E.; Homberg, J.R.; Nguyen, H.P. The BACHD Rat Model of Huntington Disease Shows Specific Deficits in a Test Battery of Motor Function. Front. Behav. Neurosci. 2017, 11, 218. [Google Scholar] [CrossRef]
- Bode, F.J.; Stephan, M.; Suhling, H.; Pabst, R.; Straub, R.H.; Raber, K.A.; Bonin, M.; Nguyen, H.P.; Riess, O.; Bauer, A.; et al. Sex differences in a transgenic rat model of Huntington’s disease: Decreased 17beta-estradiol levels correlate with reduced numbers of DARPP32+ neurons in males. Hum. Mol. Genet. 2008, 17, 2595–2609. [Google Scholar] [CrossRef]
- von Hörsten, S.; Schmitt, I.; Nguyen, H.P.; Holzmann, C.; Schmidt, T.; Walther, T.; Bader, M.; Pabst, R.; Kobbe, P.; Krotova, J.; et al. Transgenic rat model of Huntington’s disease. Hum. Mol. Genet. 2003, 12, 617–624. [Google Scholar] [CrossRef]
- Kantor, O.; Temel, Y.; Holzmann, C.; Raber, K.; Nguyen, H.P.; Cao, C.; Turkoglu, H.O.; Rutten, B.P.; Visser-Vandewalle, V.; Steinbusch, H.W.; et al. Selective striatal neuron loss and alterations in behavior correlate with impaired striatal function in Huntington’s disease transgenic rats. Neurobiol. Dis. 2006, 22, 538–547. [Google Scholar] [CrossRef]
- Lin, C.-H.; Tallaksen-Greene, S.; Chien, W.-M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.-J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef]
- Brooks, S.; Higgs, G.; Jones, L.; Dunnett, S.B. Longitudinal analysis of the behavioural phenotype in HdhQ92 Huntington’s disease knock-in mice. Brain Res. Bull. 2012, 88, 148–155. [Google Scholar] [CrossRef]
- Yhnell, E.; Dunnett, S.B.; Brooks, S.P. A Longitudinal Motor Characterisation of the HdhQ111 Mouse Model of Huntington’s Disease. J. Huntingt. Dis. 2016, 5, 149–161. [Google Scholar] [CrossRef]
- Menalled, L.B.; Kudwa, A.E.; Miller, S.; Fitzpatrick, J.; Watson-Johnson, J.; Keating, N.; Ruiz, M.; Mushlin, R.; Alosio, W.; McConnell, K.; et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS ONE 2012, 7, e49838. [Google Scholar] [CrossRef]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef]
- Handley, R.R.; Reid, S.J.; Patassini, S.; Rudiger, S.R.; Obolonkin, V.; McLaughlan, C.J.; Jacobsen, J.C.; Gusella, J.F.; MacDonald, M.E.; Waldvogel, H.J.; et al. Metabolic disruption identified in the Huntington’s disease transgenic sheep model. Sci. Rep. 2016, 6, 20681. [Google Scholar] [CrossRef]
- Huntington’s Disease Sheep Collaborative Research, G.; Reid, S.J.; Patassini, S.; Handley, R.R.; Rudiger, S.R.; McLaughlan, C.J.; Osmand, A.; Jacobsen, J.C.; Morton, A.J.; Weiss, A.; et al. Further molecular characterisation of the OVT73 transgenic sheep model of Huntington’s disease identifies cortical aggregates. J. Huntingt. Dis. 2013, 2, 279–295. [Google Scholar] [CrossRef]
- Vidinska, D.; Vochozkova, P.; Smatlikova, P.; Ardan, T.; Klima, J.; Juhas, S.; Juhasova, J.; Bohuslavova, B.; Baxa, M.; Valekova, I.; et al. Gradual Phenotype Development in Huntington Disease Transgenic Minipig Model at 24 Months of Age. Neurodegener. Dis. 2018, 18, 107–119. [Google Scholar] [CrossRef]
- Morton, A.J. and Howland, D. S. Large genetic animal models of Huntington’s Disease. J. Huntingt. Dis. 2013, 2, 3–19. [Google Scholar] [CrossRef]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Salinska, E.; Danysz, W.; Lazarewicz, J.W. The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 2005, 43, 322–339. [Google Scholar] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef]
- Sepers, M.D.; Raymond, L.A. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov. Today 2014, 19, 990–996. [Google Scholar]
- Coyle, J.T.; Schwarcz, R. Lesion of striatal neurones with kainic acid provides a model for Huntington’s chorea. Nature 1976, 263, 244–246. [Google Scholar]
- McGeer, E.G.; McGeer, P.L. Duplication of biochemical changes of Huntington’s chorea by intrastriatal injections of glutamic and kainic acids. Nature 1976, 263, 517–519. [Google Scholar]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R.; et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Wu, N.P.; Andre, V.M.; Cummings, D.M.; Cepeda, C.; Joyce, J.A.; Carroll, J.B.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S.; et al. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J. Neurosci. 2009, 29, 2414–2427. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.Y.; Detloff, P.J.; Wang, P.L.; Tsien, J.Z.; Albin, R.L. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J. Neurosci. 2009, 29, 3200–3205. [Google Scholar] [CrossRef] [PubMed]
- Rigby, M.; Le Bourdelles, B.; Heavens, R.P.; Kelly, S.; Smith, D.; Butler, A.; Hammans, R.; Hills, R.; Xuereb, J.H.; Hill, R.G.; et al. The messenger RNAs for the N-methyl-D-aspartate receptor subunits show region-specific expression of different subunit composition in the human brain. Neuroscience 1996, 73, 429–447. [Google Scholar] [CrossRef] [PubMed]
- van Marum, R.J. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 237–247. [Google Scholar] [CrossRef]
- Lee, S.-T.; Chu, K.; Park, J.-E.; Kang, L.; Ko, S.-Y.; Jung, K.-H.; Kim, M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006, 1118, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ondo, W.G.; Mejia, N.I.; Hunter, C.B. A pilot study of the clinical efficacy and safety of memantine for Huntington’s disease. Park. Relat. Disord. 2007, 13, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; McDermott, M.P.; Voss, T.S.; Corey-Bloom, J.; Deuel, L.M.; Dorsey, E.R.; Factor, S.; Geschwind, M.D.; Hodgeman, K.; Kayson, E.; et al. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch Neurol. 2010, 67, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Crosby, N.J.; Deane, K.H.; Clarke, C.E. Amantadine for dyskinesia in Parkinson’s disease. Cochrane Database Syst. Rev. 2003, 2003, CD003467. [Google Scholar] [CrossRef]
- Luginger, E.; Wenning, G.K.; Bosch, S.; Poewe, W. Beneficial effects of amantadine on L-dopa-induced dyskinesias in Parkinson’s disease. Mov. Disord. 2000, 15, 873–878. [Google Scholar] [CrossRef]
- Verhagen Metman, L.; Morris, M.J.; Farmer, C.; Gillespie, M.; Mosby, K.; Wuu, J.; Chase, T.N. Huntington’s disease: A randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002, 59, 694–699. [Google Scholar] [CrossRef]
- Lucetti, C.; Del Dotto, P.; Gambaccini, G.; Dell’ Agnello, G.; Bernardini, S.; Rossi, G.; Murri, L.; Bonuccelli, U. IV amantadine improves chorea in Huntington’s disease: An acute randomized, controlled study. Neurology 2003, 60, 1995–1997. [Google Scholar] [CrossRef]
- O’Suilleabhain, P.; Dewey, J.; Richard, B. A Randomized Trial of Amantadine in Huntington Disease. Arch. Neurol. 2003, 60, 996–998. [Google Scholar]
- Landwehrmeyer, G.B.; Dubois, B.; de Yebenes, J.G.; Kremer, B.; Gaus, W.; Kraus, P.H.; Przuntek, H.; Dib, M.; Doble, A.; Fischer, W.; et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann. Neurol. 2007, 62, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Ferrante, R.J.; Gardian, G.; Browne, S.; Chabrier, P.E.; Beal, M.F. Increased survival and neuroprotective effects of BN82451 in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2003, 86, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Klapstein, G.J.; Koppel, A.; Gruen, E.; Cepeda, C.; Vargas, M.E.; Jokel, E.S.; Carpenter, E.M.; Zanjani, H.; Hurst, R.S.; et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J. Neurosci. Res. 1999, 58, 515–532. [Google Scholar] [CrossRef]
- Cepeda, C.; Ariano, M.A.; Calvert, C.R.; Flores-Hernandez, J.; Chandler, S.H.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S. NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 2001, 66, 525–539. [Google Scholar] [CrossRef]
- Starling, A.J.; Andre, V.M.; Cepeda, C.; de Lima, M.; Chandler, S.H.; Levine, M.S. Alterations in N-methyl-D-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington’s disease. J. Neurosci. Res. 2005, 82, 377–386. [Google Scholar] [CrossRef]
- Cepeda, C.; Murphy, K.P.; Parent, M.; Levine, M.S. The role of dopamine in Huntington’s disease. Prog. Brain Res. 2014, 211, 235–254. [Google Scholar]
- Garrett, M.C.; Soares-da-Silva, P. Increased cerebrospinal fluid dopamine and 3,4-dihydroxyphenylacetic acid levels in Huntington’s disease: Evidence for an overactive dopaminergic brain transmission. J. Neurochem. 1992, 58, 101–106. [Google Scholar]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 1987, 22, 386–389. [Google Scholar] [CrossRef]
- Richfield, E.K.; O’Brien, C.F.; Eskin, T.; Shoulson, I. Heterogeneous dopamine receptor changes in early and late Huntington’s disease. Neurosci. Lett. 1991, 132, 121–126. [Google Scholar] [CrossRef]
- van Oostrom, J.C.; Dekker, M.; Willemsen, A.T.; de Jong, B.M.; Roos, R.A.; Leenders, K.L. Changes in striatal dopamine D2 receptor binding in pre-clinical Huntington’s disease. Eur. J. Neurol. 2009, 16, 226–231. [Google Scholar] [CrossRef]
- Ginovart, N.; Lundin, A.; Farde, L.; Halldin, C.; Backman, L.; Swahn, C.G.; Pauli, S.; Sedvall, G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain 1997, 120 Pt 3, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Koeppe, R.A.; Meyer, P.; Ficaro, E.; Wernette, K.; Kilbourn, M.R.; Kuhl, D.E.; Frey, K.A.; Albin, R.L. Decreased striatal monoaminergic terminals in Huntington disease. Neurology 2000, 54, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.A.; Reynolds, G.P.; Morton, A.J. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington’s disease. J. Neurochem. 2002, 81, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Rajan, V.; Miller, C.E.; Wightman, R.M. Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 2006, 97, 737–746. [Google Scholar] [CrossRef]
- Callahan, J.W.; Abercrombie, E.D. In vivo Dopamine Efflux is Decreased in Striatum of both Fragment (R6/2) and Full-Length (YAC128) Transgenic Mouse Models of Huntington’s Disease. Front. Syst. Neurosci. 2011, 5, 61. [Google Scholar]
- Ortiz, A.N.; Kurth, B.J.; Osterhaus, G.L.; Johnson, M.A. Dysregulation of intracellular dopamine stores revealed in the R6/2 mouse striatum. J Neurochem. 2010, 112, 755–761. [Google Scholar] [CrossRef]
- Koch, E.T.; Raymond, L.A. Dysfunctional striatal dopamine signaling in Huntington’s disease. J. Neurosci. Res. 2019, 97, 1636–1654. [Google Scholar]
- Wang, H.; Chen, X.; Li, Y.; Tang, T.S.; Bezprozvanny, I. Tetrabenazine is neuroprotective in Huntington’s disease mice. Mol. Neurodegener. 2010, 5, 18. [Google Scholar] [CrossRef]
- Kegelmeyer, D.A.; Kloos, A.D.; Fritz, N.E.; Fiumedora, M.M.; White, S.E.; Kostyk, S.K. Impact of tetrabenazine on gait and functional mobility in individuals with Huntington’s disease. J. Neurol. Sci. 2014, 347, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.S.; Chen, X.; Liu, J.; Bezprozvanny, I. Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J. Neurosci. 2007, 27, 7899–7910. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Testa, C.M.; Stamler, D.; Kayson, E.; Davis, C.; Edmondson, M.C.; Kinel, S.; Leavitt, B.; Oakes, D.; O’neill, C. Effect of deutetrabenazine on chorea among patients with Huntington disease: A randomized clinical trial. Jama 2016, 316, 40–50. [Google Scholar] [PubMed]
- Reilmann, R. Deutetrabenazine-Not a Revolution but Welcome Evolution for Treating Chorea in Huntington Disease. JAMA Neurol. 2016, 73, 1404–1406. [Google Scholar] [CrossRef] [PubMed]
- Furr Stimming, E.; Claassen, D.O.; Kayson, E.; Goldstein, J.; Mehanna, R.; Zhang, H.; Liang, G.S.; Haubenberger, D.; Huntington Study Group, K.-H.D.C. Safety and efficacy of valbenazine for the treatment of chorea associated with Huntington’s disease (KINECT-HD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2023, 22, 494–504. [Google Scholar] [CrossRef]
- Unti, E.; Mazzucchi, S.; Palermo, G.; Bonuccelli, U.; Ceravolo, R. Antipsychotic drugs in Huntington’s disease. Expert Rev. Neurother. 2017, 17, 227–237. [Google Scholar] [CrossRef]
- Brown, C.S.; Markowitz, J.S.; Moore, T.R.; Parker, N.G. Atypical antipsychotics: Part II: Adverse effects, drug interactions, and costs. Ann. Pharmacother. 1999, 33, 210–217. [Google Scholar] [CrossRef]
- DeMarch, Z.; Giampa, C.; Patassini, S.; Bernardi, G.; Fusco, F.R. Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2008, 30, 375–387. [Google Scholar] [CrossRef]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef]
- Trushina, E.; Dyer, R.B.; Badger, J.D., 2nd; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef]
- Chang, D.T.; Rintoul, G.L.; Pandipati, S.; Reynolds, I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006, 22, 388–400. [Google Scholar] [CrossRef]
- Antonini, A.; Leenders, K.L.; Spiegel, R.; Meier, D.; Vontobel, P.; Weigell-Weber, M.; Sanchez-Pernaute, R.; de Yebenez, J.G.; Boesiger, P.; Weindl, A.; et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain 1996, 119 Pt 6, 2085–2095. [Google Scholar] [CrossRef]
- Mazziotta, J.C.; Phelps, M.E.; Pahl, J.J.; Huang, S.C.; Baxter, L.R.; Riege, W.H.; Hoffman, J.M.; Kuhl, D.E.; Lanto, A.B.; Wapenski, J.A.; et al. Reduced cerebral glucose metabolism in asymptomatic subjects at risk for Huntington’s disease. N. Engl. J. Med. 1987, 316, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.G.; Koroshetz, W.J.; Beal, M.F.; Rosen, B.R. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 1993, 43, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; N’Guyen, T.-M.; Deelchand, D.; Rinaldi, D.; Valabregue, R.; Wary, C.; Carlier, P.G.; Durr, A.; Henry, P.-G. Abnormal response to cortical activation in early stages of Huntington disease. Mov. Disord. 2012, 27, 907–910. [Google Scholar] [CrossRef]
- Mochel, F.; Durant, B.; Meng, X.; O’Callaghan, J.; Yu, H.; Brouillet, E.; Wheeler, V.C.; Humbert, S.; Schiffmann, R.; Durr, A. Early alterations of brain cellular energy homeostasis in Huntington disease models. J. Biol. Chem. 2012, 287, 1361–1370. [Google Scholar] [CrossRef]
- Seong, I.S.; Ivanova, E.; Lee, J.M.; Choo, Y.S.; Fossale, E.; Anderson, M.; Gusella, J.F.; Laramie, J.M.; Myers, R.H.; Lesort, M.; et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005, 14, 2871–2880. [Google Scholar] [CrossRef]
- Milakovic, T.; Quintanilla, R.A.; Johnson, G.V. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: Functional consequences. J. Biol. Chem. 2006, 281, 34785–34795. [Google Scholar] [CrossRef]
- Sawa, A.; Wiegand, G.W.; Cooper, J.; Margolis, R.L.; Sharp, A.H.; Lawler, J.F., Jr.; Greenamyre, J.T.; Snyder, S.H.; Ross, C.A. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat. Med. 1999, 5, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. In Polyglutamine Disorders; Nóbrega, C., Pereira de Almeida, L., Eds.; Springer International Publishing: Cham, Swizerland, 2018; pp. 59–83. [Google Scholar]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Rogers, D.A.; Lu, G.; Barakauskas, V.E.; Barr, A.M.; Honer, W.G.; Hayden, M.R.; Leavitt, B.R. Ethyl-EPA treatment improves motor dysfunction, but not neurodegeneration in the YAC128 mouse model of Huntington disease. Exp. Neurol. 2005, 196, 266–272. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Rosser, A.; Craufurd, D.; Squitieri, F.; Mallard, N.; Landwehrmeyer, B. Ethyl-eicosapentaenoic acid treatment in Huntington’s disease: A placebo-controlled clinical trial. Mov. Disord. 2015, 30, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study, G. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology 2001, 57, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Annoucement of 2CARE Early Study Closure. 2014. Available online: http://hdsa.org/wp-content/uploads/2015/01/Announcement-of-2CARE-Early-Study-Closure.pdf (accessed on 17 August 2023).
- Ferrante, R.J.; Andreassen, O.A.; Jenkins, B.G.; Dedeoglu, A.; Kuemmerle, S.; Kubilus, J.K.; Kaddurah-Daouk, R.; Hersch, S.M.; Beal, M.F. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J. Neurosci. 2000, 20, 4389–4397. [Google Scholar] [CrossRef]
- Hersch, S.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2′ dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Schifitto, G.; Oakes, D.; Bredlau, A.L.; Meyers, C.M.; Nahin, R.; Rosas, H.D.; Huntington Study Group, C.-E.I. Coordinators The CREST-E study of creatine for Huntington disease: A randomized controlled trial. Neurology 2017, 89, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1739–1753. [Google Scholar] [CrossRef]
- van Diemen, M.P.J.; Hart, E.P.; Abbruscato, A.; Mead, L.; van Beelen, I.; Bergheanu, S.C.; Hameeteman, P.W.; Coppen, E.; Winder, J.Y.; Moerland, M.; et al. Safety, pharmacokinetics and pharmacodynamics of SBT-020 in patients with early stage Huntington’s disease, a 2-part study. Br. J. Clin. Pharmacol. 2021, 87, 2290–2302. [Google Scholar] [CrossRef]
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495. [Google Scholar] [CrossRef]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A Focus on Several Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2015, 2015, 392169. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar]
- Kassubek, J.; Juengling, F.D.; Kioschies, T.; Henkel, K.; Karitzky, J.; Kramer, B.; Ecker, D.; Andrich, J.; Saft, C.; Kraus, P.; et al. Topography of cerebral atrophy in early Huntington’s disease: A voxel based morphometric MRI study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 213–220. [Google Scholar] [PubMed]
- Hobbs, N.Z.; Barnes, J.; Frost, C.; Henley, S.M.; Wild, E.J.; Macdonald, K.; Barker, R.A.; Scahill, R.I.; Fox, N.C.; Tabrizi, S.J. Onset and progression of pathologic atrophy in Huntington disease: A longitudinal MR imaging study. AJNR Am. J. Neuroradiol. 2010, 31, 1036–1041. [Google Scholar] [CrossRef]
- Pagano, G.; Niccolini, F.; Politis, M. Current status of PET imaging in Huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1171–1182. [Google Scholar] [CrossRef]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 2007, 72, 148–151. [Google Scholar] [CrossRef]
- Politis, M.; Pavese, N.; Tai, Y.F.; Tabrizi, S.J.; Barker, R.A.; Piccini, P. Hypothalamic involvement in Huntington’s disease: An in vivo PET study. Brain 2008, 131, 2860–2869. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Fujita, M.; Fujimura, Y.; Kimura, N.; Jenko, K.J.; Kannan, P.; Hong, J.; Morse, C.L.; Zoghbi, S.S.; Gladding, R.L.; et al. Comparison of [(11)C]-(R)-PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage 2010, 49, 2924–2932. [Google Scholar] [CrossRef]
- Collste, K.; Forsberg, A.; Varrone, A.; Amini, N.; Aeinehband, S.; Yakushev, I.; Halldin, C.; Farde, L.; Cervenka, S. Test-retest reproducibility of [(11)C]PBR28 binding to TSPO in healthy control subjects. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 173–183. [Google Scholar] [CrossRef]
- Datta, G.; Colasanti, A.; Kalk, N.; Owen, D.; Scott, G.; Rabiner, E.A.; Gunn, R.N.; Lingford-Hughes, A.; Malik, O.; Ciccarelli, O.; et al. (11)C-PBR28 and (18)F-PBR111 Detect White Matter Inflammatory Heterogeneity in Multiple Sclerosis. J. Nucl. Med. 2017, 58, 1477–1482. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.M.; Yang, S.; Huang, S.S.; Tang, B.S.; Guo, J.F. Microglial Activation in the Pathogenesis of Huntington’s Disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Kraft, A.D.; Kaltenbach, L.S.; Lo, D.C.; Harry, G.J. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol. Aging 2012, 33, 621.e17–621.e33. [Google Scholar] [CrossRef]
- Khoshnan, A.; Ko, J.; Watkin, E.E.; Paige, L.A.; Reinhart, P.H.; Patterson, P.H. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci. 2004, 24, 7999–8008. [Google Scholar] [CrossRef]
- Khoshnan, A.; Patterson, P.H. The role of IkappaB kinase complex in the neurobiology of Huntington’s disease. Neurobiol. Dis. 2011, 43, 305–311. [Google Scholar]
- Schwarcz, R.; Guidetti, P.; Sathyasaikumar, K.V.; Muchowski, P.J. Of mice, rats and men: Revisiting the quinolinic acid hypothesis of Huntington’s disease. Prog. Neurobiol. 2010, 90, 230–245. [Google Scholar] [CrossRef]
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–531. [Google Scholar] [CrossRef]
- Liddelow, S.A. and Barres, B. A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [PubMed]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Liddelow, S.A. Play It Again, SAM: Macrophages Control Peripheral Fat Metabolism. Trends Immunol. 2018, 39, 81–82. [Google Scholar] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Robel, S.; Berninger, B.; Gotz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104. [Google Scholar] [CrossRef]
- Alam, J.; Blackburn, K.; Patrick, D. Neflamapimod: Clinical Phase 2b-Ready Oral Small Molecule Inhibitor of p38alpha to Reverse Synaptic Dysfunction in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2017, 4, 273–278. [Google Scholar]
- Prins, N.D.; Harrison, J.E.; Chu, H.-M.; Blackburn, K.; Alam, J.J.; Scheltens, P.; Arnold; Coskinas; Gonzales; Joseph; et al. A phase 2 double-blind placebo-controlled 24-week treatment clinical study of the p38 alpha kinase inhibitor neflamapimod in mild Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 106. [Google Scholar]
- Feigin, A.; Evans, E.E.; Fisher, T.L.; Leonard, J.E.; Smith, E.S.; Reader, A.; Mishra, V.; Manber, R.; Walters, K.A.; Kowarski, L.; et al. Pepinemab antibody blockade of SEMA4D in early Huntington’s disease: A randomized, placebo-controlled, phase 2 trial. Nat. Med. 2022, 28, 2183–2193. [Google Scholar] [CrossRef]
- Fernandes, J. Treatment with VX15 may protect brain in early phase Huntington’s, data shows. Huntington’s Disease News. 2017. Available online: https://huntingtonsdiseasenews.com/news/vx15-protect-brain-early-phase-huntingtons/ (accessed on 17 August 2023).
- Ehrnhoefer, D.E.; Caron, N.S.; Deng, Y.; Qiu, X.; Tsang, M.; Hayden, M.R. Laquinimod decreases Bax expression and reduces caspase-6 activation in neurons. Exp. Neurol. 2016, 283, 121–128. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Hong, X.; Tan, L.J.; Caron, N.S.; Huang, Y.; To, X.V.; Lin, R.Y.; Franciosi, S.; Papapetropoulos, S.; Hayardeny, L.; et al. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the YAC128 model of Huntington disease. Sci. Rep. 2016, 6, 31652. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Yusof, N.; Tan, J.Y.; Radulescu, C.I.; Sidik, H.; Tan, L.J.; Belinson, H.; Zach, N.; Hayden, M.R.; Pouladi, M.A. Laquinimod Treatment Improves Myelination Deficits at the Transcriptional and Ultrastructural Levels in the YAC128 Mouse Model of Huntington Disease. Mol. Neurobiol. 2019, 56, 4464–4478. [Google Scholar] [CrossRef] [PubMed]
- Roussakis, A.A.; Gennaro, M.; Gordon, M.F.; Reilmann, R.; Borowsky, B.; Rynkowski, G.; Lao-Kaim, N.P.; Papoutsou, Z.; Savola, J.M.; Hayden, M.R.; et al. A PET-CT study on neuroinflammation in Huntington’s disease patients participating in a randomized trial with laquinimod. Brain Commun. 2023, 5, fcad084. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Heuberger, C.; Reisecker, F. Minocycline for Huntington’s disease: An open label study. Neurology 2003, 60, 883–884. [Google Scholar] [CrossRef]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef]
- Huntington Study Group, D.I. A futility study of minocycline in Huntington’s disease. Mov. Disord. 2010, 25, 2219–2224. [Google Scholar]
- Leitman, J.; Barak, B.; Benyair, R.; Shenkman, M.; Ashery, U.; Hartl, F.U.; Lederkremer, G.Z. ER Stress-Induced eIF2-alpha Phosphorylation Underlies Sensitivity of Striatal Neurons to Pathogenic Huntingtin. PLoS ONE 2014, 9, e90803. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Lindquist, S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008, 22, 3308–3319. [Google Scholar]
- Carnemolla, A.; Fossale, E.; Agostoni, E.; Michelazzi, S.; Calligaris, R.; De Maso, L.; Del Sal, G.; MacDonald, M.E.; Persichetti, F. Rrs1 Is Involved in Endoplasmic Reticulum Stress Response in Huntington Disease. J. Biol. Chem. 2009, 284, 18167–18173. [Google Scholar] [CrossRef]
- Cho, K.J.; Lee, B.I.; Cheon, S.Y.; Kim, H.W.; Kim, H.J.; Kim, G.W. Inhibition of apoptosis signal-regulating kinase 1 reduces endoplasmic reticulum stress and nuclear huntingtin fragments in a mouse model of Huntington disease. Neuroscience 2009, 163, 1128–1134. [Google Scholar] [CrossRef]
- Noh, J.-Y.; Lee, H.; Song, S.; Kim, N.S.; Im, W.; Kim, M.; Seo, H.; Chung, C.-W.; Chang, J.-W.; Ferrante, R.J.; et al. SCAMP5 Links Endoplasmic Reticulum Stress to the Accumulation of Expanded Polyglutamine Protein Aggregates via Endocytosis Inhibition. J. Biol. Chem. 2009, 284, 11318–11325. [Google Scholar] [CrossRef]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef]
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington’s disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Hervas, D.; Fornes-Ferrer, V.; Gomez-Escribano, A.P.; Sequedo, M.D.; Peiro, C.; Millan, J.M.; Vazquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef]
- Su, T.-P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.A.; Cavendish, J.Z.; Robson, M.J.; Scandinaro, A.L.; Matsumoto, R.R. Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 2015, 127, 17–29. [Google Scholar] [CrossRef]
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J Cell Mol Med. 2015, 19, 2540–2548. [Google Scholar] [CrossRef]
- de Yebenes, J.G.; Landwehrmeyer, B.; Squitieri, F.; Reilmann, R.; Rosser, A.; Barker, R.A.; Saft, C.; Magnet, M.K.; Sword, A.; Rembratt, A.; et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011, 10, 1049–1057. [Google Scholar] [CrossRef]
- Huntington Study Group, H.I. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov. Disord. 2013, 28, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Lundin, A.; Dietrichs, E.; Haghighi, S.; Goller, M.L.; Heiberg, A.; Loutfi, G.; Widner, H.; Wiktorin, K.; Wiklund, L.; Svenningsson, A.; et al. Efficacy and safety of the dopaminergic stabilizer Pridopidine (ACR16) in patients with Huntington’s disease. Clin. Neuropharmacol. 2010, 33, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Reilmann, R.; McGarry, A.; Grachev, I.D.; Savola, J.-M.; Borowsky, B.; Eyal, E.; Gross, N.; Langbehn, D.; Schubert, R.; Wickenberg, A.T.; et al. Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE-HD): A phase 2, randomised, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. 2019, 18, 165–176. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751. [Google Scholar] [CrossRef]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [PubMed]
- Dedeoglu, A.; Kubilus, J.K.; Jeitner, T.M.; Matson, S.A.; Bogdanov, M.; Kowall, N.W.; Matson, W.R.; Cooper, A.J.; Ratan, R.R.; Beal, M.F.; et al. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J. Neurosci. 2002, 22, 8942–8950. [Google Scholar] [CrossRef]
- Verny, C.; Bachoud-Levi, A.C.; Durr, A.; Goizet, C.; Azulay, J.P.; Simonin, C.; Tranchant, C.; Calvas, F.; Krystkowiak, P.; Charles, P.; et al. A randomized, double-blind, placebo-controlled trial evaluating cysteamine in Huntington’s disease. Mov. Disord. 2017, 32, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Ayton, S.; Finkelstein, D.I.; Bush, A.I.; McColl, G.; Massa, S.M. PBT2 Reduces Toxicity in a C. elegans Model of polyQ Aggregation and Extends Lifespan, Reduces Striatal Atrophy and Improves Motor Performance in the R6/2 Mouse Model of Huntington’s Disease. J. Huntingt. Dis. 2012, 1, 211–219. [Google Scholar]
- Huntington Study Group Reach, H.D.I. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 39–47. [Google Scholar]
- Wojtecki, L.; Groiss, S.J.; Hartmann, C.J.; Elben, S.; Omlor, S.; Schnitzler, A.; Vesper, J. Deep Brain Stimulation in Huntington’s Disease-Preliminary Evidence on Pathophysiology, Efficacy and Safety. Brain Sci. 2016, 6, 38. [Google Scholar] [CrossRef]
- Wojtecki, L.; Groiss, S.J.; Ferrea, S.; Elben, S.; Hartmann, C.J.; Dunnett, S.B.; Rosser, A.; Saft, C.; Sudmeyer, M.; Ohmann, C.; et al. A Prospective Pilot Trial for Pallidal Deep Brain Stimulation in Huntington’s Disease. Front. Neurol. 2015, 6, 177. [Google Scholar] [CrossRef]
- Fasano, A.; Llinas, M.; Munhoz, R.P.; Hlasny, E.; Kucharczyk, W.; Lozano, A.M. MRI-guided focused ultrasound thalamotomy in non-ET tremor syndromes. Neurology 2017, 89, 771–775. [Google Scholar] [CrossRef]
- Kapadia, A.N.; Elias, G.J.B.; Boutet, A.; Germann, J.; Pancholi, A.; Chu, P.; Zhong, J.; Fasano, A.; Munhoz, R.; Chow, C.; et al. Multimodal MRI for MRgFUS in essential tremor: Post-treatment radiological markers of clinical outcome. J. Neurol. Neurosurg. Psychiatry 2020, 91, 921–927. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Vescovi, A.L.; Gritti, A.; Galli, R.; Parati, E.A. Isolation and intracerebral grafting of nontransformed multipotential embryonic human CNS stem cells. J. Neurotrauma 1999, 16, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Seredenina, T. and Luthi-Carter, R. What have we learned from gene expression profiles in Huntington’s disease? Neurobiol. Dis. 2012, 45, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, D.; Mori, E.; Incarnato, D.; Malusa, F.; Fondelli, C.; Magnoni, L.; Pollio, G. Whole gene expression profile in blood reveals multiple pathways deregulation in R6/2 mouse model. Biomark Res. 2013, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum. Mol. Genet. 2002, 11, 1911–1926. [Google Scholar] [CrossRef]
- Strand, A.D.; Aragaki, A.K.; Shaw, D.; Bird, T.; Holton, J.; Turner, C.; Tapscott, S.J.; Tabrizi, S.J.; Schapira, A.H.; Kooperberg, C.; et al. Gene expression in Huntington’s disease skeletal muscle: A potential biomarker. Hum. Mol. Genet. 2005, 14, 1863–1876. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Mitchell, C.T.; Krier, I.; Arjomand, J.; Borowsky, B.; Tabrizi, S.J.; Leavitt, B.R.; Investigators, T.-H.; Luthi-Carter, R. Longitudinal expression changes are weak correlates of disease progression in Huntington’s disease. Brain Commun. 2020, 2, fcaa172. [Google Scholar] [CrossRef]
- Malla, B.; Guo, X.; Senger, G.; Chasapopoulou, Z.; Yildirim, F. A Systematic Review of Transcriptional Dysregulation in Huntington’s Disease Studied by RNA Sequencing. Front. Genet. 2021, 12, 751033. [Google Scholar] [CrossRef]
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020, 8, 19. [Google Scholar] [CrossRef]
- Malaiya, S.; Cortes-Gutierrez, M.; Herb, B.R.; Coffey, S.R.; Legg, S.R.W.; Cantle, J.P.; Colantuoni, C.; Carroll, J.B.; Ament, S.A. Single-Nucleus RNA-Seq Reveals Dysregulation of Striatal Cell Identity Due to Huntington’s Disease Mutations. J. Neurosci. 2021, 41, 5534–5552. [Google Scholar] [CrossRef]
- Lee, H.; Fenster, R.J.; Pineda, S.S.; Gibbs, W.S.; Mohammadi, S.; Davila-Velderrain, J.; Garcia, F.J.; Therrien, M.; Novis, H.S.; Gao, F.; et al. Cell Type-Specific Transcriptomics Reveals that Mutant Huntingtin Leads to Mitochondrial RNA Release and Neuronal Innate Immune Activation. Neuron 2020, 107, 891–908.e8. [Google Scholar] [CrossRef]
- Rudinskiy, N.; Kaneko, Y.A.; Beesen, A.A.; Gokce, O.; Regulier, E.; Deglon, N.; Luthi-Carter, R. Diminished hippocalcin expression in Huntington’s disease brain does not account for increased striatal neuron vulnerability as assessed in primary neurons. J. Neurochem. 2009, 111, 460–472. [Google Scholar] [CrossRef]
- Seredenina, T.; Gokce, O.; Luthi-Carter, R. Decreased striatal RGS2 expression is neuroprotective in Huntington’s disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation. PLoS ONE 2011, 6, e22231. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar]
- Lu, B. BDNF and activity-dependent synaptic modulation. Learn. Mem. 2003, 10, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hayden, M.R.; Xu, B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 2010, 30, 14708–14718. [Google Scholar] [CrossRef]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S.; et al. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Todd, D.; Gowers, I.; Dowler, S.J.; Wall, M.D.; McAllister, G.; Fischer, D.F.; Dijkstra, S.; Fratantoni, S.A.; van de Bospoort, R.; Veenman-Koepke, J.; et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS ONE 2014, 9, e87923. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Lee, B.; Cho, H.Y.; Reyes, I.B.; Pu, X.A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington’s disease. Neurobiol. Dis. 2009, 36, 259–268. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef]
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar]
- Gines, S.; Seong, I.S.; Fossale, E.; Ivanova, E.; Trettel, F.; Gusella, J.F.; Wheeler, V.C.; Persichetti, F.; MacDonald, M.E. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum. Mol. Genet. 2003, 12, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Swartz, J.; Kita, H.; Thykjaer, T.; Carmichael, J.; Bradley, J.; Brown, R.; Maxwell, M.; Schapira, A.; Orntoft, T.F.; et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1829–1845. [Google Scholar] [CrossRef]
- Nucifora, F.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L.; et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001, 291, 2423–2428. [Google Scholar] [CrossRef]
- Adachi, M.; Monteggia, L.M. Decoding transcriptional repressor complexes in the adult central nervous system. Neuropharmacology 2014, 80, 45–52. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buckley, N.J.; Johnson, R.; Zuccato, C.; Bithell, A.; Cattaneo, E. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis. 2010, 39, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Chandra, A.; Flint Beal, M. PGC-1alpha, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jager, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, e101. [Google Scholar] [CrossRef]
- Weydt, P.; Soyal, S.M.; Gellera, C.; Didonato, S.; Weidinger, C.; Oberkofler, H.; Landwehrmeyer, G.B.; Patsch, W. The gene coding for PGC-1alpha modifies age at onset in Huntington’s Disease. Mol. Neurodegener. 2009, 4, 3. [Google Scholar] [CrossRef]
- Taherzadeh-Fard, E.; Saft, C.; Andrich, J.; Wieczorek, S.; Arning, L. PGC-1alpha as modifier of onset age in Huntington disease. Mol. Neurodegener. 2009, 4, 10. [Google Scholar] [CrossRef]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Ryu, H.; Kubilus, J.K.; D’Mello, S.; Sugars, K.L.; Lee, J.; Lu, P.; Smith, K.; Browne, S.; Beal, M.F.; et al. Chemotherapy for the Brain: The Antitumor Antibiotic Mithramycin Prolongs Survival in a Mouse Model of Huntington’s Disease. J. Neurosci. 2004, 24, 10335–10342. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef]
- Vashishtha, M.; Ng, C.W.; Yildirim, F.; Gipson, T.A.; Kratter, I.H.; Bodai, L.; Song, W.; Lau, A.; Labadorf, A.; Vogel-Ciernia, A.; et al. Targeting H3K4 trimethylation in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, E3027–E3036. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Sweatt, J.D. Epigenetic Mechanisms in Cognition. Neuron 2011, 70, 813–829. [Google Scholar]
- Guibert, S.; Weber, M. Chapter Two—Functions of DNA Methylation and Hydroxymethylation in Mammalian Development. In Current Topics in Developmental Biology; Heard, E., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 47–83. [Google Scholar]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Y.; Lin, X.; Wang, J.Q.; Wu, Y.S.; Xie, W.; Wang, D.; Zhu, S.; Liao, Y.Q.; Sun, Q.; et al. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 3641–3653. [Google Scholar] [CrossRef]
- Johnson, R. Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol Dis. 2012, 46, 245–254. [Google Scholar] [CrossRef]
- Molasy, M.; Walczak, A.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. MicroRNAs in glaucoma and neurodegenerative diseases. J. Hum. Genet. 2017, 62, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008, 28, 14341–14346. [Google Scholar] [CrossRef]
- Johnson, R.; Buckley, N.J. Gene Dysregulation in Huntington’s Disease: REST, MicroRNAs and Beyond. NeuroMol. Med. 2009, 11, 183–199. [Google Scholar]
- Soldati, C.; Bithell, A.; Johnston, C.; Wong, K.Y.; Stanton, L.W.; Buckley, N.J. Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington’s disease. J. Neurochem. 2013, 124, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.W.; Rudnicki, D.D.; Yu, L.; Margolis, R.L. A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum. Mol. Genet. 2011, 20, 3467–3477. [Google Scholar] [CrossRef]
- Sadri-Vakili, G.; Bouzou, B.; Benn, C.L.; Kim, M.O.; Chawla, P.; Overland, R.P.; Glajch, K.E.; Xia, E.; Qiu, Z.; Hersch, S.M.; et al. Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet. 2007, 16, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Browne, S.E.; Choi, D.K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef]
- Chiu, C.-T.; Liu, G.; Leeds, P.; Chuang, D.-M. Combined Treatment with the Mood Stabilizers Lithium and Valproate Produces Multiple Beneficial Effects in Transgenic Mouse Models of Huntington’s Disease. Neuropsychopharmacology 2011, 36, 2406–2421. [Google Scholar] [CrossRef]
- Scheuing, L.; Chiu, C.T.; Liao, H.M.; Linares, G.R.; Chuang, D.M. Preclinical and clinical investigations of mood stabilizers for Huntington’s disease: What have we learned? Int. J. Biol. Sci. 2014, 10, 1024–1038. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Kast, R.J.; Steffan, J.S.; Thomas, E.A. Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: Implications for the ubiquitin–proteasomal and autophagy systems. Hum. Mol. Genet. 2012, 21, 5280–5293. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef]
- Zeitler, B.; Froelich, S.; Marlen, K.; Shivak, D.A.; Yu, Q.; Li, D.; Pearl, J.R.; Miller, J.C.; Zhang, L.; Paschon, D.E.; et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019, 25, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar]
- Fink, K.D.; Deng, P.; Gutierrez, J.; Anderson, J.S.; Torrest, A.; Komarla, A.; Kalomoiris, S.; Cary, W.; Anderson, J.D.; Gruenloh, W.; et al. Allele-Specific Reduction of the Mutant Huntingtin Allele Using Transcription Activator-Like Effectors in Human Huntington’s Disease Fibroblasts. Cell Transpl. 2016, 25, 677–686. [Google Scholar] [CrossRef]
- Shin, J.W.; Kim, K.H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar]
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef]
- Rodriguez-Lebron, E.; Denovan-Wright, E.M.; Nash, K.; Lewin, A.S.; Mandel, R.J. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol. Ther. 2005, 12, 618–633. [Google Scholar] [CrossRef]
- Boudreau, R.L.; Martins, I.; Davidson, B.L. Artificial microRNAs as siRNA shuttles: Improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. 2009, 17, 169–175. [Google Scholar] [CrossRef]
- Machida, Y.; Okada, T.; Kurosawa, M.; Oyama, F.; Ozawa, K.; Nukina, N. rAAV-mediated shRNA ameliorated neuropathology in Huntington disease model mouse. Biochem. Biophys. Res. Commun. 2006, 343, 190–197. [Google Scholar] [CrossRef]
- Miniarikova, J.; Zanella, I.; Huseinovic, A.; van der Zon, T.; Hanemaaijer, E.; Martier, R.; Koornneef, A.; Southwell, A.L.; Hayden, M.R.; van Deventer, S.J.; et al. Design, Characterization, and Lead Selection of Therapeutic miRNAs Targeting Huntingtin for Development of Gene Therapy for Huntington’s Disease. Mol. Ther. Nucleic Acids. 2016, 5, e297. [Google Scholar] [CrossRef]
- Samaranch, L.; Blits, B.; San Sebastian, W.; Hadaczek, P.; Bringas, J.; Sudhakar, V.; Macayan, M.; Pivirotto, P.J.; Petry, H.; Bankiewicz, K.S. MR-guided parenchymal delivery of adeno-associated viral vector serotype 5 in non-human primate brain. Gene Ther. 2017, 24, 253–261. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018, 18, 126–131. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves First Drug for Spinal Muscular Atrophy. 2016. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy (accessed on 17 August 2023).
- US Food and Drug Administration. FDA Grants Accelerated Approval to First Drug for Duchenne Muscular Dystrophy. 2016. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular-dystrophy (accessed on 17 August 2023).
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Roche, Roche-Genentech-HD-Global-Community-Letter-January-2022, 2022. Available online: https://hdsa.org/wp-content/uploads/2022/09/Roche-Global-Patient-Community-letter_EHDN-2022.pdf (accessed on 17 August 2023).
- Bezprozvanny, I. The rise and fall of Dimebon. Drug News Perspect. 2010, 23, 518–523. [Google Scholar] [CrossRef]
- Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. J. Neural. Transm. Suppl. 2004, 68, 117–122. [Google Scholar]
- Gelderblom, H.; Wustenberg, T.; McLean, T.; Mutze, L.; Fischer, W.; Saft, C.; Hoffmann, R.; Sussmuth, S.; Schlattmann, P.; van Duijn, E.; et al. Bupropion for the treatment of apathy in Huntington’s disease: A multicenter, randomised, double-blind, placebo-controlled, prospective crossover trial. PLoS ONE 2017, 12, e0173872. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study, G. Tetrabenazine as antichorea therapy in Huntington disease: A randomized controlled trial. Neurology 2006, 66, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Der, N.P.; Zsindely, N.; Bodai, L. Green tea infusion alleviates neurodegeneration induced by mutant Huntingtin in Drosophila. Nutr. Neurosci. 2020, 23, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef]
- Reilmann, R.; Squitieri, F.; Priller, J.; Saft, C.; Mariotti, C.; Süssmuth, S.; Nemeth, A.; Tabrizi, S.; Quarrell, O.; Craufurd, D.; et al. N02 Safety And Tolerability Of Selisistat For The Treatment Of Huntington’s Disease: Results From A Randomised, Double-blind, Placebo-controlled Phase Ii Trial. J. Neurol. Neurosurg. Psychiatry 2014, 85, A102-A02. [Google Scholar] [CrossRef]
- Hogarth, P.; Lovrecic, L.; Krainc, D. Sodium phenylbutyrate in Huntington’s disease: A dose-finding study. Mov. Disord. 2007, 22, 1962–1964. [Google Scholar] [CrossRef]
- Keller, C.G.; Shin, Y.; Monteys, A.M.; Renaud, N.; Beibel, M.; Teider, N.; Peters, T.; Faller, T.; St-Cyr, S.; Knehr, J.; et al. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat. Commun. 2022, 13, 1150. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S. Effects of IONIS-HTTRx in patients with early Huntington’s disease, results of the first HTT-lowering drug trial (CT. 002). Neurology 2018, 90, CT.002. [Google Scholar]
- Hersch, S.; Claassen, D.; Edmondson, M.; Wild, E.; Guerciolini, R.; Panzara, M. Multicenter, Randomized, Double-blind, Placebo-controlled Phase 1b/2a Studies of WVE-120101 and WVE-120102 in Patients with Huntington’s Disease (P2.006). Neurology 2017, 88, P2.006. [Google Scholar]
- Brownstein, M.J.; Simon, N.G.; Long, J.D.; Yankey, J.; Maibach, H.T.; Cudkowicz, M.; Coffey, C.; Conwit, R.A.; Lungu, C.; Anderson, K.E.; et al. Safety and Tolerability of SRX246, a Vasopressin 1a Antagonist, in Irritable Huntington’s Disease Patients-A Randomized Phase 2 Clinical Trial. J. Clin. Med. 2020, 9, 3682. [Google Scholar] [CrossRef]
- Travessa, A.M.; Rodrigues, F.B.; Mestre, T.A.; Ferreira, J.J. Fifteen Years of Clinical Trials in Huntington’s Disease: A Very Low Clinical Drug Development Success Rate. J. Huntingt. Dis. 2017, 6, 157–163. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Scahill, R.I.; Durr, A.; Roos, R.A.; Leavitt, B.R.; Jones, R.; Landwehrmeyer, G.B.; Fox, N.C.; Johnson, H.; Hicks, S.L. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: The 12-month longitudinal analysis. Lancet Neurol. 2011, 10, 31–42. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Reilmann, R.; Roos, R.A.; Durr, A.; Leavitt, B.; Owen, G.; Jones, R.; Johnson, H.; Craufurd, D.; Hicks, S.L. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: Analysis of 24 month observational data. Lancet Neurol. 2012, 11, 42–53. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Scahill, R.I.; Owen, G.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Borowsky, B.; Landwehrmeyer, B.; Frost, C.; Johnson, H.; et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: Analysis of 36-month observational data. Lancet Neurol. 2013, 12, 637–649. [Google Scholar] [CrossRef]
- Sathe, S.; Ware, J.; Levey, J.; Neacy, E.; Blumenstein, R.; Noble, S.; Muhlback, A.; Rosser, A.; Landwehrmeyer, G.B.; Sampaio, C. Enroll-HD: An Integrated Clinical Research Platform and Worldwide Observational Study for Huntington’s Disease. Front. Neurol. 2021, 12, 667420. [Google Scholar] [CrossRef]




| Country | Region | Prevalence (per 100,000) | Year Studied | Reference |
|---|---|---|---|---|
| Europe | ||||
| Russia | Volgograd and Volzhsky | 0.6 | 2000s | [20] |
| Iceland | - | 1.0 | 2007 | [21] |
| Finland | - | 2.12 | 2010 | [22] |
| Greece | - | 3.95 | 2008 | [23] |
| Croatia | Rijeka | 4.5 | 1981 | [24] |
| Germany | Franconia | 4.7 | 1987 | [25] |
| France | Haute Vienne | 4.8 | 1970s | [26] |
| Nord and Pas-de-Calais | 5.0 | 1980s | [27] | |
| Slovenia | - | 5.2 | 2006 | [28] |
| Sweden | - | 4.5 | 1974 | [29] |
| - | 5.6 | 1985 | [30] | |
| Netherlands | - | 6.5 | 1999 | [31] |
| Norway | - | 5.8 | 1950 | [32] |
| - | 6.7 | 1940 | [32] | |
| - | 6.9 | 1930 | [32] | |
| Spain | Valencia | 5.38 | 1992 | [33] |
| Salamanca | 8.4 | 1980s | [34] | |
| Italy | Molise | 10.85 | 2010s | [35] |
| Malta | - | 11.8 | 1994 | [36] |
| United Kingdom | Northern Ireland | 6.4 | 1991 | [37] |
| Northern Ireland | 10.4 | 2001 | [16] | |
| - | 11.2 | 1990 | [7] | |
| - | 12.3 | 2010 | [7] | |
| Americas | ||||
| Venezuela | Nationwide excluding Zulia | 0.5 | 2006 | [38] |
| Mexico | Mexico City | 4.0 | 2008 | [39] |
| United States of America | Michigan | 5.0 | 1940 | [40] |
| South Carolina | 5.0 | 1980 | [41] | |
| Maryland | 5.15 | 1980s | [42] | |
| Minnesota | 6.3 | 1989 | [43] | |
| Canada | Quebec | 3.4 | 1963 | [44] |
| Saskatchewan and Manitoba | 8.4 | 1975 | [45] | |
| British Columbia | 13.7 | 2012 | [14] | |
| Oceania | ||||
| Australia | New South Wales | 6.3 | 1996 | [46] |
| Tasmania | 12.1 | 1990 | [19] | |
| Africa | ||||
| South Africa | - | 0.01 | 1970s | [12] |
| Zimbabwe | - | 0.5–1 | 1980s | [47] |
| Kilimanjaro | 7 | 1970s | [48] | |
| Asia | ||||
| China | Hong Kong | 0.37 | 1991 | [10,11] |
| Taiwan | 0.42 | 2007 | [49] | |
| Japan | San-in | 0.65 | 1997 | [9] |
| Western Japan | 0.72 | 1997 | [9] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, A.; Handley, R.R.; Lehnert, K.; Snell, R.G. From Pathogenesis to Therapeutics: A Review of 150 Years of Huntington’s Disease Research. Int. J. Mol. Sci. 2023, 24, 13021. https://doi.org/10.3390/ijms241613021
Jiang A, Handley RR, Lehnert K, Snell RG. From Pathogenesis to Therapeutics: A Review of 150 Years of Huntington’s Disease Research. International Journal of Molecular Sciences. 2023; 24(16):13021. https://doi.org/10.3390/ijms241613021
Chicago/Turabian StyleJiang, Andrew, Renee R. Handley, Klaus Lehnert, and Russell G. Snell. 2023. "From Pathogenesis to Therapeutics: A Review of 150 Years of Huntington’s Disease Research" International Journal of Molecular Sciences 24, no. 16: 13021. https://doi.org/10.3390/ijms241613021
APA StyleJiang, A., Handley, R. R., Lehnert, K., & Snell, R. G. (2023). From Pathogenesis to Therapeutics: A Review of 150 Years of Huntington’s Disease Research. International Journal of Molecular Sciences, 24(16), 13021. https://doi.org/10.3390/ijms241613021