
The Role of Hydrogen Incorporation into Amorphous Carbon Films in the Change of the Secondary Electron Yield

 ,
,  ,
,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
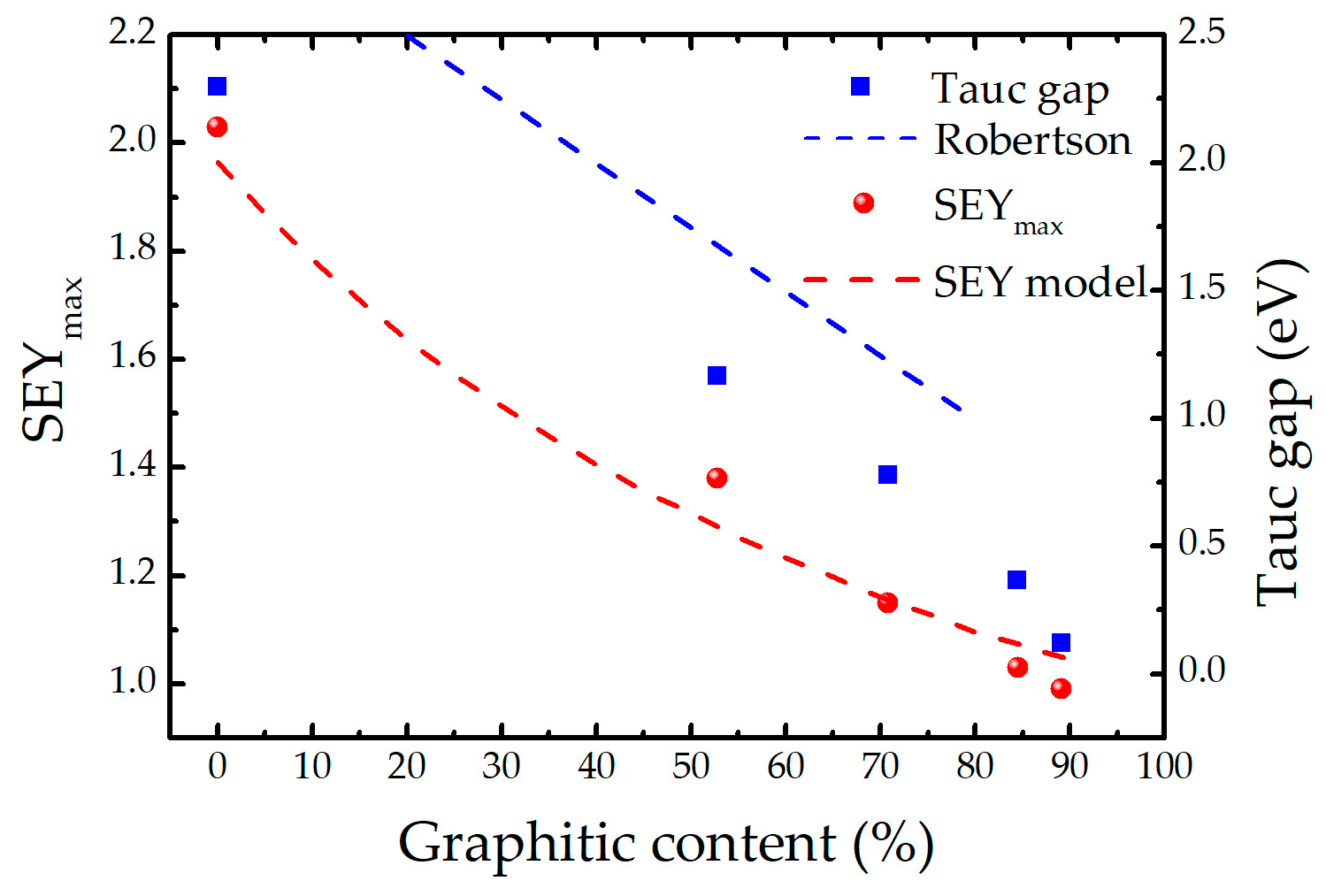
2.1. Composition, Electron Emission and Optical Properties of the Coatings
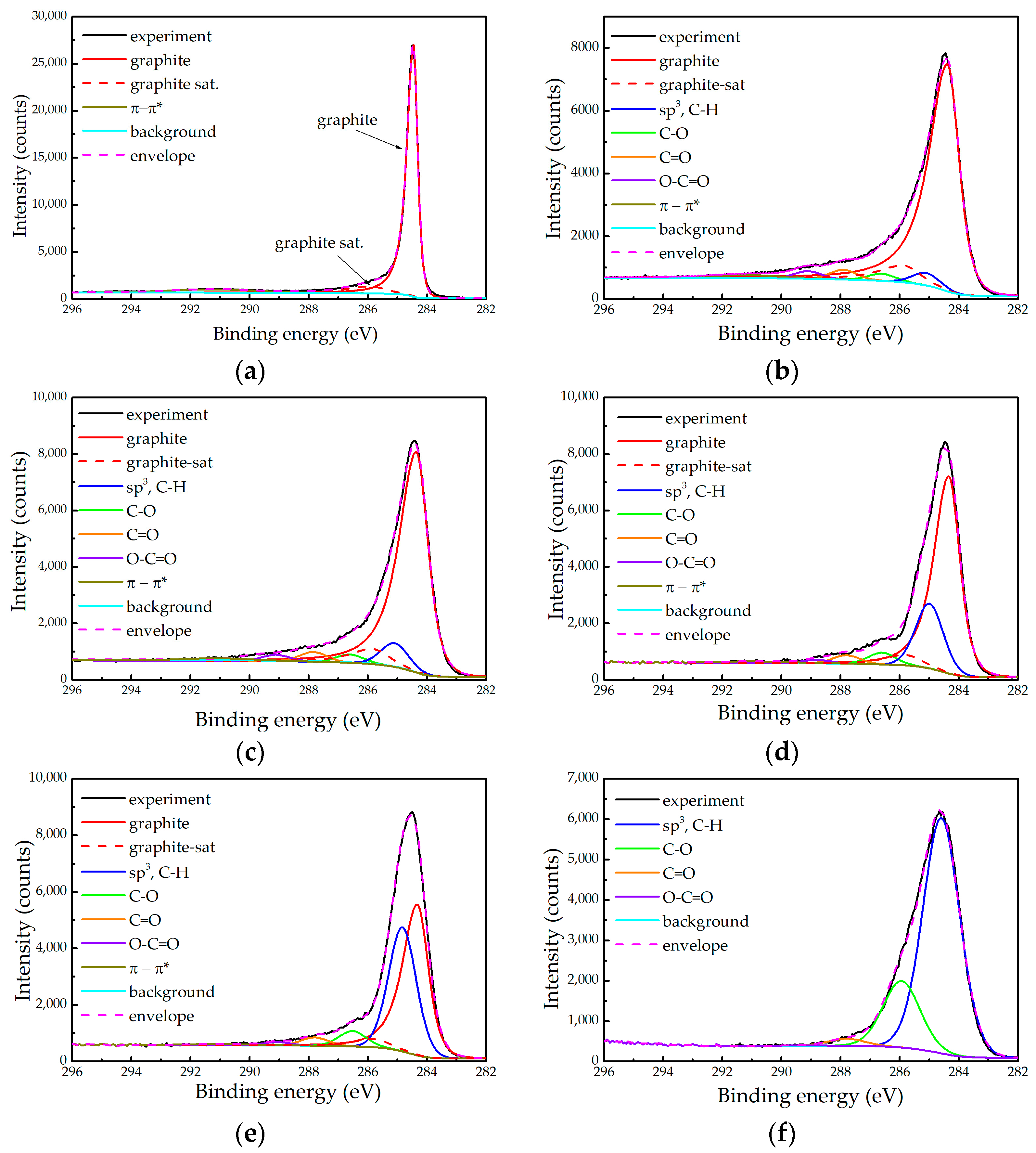
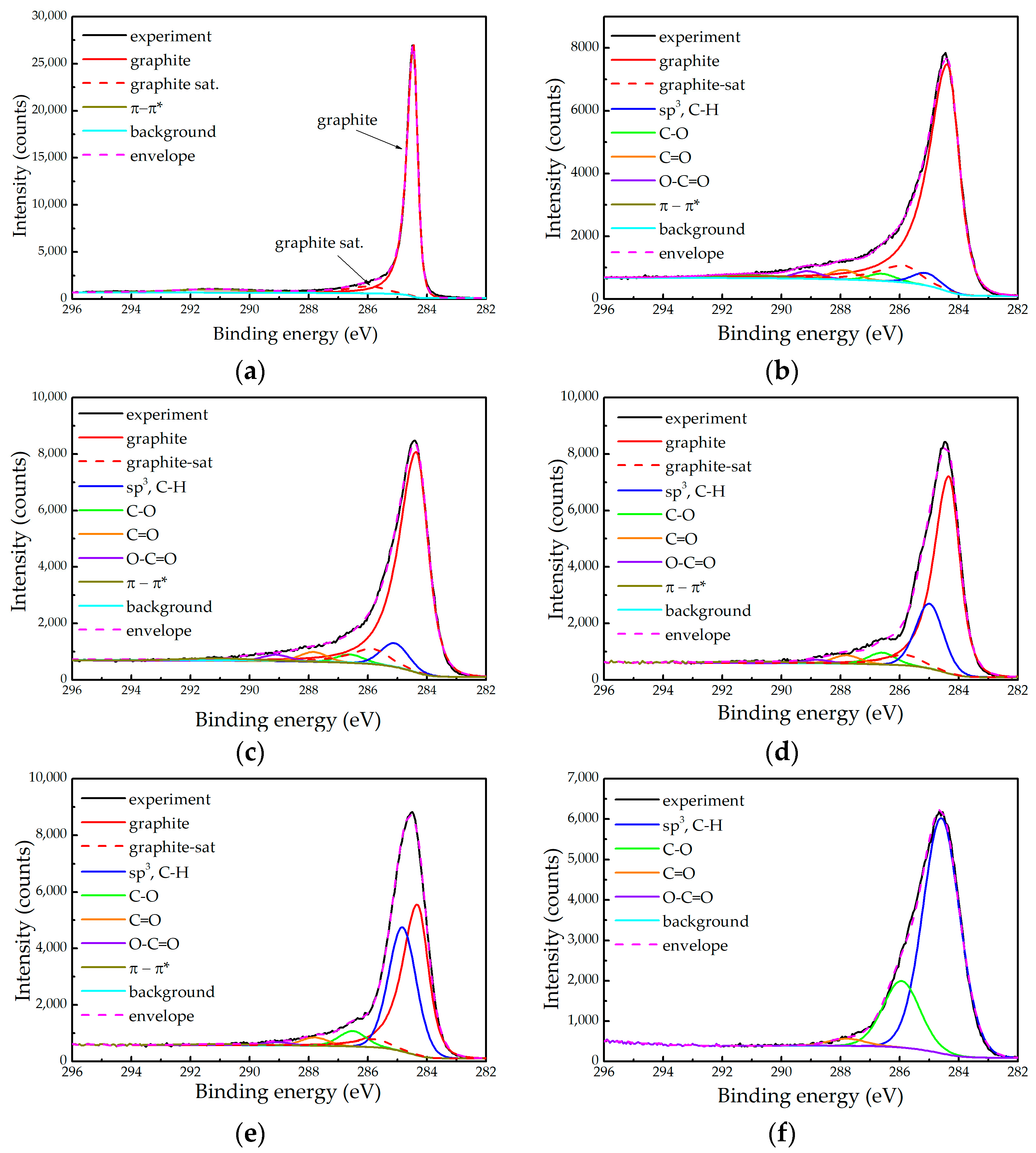
2.2. XPS Measurements
- The relative contribution of the graphitic component decreases and even disappears in the sample 10D;
- The relative contribution attributed to the sp3 carbon and the hydrocarbons increases;
- The asymmetry of the graphitic component becomes less pronounced.
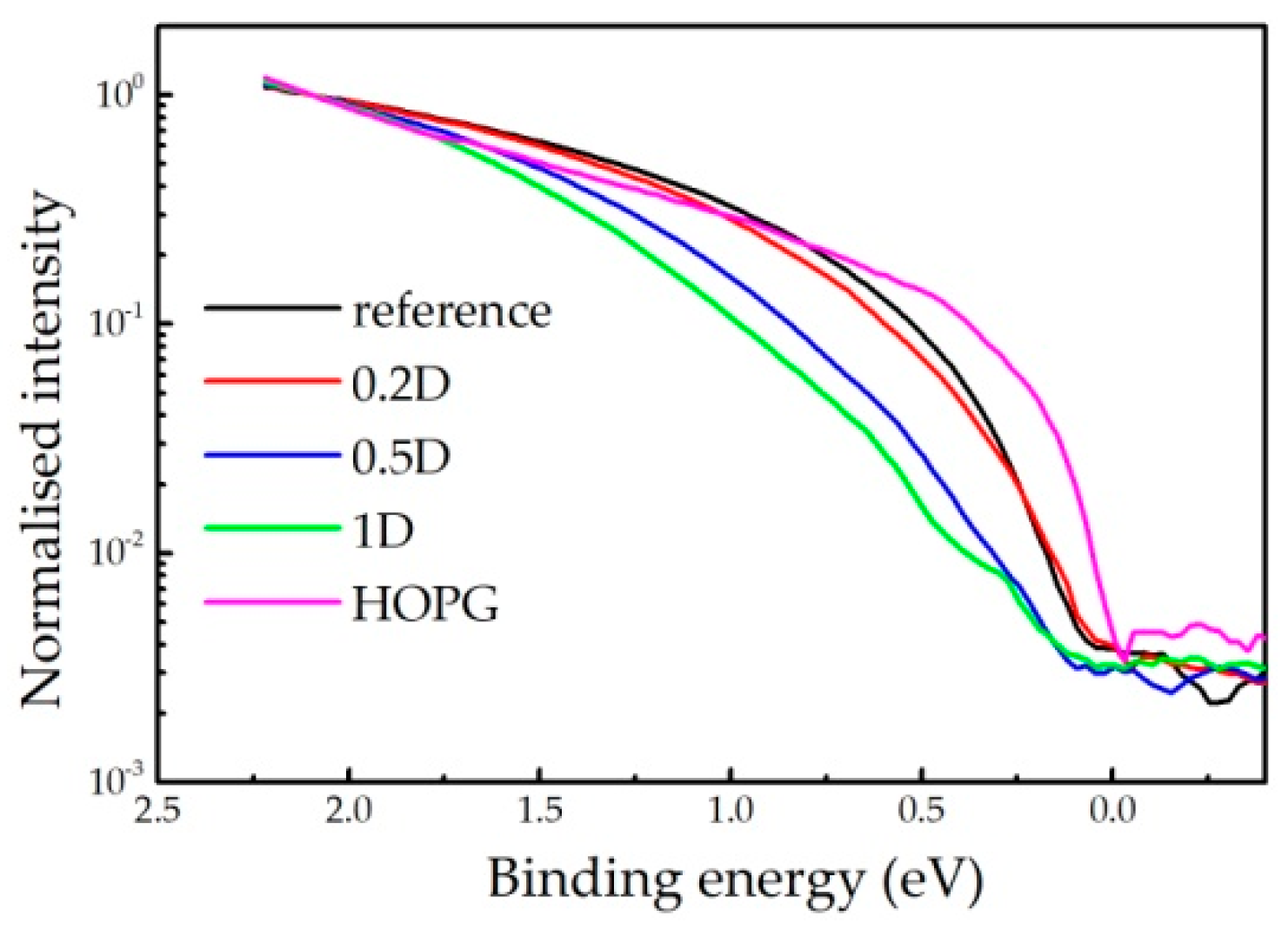
2.3. UPS Measurements
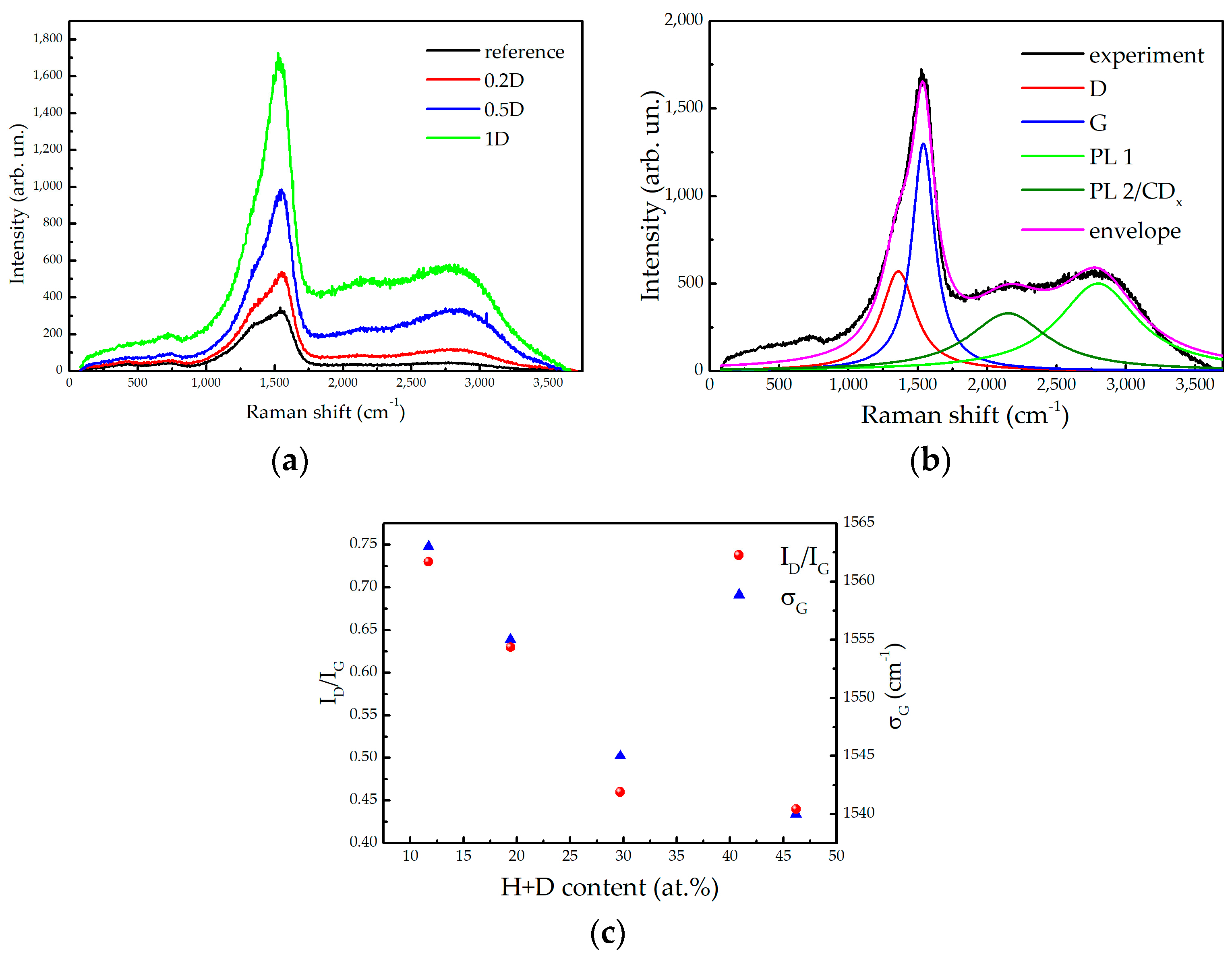
2.4. Raman Spectroscopy
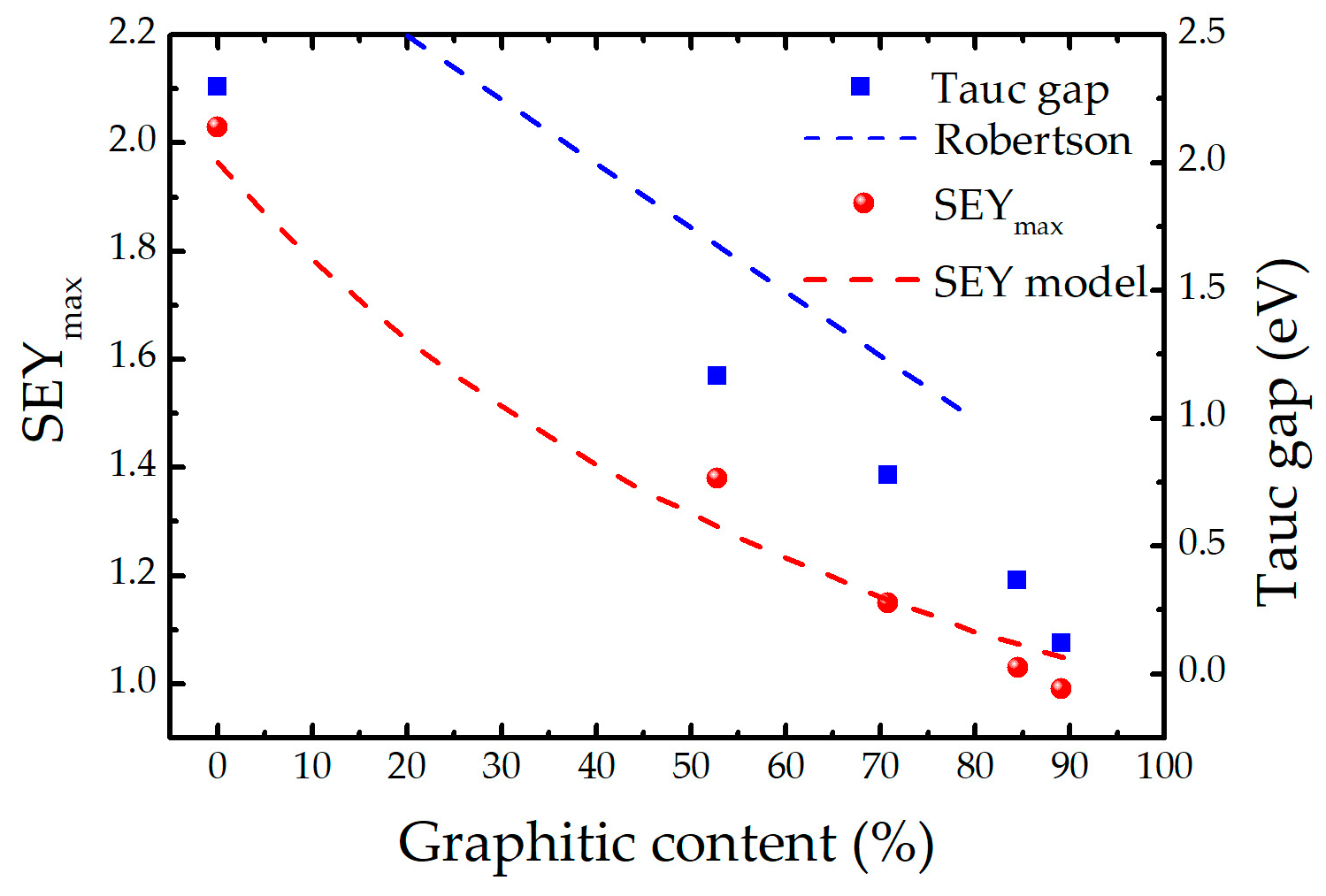
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
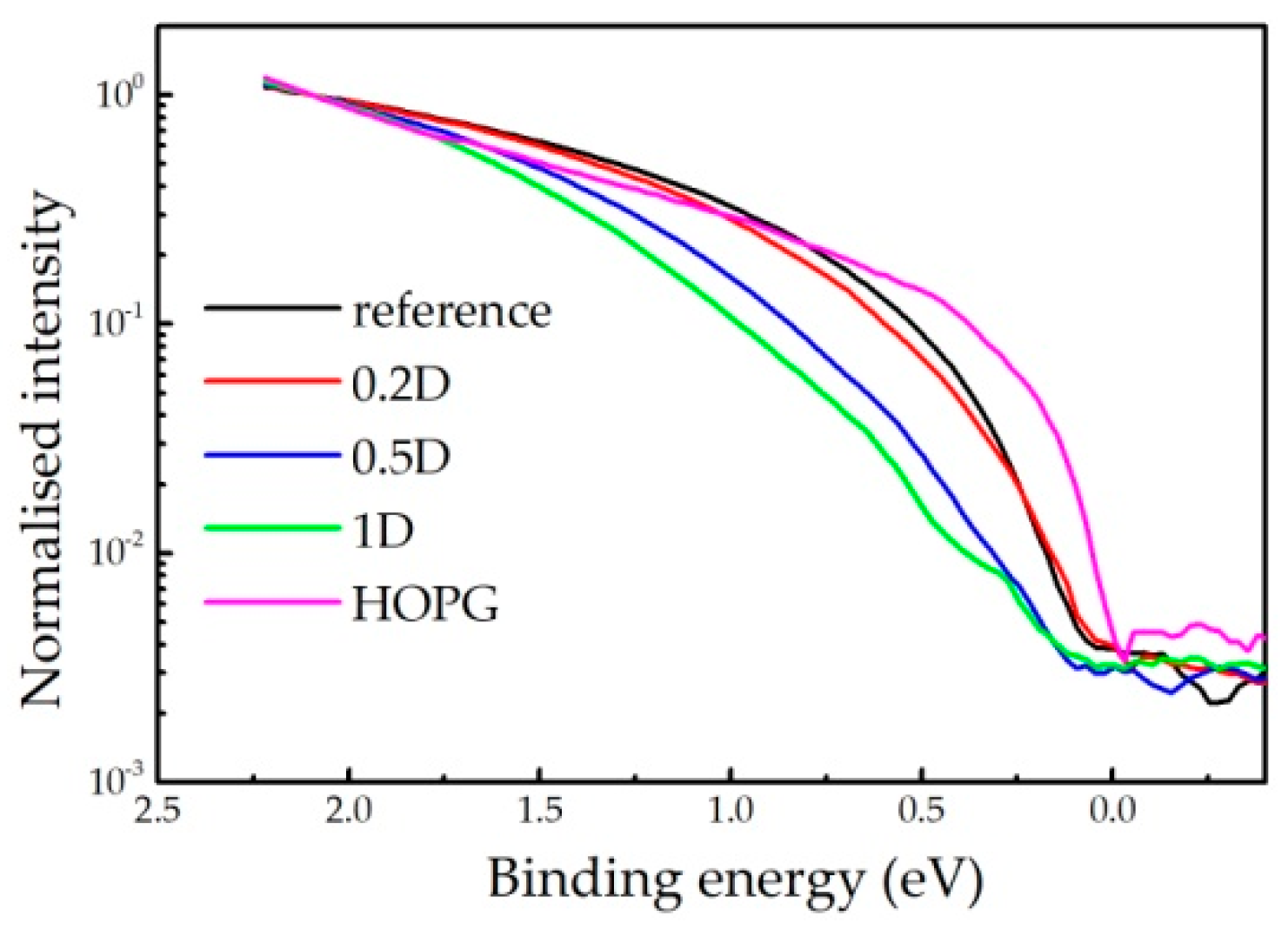
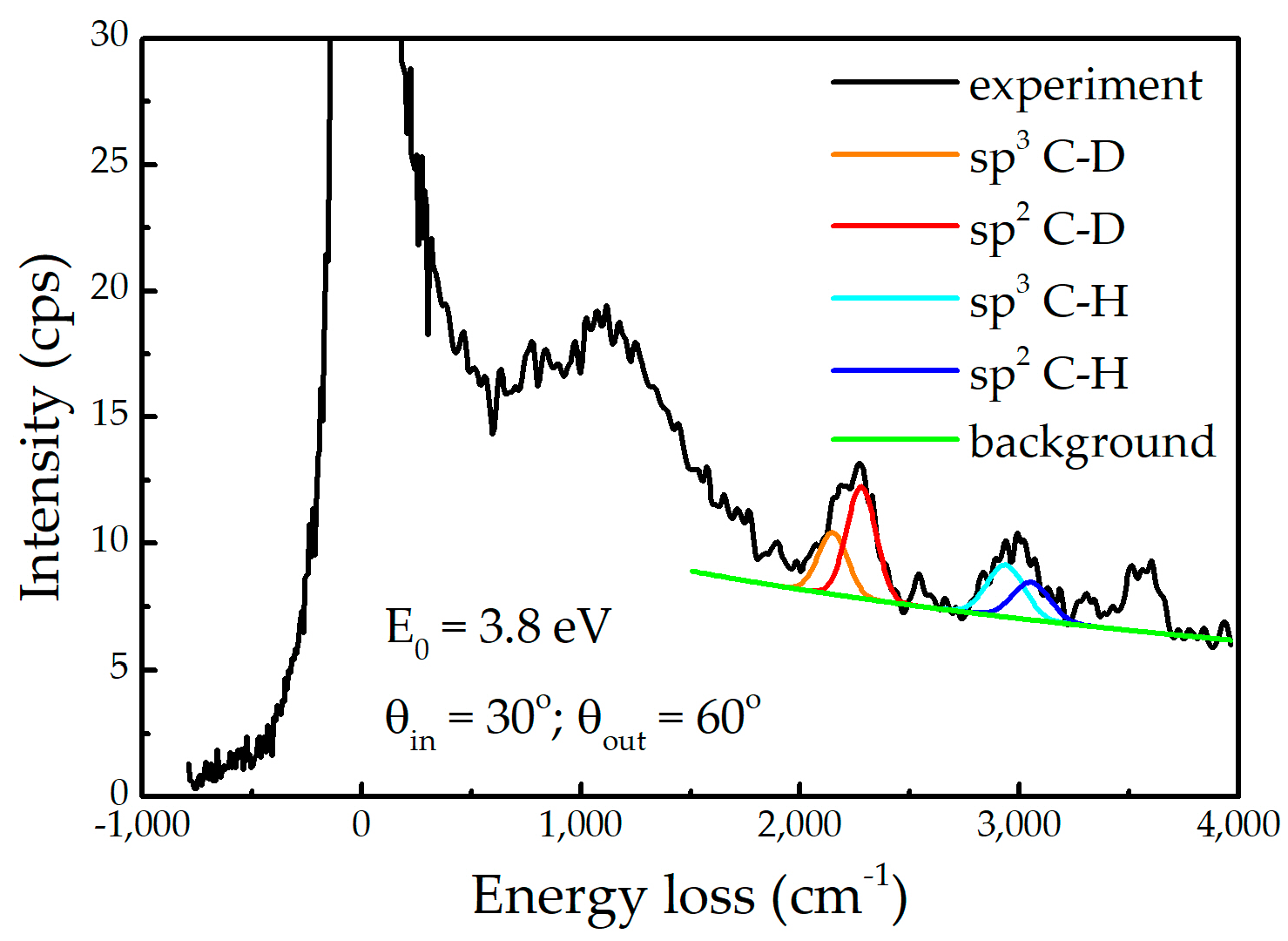
Appendix A. HREELS Measurements
Appendix A.1. Experimental Setup
Appendix A.2. Results
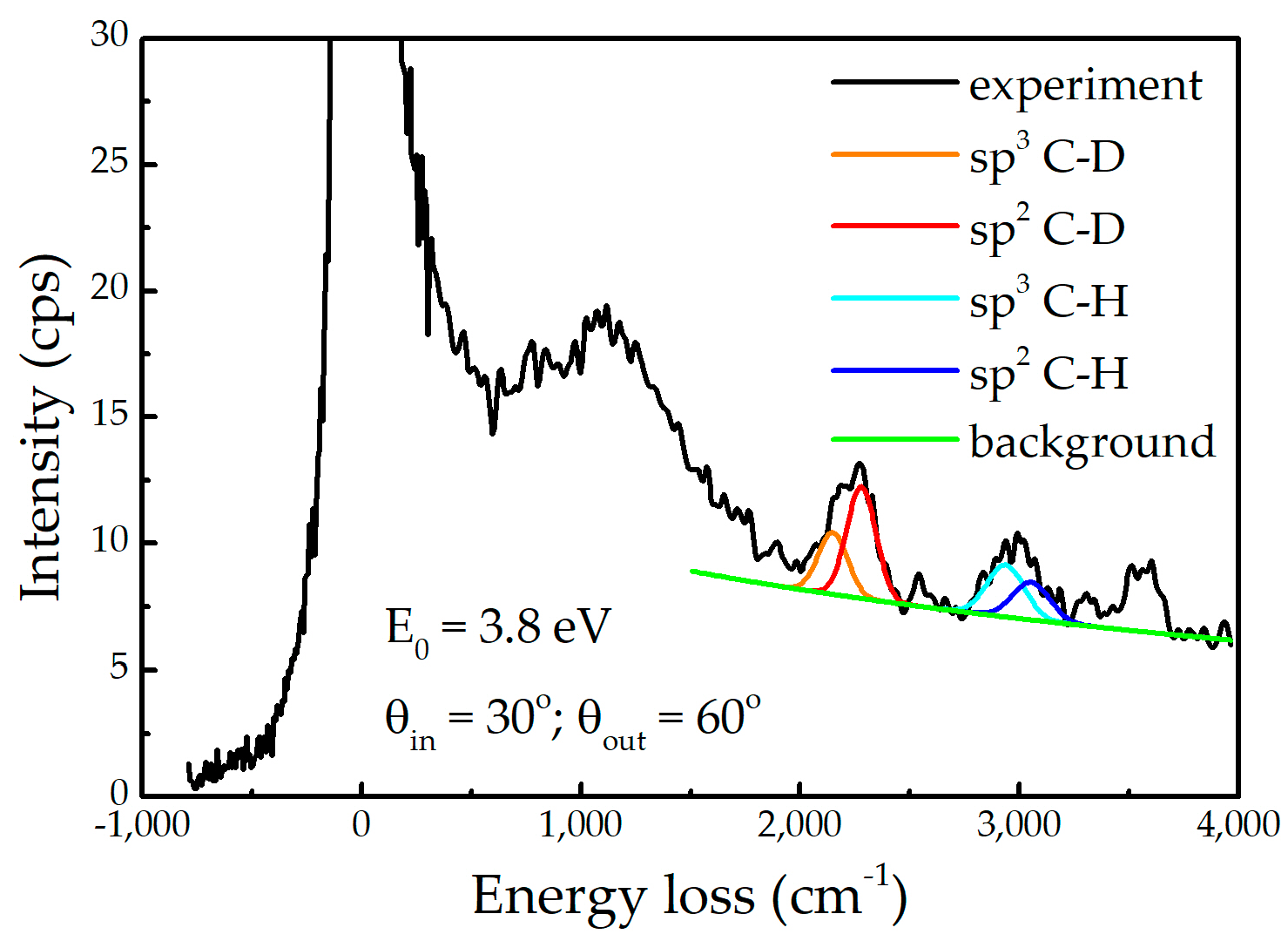
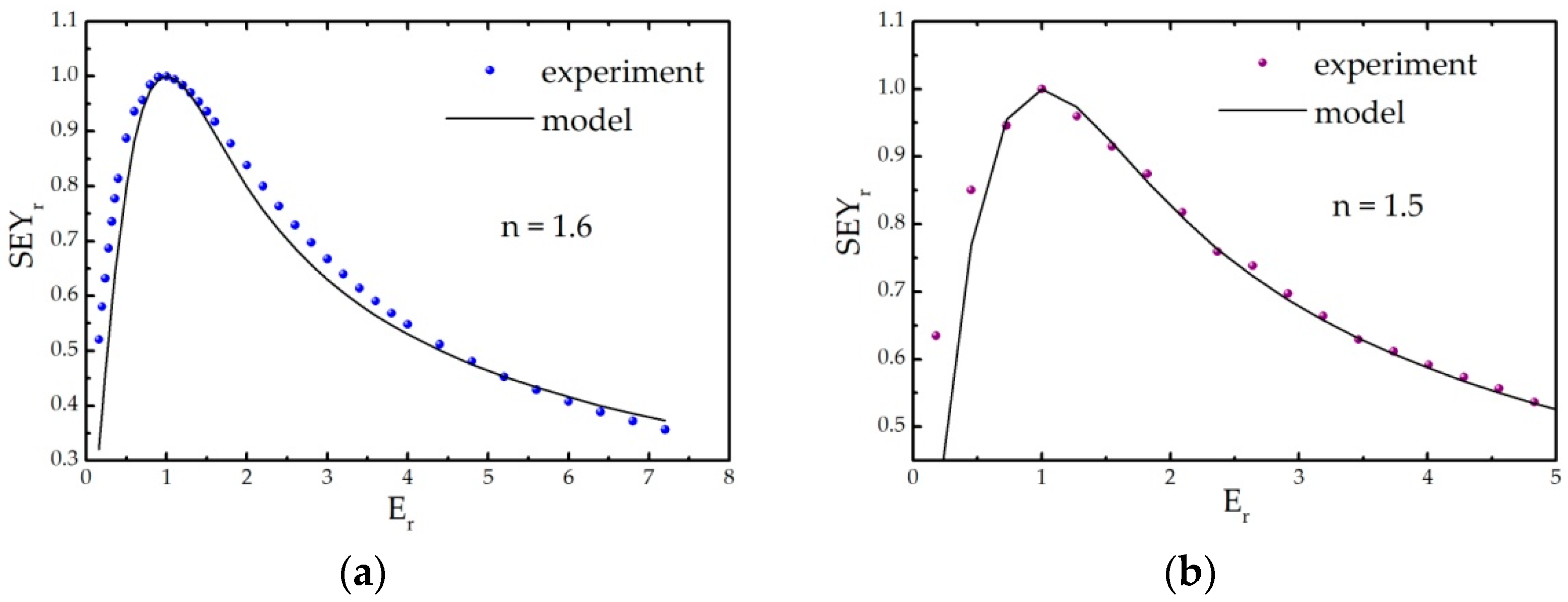
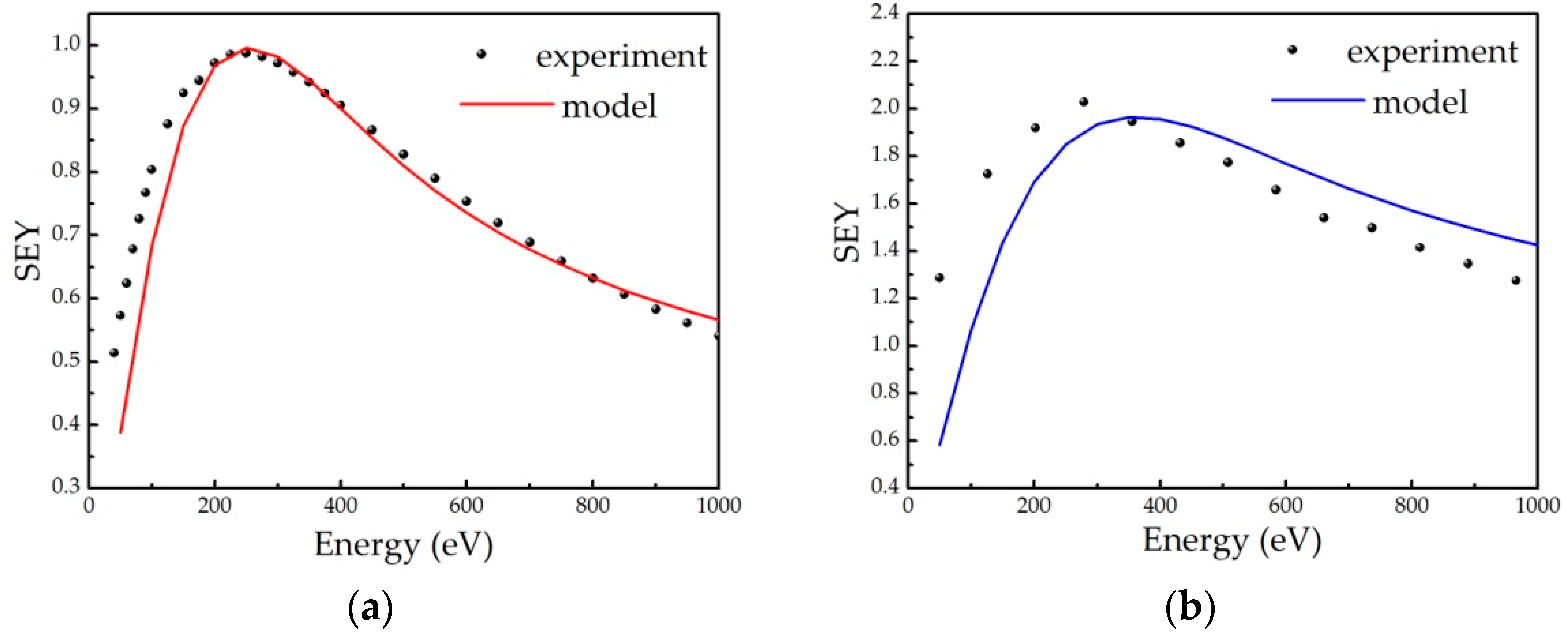
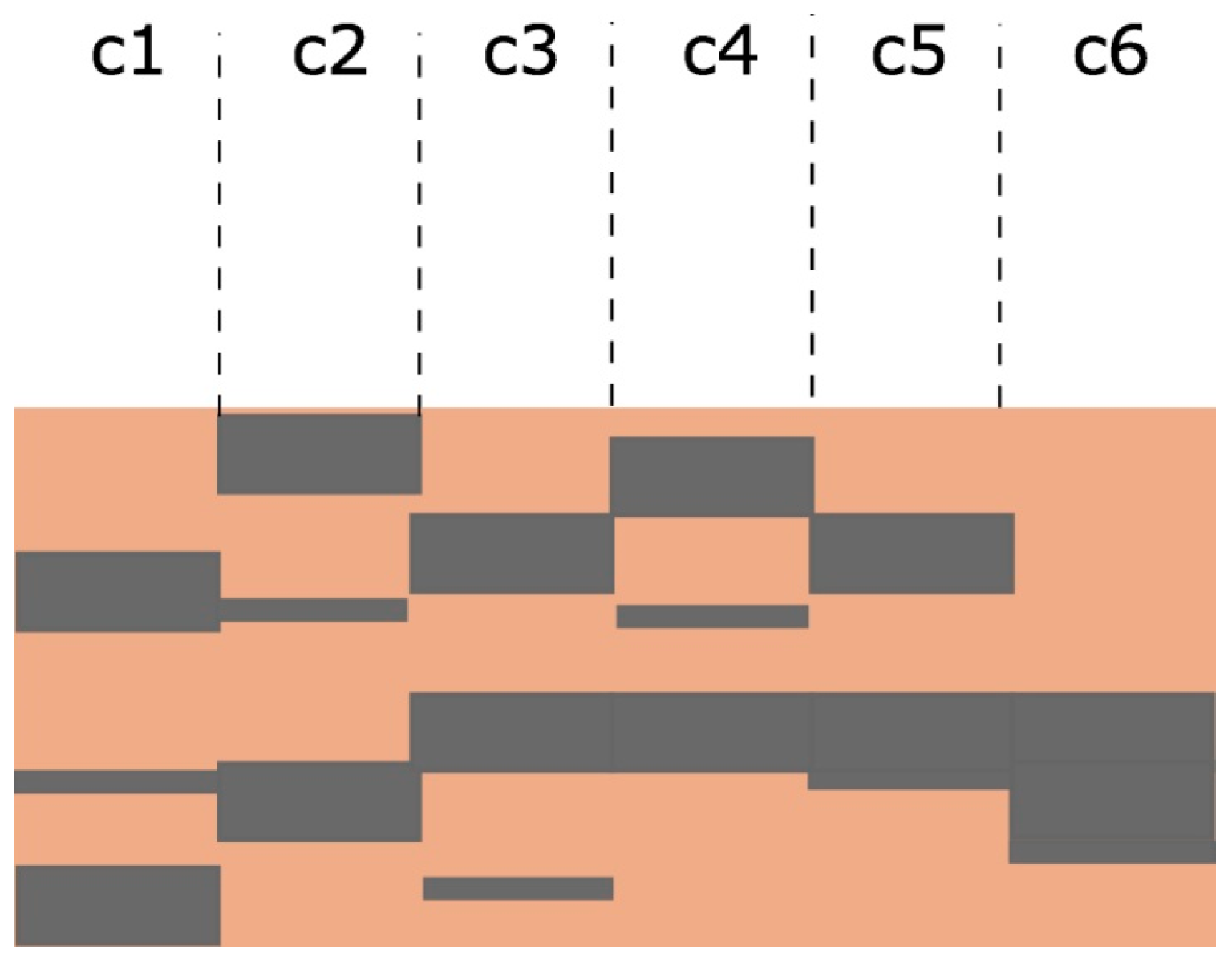
Appendix B. Modelling SEY from Non-Uniform A-Carbon Samples
Appendix B.1. Semi-Empirical Theory of Secondary Electron Emission and Its Application to Graphitic and Polymeric Samples
- all primary electrons have the same range R in a material, which can be described by the power law R = b∙En, with E being the energy of incident electrons, while b is the material constant;
- the number of internal secondary electrons generated in the depth interval (z, z + Δz) is directly proportional to the stopping power of primary electrons S(z) = −dE/dz, ΔN(z) = S(z)Δz/ε, where ε is the effective energy required to create one internal secondary electron;
- the electron stopping power is assumed to be constant, and its average value along the trajectory E/R(E) is frequently used;
- the probability that an internal secondary electron will be emitted is 0.5∙exp(−z/λ), where λ is an electron escape depth (a material dependent parameter).
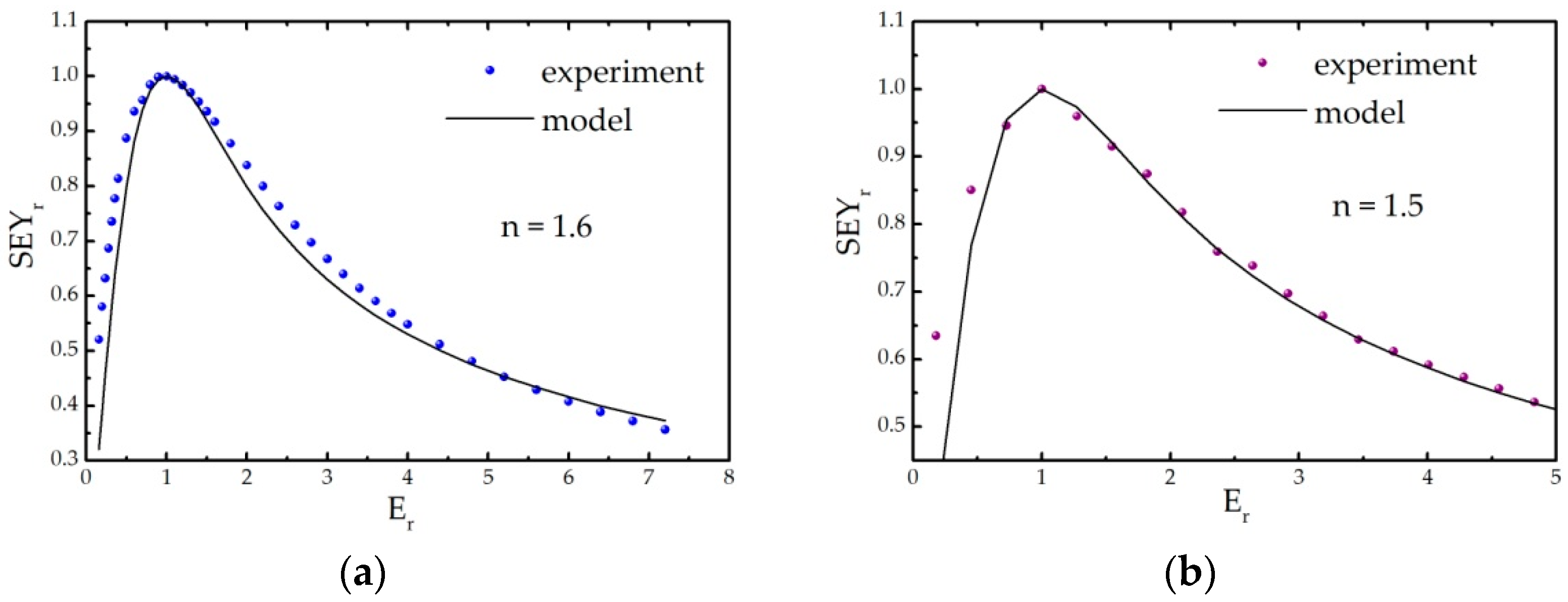
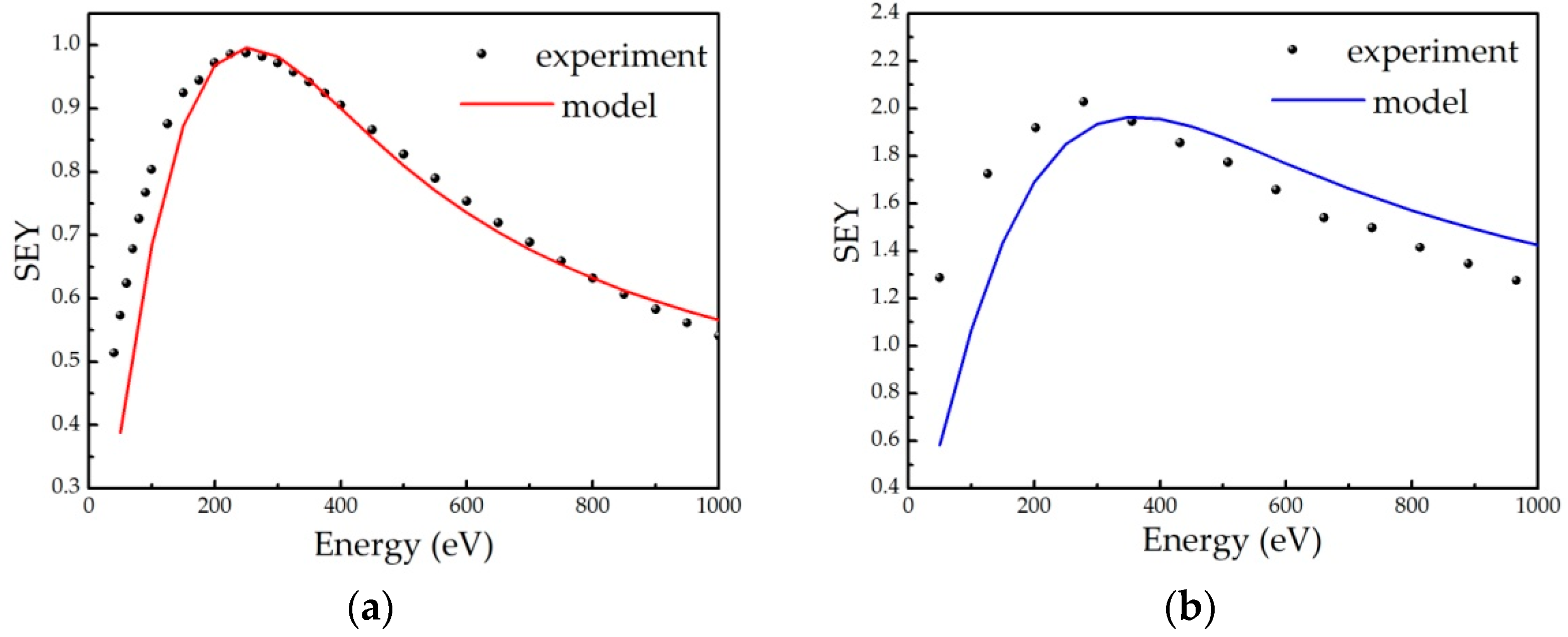
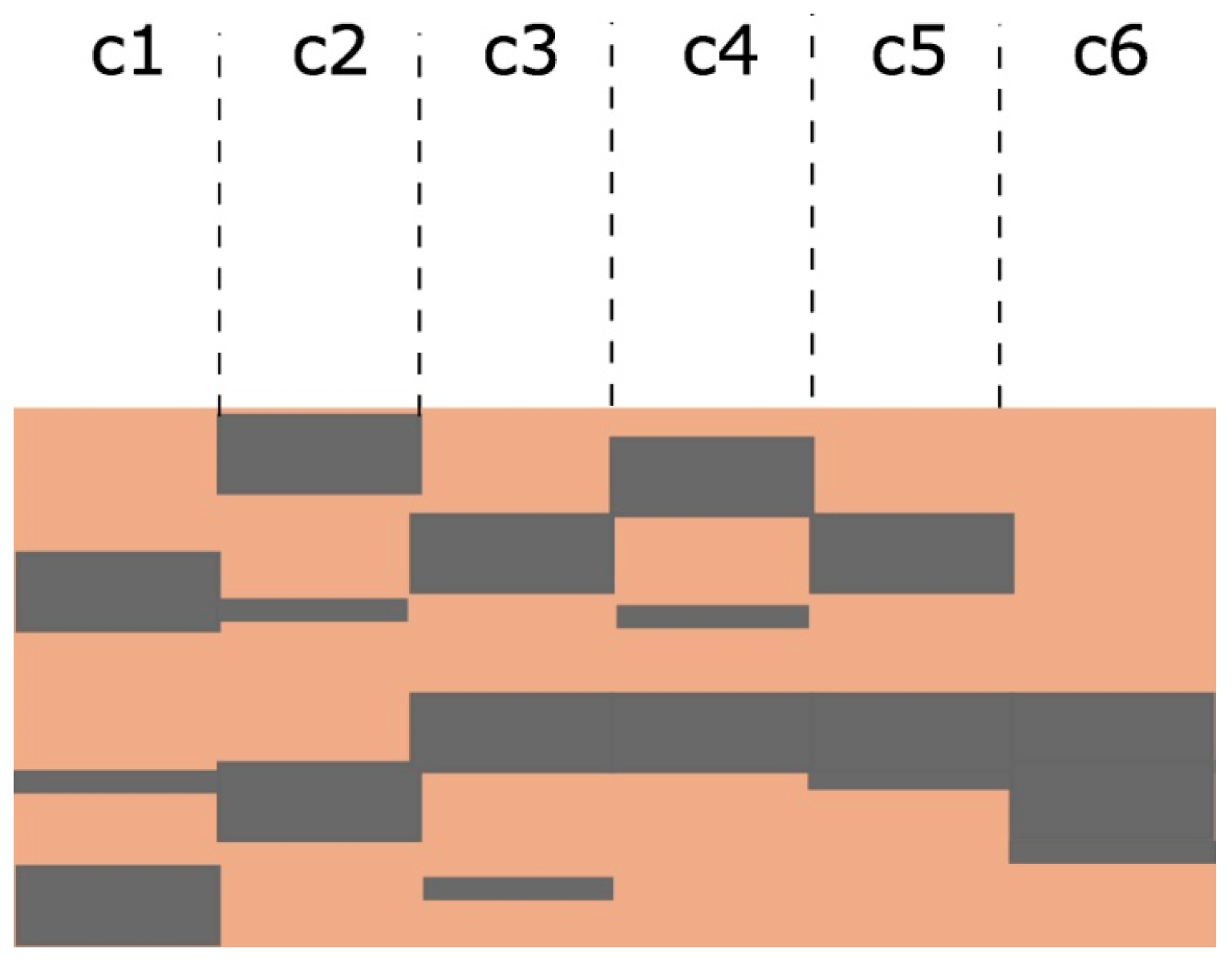
Appendix B.2. Model Description
References
- Gröbner, O. Bunch induced multipactoring. In Proceedings of the 10th International Conference on High-Energy Accelerators, Serpukhov, Russia, 11–17 July 1977; CM-P0064850. CERN Libraries: Geneva, Switzerland, 1977. [Google Scholar]
- Cimino, R.; Collins, I.R.; Furman, M.A.; Pivi, M.; Ruggiero, F.; Rumolo, G.; Zimmermann, F. Can low-energy electrons affect high-energy physics accelerators? Phys. Rev. Lett. 2004, 93, 014801. [Google Scholar] [CrossRef]
- Kishek, R.A.; Lau, Y.Y.; Ang, L.K.; Valfells, A.; Gilgenbach, R.M. Multipactor discharge on metals and dielectrics: Historical review and recent theories. Phys. Plasmas 1998, 5, 2120–2126. [Google Scholar] [CrossRef]
- Puech, J.; Lapierre, L.; Sombrin, J.; Semenov, V.; Sazontov, A.; Buyanova, M.; Vdovicheva, N.; Jordan, U.; Udiljak, R.; Anderson, D.; et al. A multipactor threshold in waveguides: Theory and experiment. In Quasi-Optical Control of Intense Microwave Transmission; NATO Science Series II: Mathematics, Physics, and Chemistry; Hirshfield, J.L., Petelin, M.I., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 203. [Google Scholar]
- Hillairet, J.; Goniche, M.; Fil, N.; Belhaj, M.; Puech, J. Multipactor in High Power Radio-Frequency Systems for Nuclear Fusion. In Proceedings of the International Workshop on Multipactor, Corona and Passive Intermodulation, MULCOPIM 2017, Noordwijk, The Netherlands, 5–7 April 2017. [Google Scholar]
- Skripka, G.; Iadarola, G.; Mether, L.; Rumolo, G. Non-monotonic dependence of heat loads induced by electron cloud on bunch population at the LHC. Eur. Phys. J. Plus 2022, 137, 849. [Google Scholar] [CrossRef]
- Vallgren, C.Y.; Arduini, G.; Bauche, J.; Calatroni, S.; Chiggiato, P.; Cornelis, K.; Pinto, P.C.; Henrist, B.; Métral, E.; Neupert, H.; et al. Amorphous carbon coatings for the mitigation of electron cloud in the CERN super proton synchrotron. Phys. Rev. Spec. Top.-Accel. Beams 2011, 14, 071001. [Google Scholar] [CrossRef]
- Willeke, F.; Beebe-Wang, J. Electron Ion Collider Conceptual Design Report 2021; Brookhaven National Lab.: Upton, NY, USA; Thomas Jefferson National Accelerator Facility (TJNAF): Newport News, VA, USA, 2021. [Google Scholar] [CrossRef]
- Verdu-Andres, S.; Verdu-Andres, S.; Brennan, J.M.; Blaskiewicz, M.; Gu, X.; Gupta, A.; Hershcovitch, A.; Mapes, M.; McIntyre, G.; Muratore, J.; et al. A beam screen to prepare the RHIC vacuum chamber for EIC hadron beams: Conceptual design and requirements. In Proceedings of the 12th International Particle Accelerator Conference—IPAC’21, Campinas, Brazil, 24–28 May 2021; Available online: https://www.osti.gov/servlets/purl/1784486 (accessed on 1 July 2023).
- Egerton, R.F. Electron-Energy Loss Spectroscopy in the Electron Microscope, 3rd ed.Springer: New York, NY, USA, 2011. [Google Scholar]
- Ono, S.; Kanaya, K. The energy dependence of secondary emission based on the range-energy retardation power formula. J. Phys. D Appl. Phys. 1979, 12, 619. [Google Scholar] [CrossRef]
- Pinto, P.C.; Calatroni, S.; Chiggiato, P.; Edwards, P.; Mensi, M.; Neupert, H.; Taborelli, M.; Yin-Vallgren, C. Carbon coating of the SPS dipole chambers. Proceedings of ECLOUD’12, La Biodola, Isola d’Elba, Italy, 5–9 June 2012; pp. 141–148. [Google Scholar]
- Santos, A.; Bundaleski, N.; Shaw, B.J.; Silva, A.G.; Teodoro, O.M.N.D. Increase of Secondary Electron Yield of Amorphous Carbon Coatings under High Vacuum Conditions. Vacuum 2013, 98, 37–40. [Google Scholar] [CrossRef]
- Fernández, H.M.; Himmerlich, M.; Pinto, P.C.; Coroa, J.; Sousa, D.; Baris, A.; Taborelli, M. The impact of H2 and N2 on the material properties and secondary electron yield of sputtered amorphous carbon films for anti-multipacting applications. Appl. Surf. Sci. 2021, 542, 148552. [Google Scholar] [CrossRef]
- Adame, C.F.; Alves, E.; Barradas, N.P.; Pinto, P.C.; Delaup, Y.; Ferreira, I.M.M.; Neupert, H.; Himmerlich, M.; Pfeiffer, S.; Rimoldi, M.; et al. Amorphous carbon thin films: Mechanisms of hydrogen incorporation during magnetron sputtering and consequences for the secondary electron emission. J. Vac. Sci. Technol. A 2023, 41, 043412. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095. [Google Scholar] [CrossRef]
- Roy, D.; Samub, G.F.; Hossain, M.K.; Janáky, C.; Rajeshwar, K. On the measured optical bandgap values of inorganic oxide semiconductors for solar fuels generation. Catal. Today 2018, 300, 136–144. [Google Scholar] [CrossRef]
- Robertson, J. Diamond-like amorphous carbon. Mater. Sci. Eng. R Rep. 2002, 37, 129–281. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, X.; Wang, F. A ternary phase diagram for amorphous carbon. Carbon 2015, 94, 202–213. [Google Scholar] [CrossRef]
- Kaciulis, S. Spectroscopy of carbon: From diamond to nitride films. Surf. Interface Anal. 2012, 44, 1155–1161. [Google Scholar] [CrossRef]
- Mérel, P.; Tabbal, M.; Chaker, M.; Moisa, S.; Morgot, J. Direct evaluation of the sp3 content in diamond-like-carbon films by XPS. Appl. Surf. Sci. 1998, 136, 105–110. [Google Scholar] [CrossRef]
- Morgan, D.J. Comments on the XPS analysis of carbon materials. C 2021, 7, 51. [Google Scholar] [CrossRef]
- Biesinger, M.C. Accessing the robustness of adventitious carbon for charge referencing (correction) purposes in XPS analysis: Insights from a multi-user facility data review. Appl. Surf. Sci. 2022, 597, 153681. [Google Scholar] [CrossRef]
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers—The Scienta ESCA300 Database; Wiley Interscience: Hoboken, NJ, USA, 1992. [Google Scholar]
- Fairley, N.; Fernandez, V.; Richard-Pluet, M.; Guillot-Deudon, C.; Walton, J.; Smith, E.; Flahaut, D.; Greiner, M.; Biesinger, M.; Tougaard, S.; et al. Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy. Appl. Surf. Sci. Adv. 2021, 5, 100112. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater subshell photoionisation cross-sections at 1254 and 1487 eV. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Law, A.; Johnson, M.; Hughes, H. Synchrotron-radiation-excited angle-resolved photoemission from single-crystal graphite. Phys. Rev. B 1986, 34, 4289. [Google Scholar] [CrossRef] [PubMed]
- Ozfidan, I.; Karkusinski, M.; Hawrylak, P. Electronic properties and electron-electron interactions in graphene quantum dots. Phys. Status Solidi RRL 2016, 10, 13–23. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Philos. Trans. R. Soc. Lond. A 2004, 362, 2477–2512. [Google Scholar] [CrossRef]
- da Costa, M.E.H.M.; Freire, F.L., Jr. Deuterated amorphous carbon films: Film growth and properties. Surf. Coat. Technol. 2010, 204, 1993–1996. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Stauss, S.; Miyazoe, H.; Shizuno, T.; Suzuki, M.; Kataoka, H.; Sasaki, T.; Terashima, K. Pulsed Laser Ablation Synthesis of Diamond Molecules in Supercritical Fluids. Appl. Phys. Express 2010, 2, 096201. [Google Scholar] [CrossRef]
- Rusli; Robertson, J.; Amaratunga, G.A.J. Amaratunga, Photoluminescence behavior of hydrogenated amorphous carbon. J. Appl. Phys. 1996, 80, 2998–3003. [Google Scholar] [CrossRef]
- Merlen, A.; Buijnsters, J.G.; Pardanaud, C. A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: From Graphene to Amorphous Carbons. Coatings 2017, 7, 153. [Google Scholar] [CrossRef]
- Pardanaud, C.; Martin, C.; Roubin, P.; Giacometti, G.; Hopf, C.; Schwarz-Selinger, T.; Jacob, W. Raman spectroscopy investigation of the H content of heated hard amorphous layers. Diam. Relat. Mater. 2013, 34, 100–104. [Google Scholar] [CrossRef]
- Robertson, J. Photoluminescence Mechanism in amorphous hydrogenated carbon. Diam. Relat. Mater. 1996, 5, 457–460. [Google Scholar] [CrossRef]
- Lye, R.G.; Dekker, A.J. Theory of Secondary Emission. Phys. Rev. 1957, 107, 977–981. [Google Scholar] [CrossRef]
- He, Y.; Shen, T.; Wang, Q.; Miao, G.; Bai, C.; Yu, B.; Yang, J.; Feng, G.; Hu, T.; Wang, X.; et al. Effect of atmospheric exposure on secondary electron yield of inert metal and its potential impact on the threshold of multipactor effect. Appl. Surf. Sci. 2020, 520, 146320. [Google Scholar] [CrossRef]
- Lin, Y.; Joy, D.C. A new examination of secondary electron yield data. Surf. Interface Anal. 2005, 37, 895–900. [Google Scholar] [CrossRef]
- Bundaleski, N.; Belhaj, M.; Gineste, T.; Teodoro, O.M.N.D. Calculation of the angular dependence of the total electron yield. Vacuum 2015, 122, 255–259. [Google Scholar] [CrossRef]
- Ibach, H.; Mills, D.L. Electron Energy Loss Spectroscopy and Surface Vibrations; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Botelho do Rego, A.M.; Rei Vilar, M.; Lopes da Silva, J. Mechanisms of vibrational and electronic excitations of polystyrene films in high resolution electron energy loss spectroscopy. J. Electron Spectrosc. Relat. Phenom. 1997, 85, 81–91. [Google Scholar] [CrossRef]
- Rei Vilar, M.; Botelho do Rego, A.M.; Lopes da Silva, J.; Abel, F.; Quillet, V.; Schott, M.; Petitjean, S.; Jerome, R. Quantitative Analysis of Polymer Surfaces and Films Using Elastic Recoil Detection Analysis (ERDA), Fourier Transform Infrared Spectroscopy (FTIRS), and High-Resolution Electron Energy Loss Spectroscopy (HREELS). Macromolecules 1994, 27, 5900–5906. [Google Scholar] [CrossRef]
- Botelho do Rego, A.M.; Ferraria, A.M.; Rei Vilar, M. Grafting of Cobaltic Protoporphyrin IX on Semiconductors toward Sensing Devices: Vibrational and Electronic High-Resolution Electron Energy Loss Spectroscopy and X-ray Photoelectron Spectroscopy Study. J. Phys. Chem. C 2013, 117, 22298–22306. [Google Scholar] [CrossRef]
- Botelho do Rego, A.M.; da Silva, J.L.; Rei Vilar, M.; Voltz, R. Resonance mechanisms in electron energy loss spectra of stearic acid Langmuir-Blodgett monolayers revealed by isotopic effects. J. Electron Spectrosc. Relat. Phenom. 1998, 87, 221–230. [Google Scholar] [CrossRef]
- Diem, M. Modern Vibrational Spectroscopy and Micro-Spectroscopy, 2nd ed.; John Wiley&Sons Ltd.: Chichester, UK, 2015. [Google Scholar]
- National Institute of Standards and Technology. NIST Standard Reference Database SRD Number 69; National Institute of Standards and Technology: Gaithersburg, MD, USA. [CrossRef]
- Rei Vilar, M.; Schott, M.; Pireaux, J.J.; Grégoire, C.; Thiry, P.A.; Caudano, R.; Lapp, A.; Botelho do Rego, A.M.; Lopes da Silva, J. Study of Polymer Film Surfaces by EELS Using Selectively Deuterated Polystyrene. Surf. Sci. 1987, 189, 927–934. [Google Scholar] [CrossRef]
- Clerc, S.; Dennison, J.R.; Hoffmann, R.; Abbott, J. On the computation of secondary electron emission models. IEEE Trans. Plasma Sci. 2006, 34, 2219–2225. [Google Scholar] [CrossRef]
- Dionne, G.F. Origin of secondary electron emission yield curve parameters. J. Appl. Phys. 1975, 46, 3347–3351. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | pD2 (Pa) | C (%) | H (%) | D (%) | O (%) | SEYmax | ET (eV) | ρ (g/cm3) |
|---|---|---|---|---|---|---|---|---|
| Reference | 0 | 83.4 | 11.7 | 0 | 4.9 | 0.99 | 0.12 | 1.44 |
| 0.2D | 3.8 × 10−3 | 77.9 | 9.8 | 9.6 | 2.7 | 1.03 | 0.37 | 1.99 |
| 0.5D | 1.1 × 10−2 | 67.8 | 6.0 | 23.7 | 2.5 | 1.15 | 0.78 | 1.91 |
| 1D | 2.6 × 10−2 | 52.2 | 6.3 | 39.9 | 1.6 | 1.38 | 1.17 | 2.34 |
| 10D | 2.2 × 10−1 | 45.2 | 2.8 | 50.4 | 1.6 | 2.03 | 2.29 | 1.15 |
| Sample | α/β | Graphitic (%) | sp3 C-H (%) | C-O (%) | C=O (%) | -(C=O)-O- (%) | H + D Content (%) |
|---|---|---|---|---|---|---|---|
| Reference | 0.45 | 88.14 | 3.29 | 1.91 | 2.55 | 2.01 | 11.7 |
| 0.2D | 0.50 | 85.17 | 6.43 | 2.41 | 2.57 | 1.73 | 19.4 |
| 0.5D | 0.58 | 71.09 | 20.4 | 3.75 | 2.46 | 1.05 | 29.7 |
| 1D | 0.58 | 52.75 | 38.11 | 4.83 | 2.46 | 0.96 | 46.2 |
| 10D | 1.00 | 0 | 75.99 | 21.53 | 2.35 | 0.13 | 53.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bundaleski, N.; Adame, C.F.; Alves, E.; Barradas, N.P.; Cerqueira, M.F.; Deuermeier, J.; Delaup, Y.; Ferraria, A.M.; Ferreira, I.M.M.; Neupert, H.; et al. The Role of Hydrogen Incorporation into Amorphous Carbon Films in the Change of the Secondary Electron Yield. Int. J. Mol. Sci. 2023, 24, 12999. https://doi.org/10.3390/ijms241612999
Bundaleski N, Adame CF, Alves E, Barradas NP, Cerqueira MF, Deuermeier J, Delaup Y, Ferraria AM, Ferreira IMM, Neupert H, et al. The Role of Hydrogen Incorporation into Amorphous Carbon Films in the Change of the Secondary Electron Yield. International Journal of Molecular Sciences. 2023; 24(16):12999. https://doi.org/10.3390/ijms241612999
Chicago/Turabian StyleBundaleski, Nenad, Carolina F. Adame, Eduardo Alves, Nuno P. Barradas, Maria F. Cerqueira, Jonas Deuermeier, Yorick Delaup, Ana M. Ferraria, Isabel M. M. Ferreira, Holger Neupert, and et al. 2023. "The Role of Hydrogen Incorporation into Amorphous Carbon Films in the Change of the Secondary Electron Yield" International Journal of Molecular Sciences 24, no. 16: 12999. https://doi.org/10.3390/ijms241612999
APA StyleBundaleski, N., Adame, C. F., Alves, E., Barradas, N. P., Cerqueira, M. F., Deuermeier, J., Delaup, Y., Ferraria, A. M., Ferreira, I. M. M., Neupert, H., Himmerlich, M., Rego, A. M. M. B. d., Rimoldi, M., Teodoro, O. M. N. D., Vasilevskiy, M., & Costa Pinto, P. (2023). The Role of Hydrogen Incorporation into Amorphous Carbon Films in the Change of the Secondary Electron Yield. International Journal of Molecular Sciences, 24(16), 12999. https://doi.org/10.3390/ijms241612999