

Effect of Siponimod on Brain and Spinal Cord Imaging Markers of Neurodegeneration in the Theiler’s Murine Encephalomyelitis Virus Model of Demyelination
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
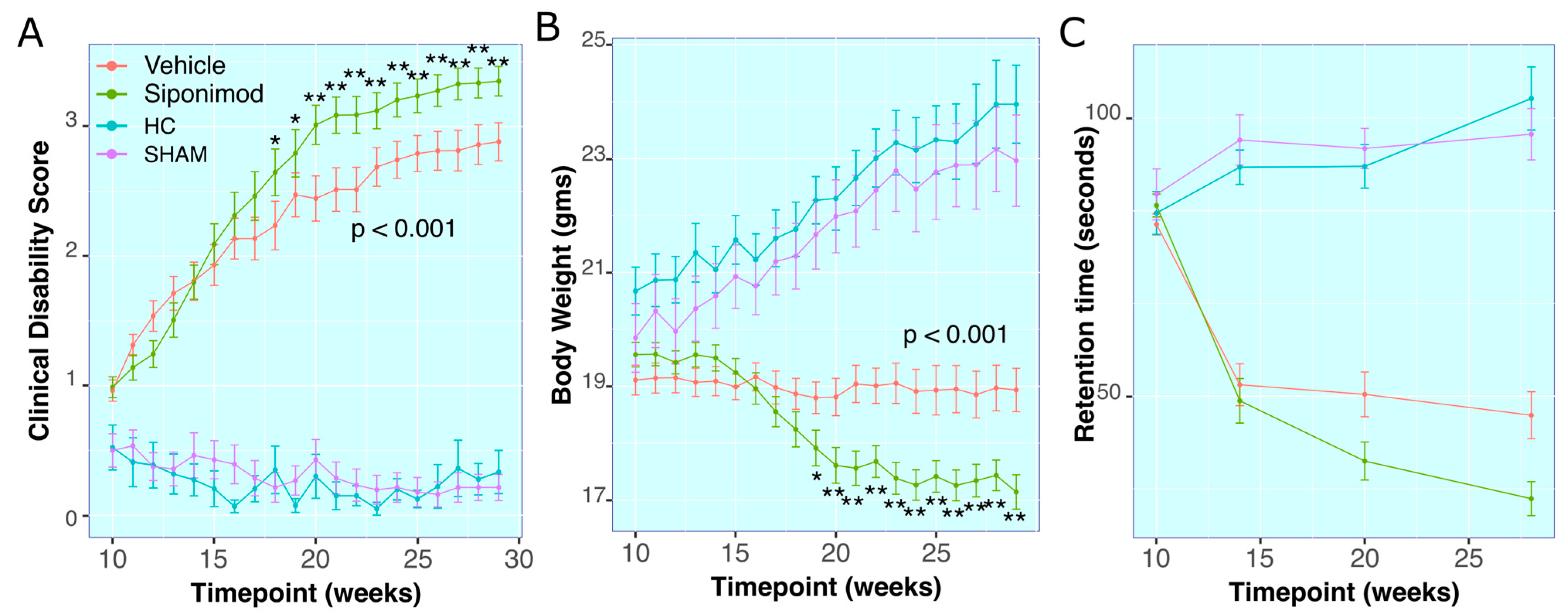
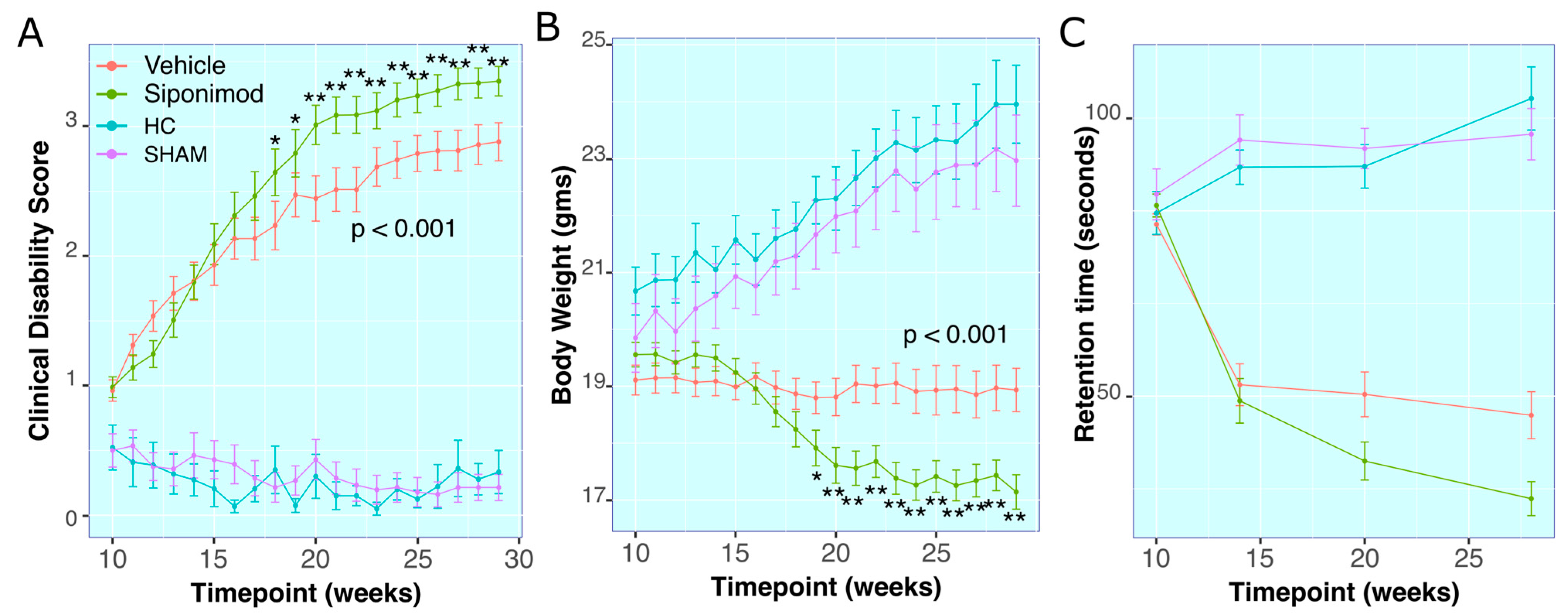
2.1. Siponimod Treatment Worsened Clinical Measures in TMEV-Induced Animals
Clinical Disability Score
2.2. Body Weight Measures
2.3. RotaRod Retention-Time
2.4. Effects of Siponimod Treatment on TMEV Disease-Induced Changes on Brain Region-Specific Volumes
2.5. Anterior Commissure
2.6. Hippocampus
2.7. Iso-Cortex
2.8. Thalamus
2.9. Striatum
2.10. Lateral Ventricles
2.11. Whole Brain
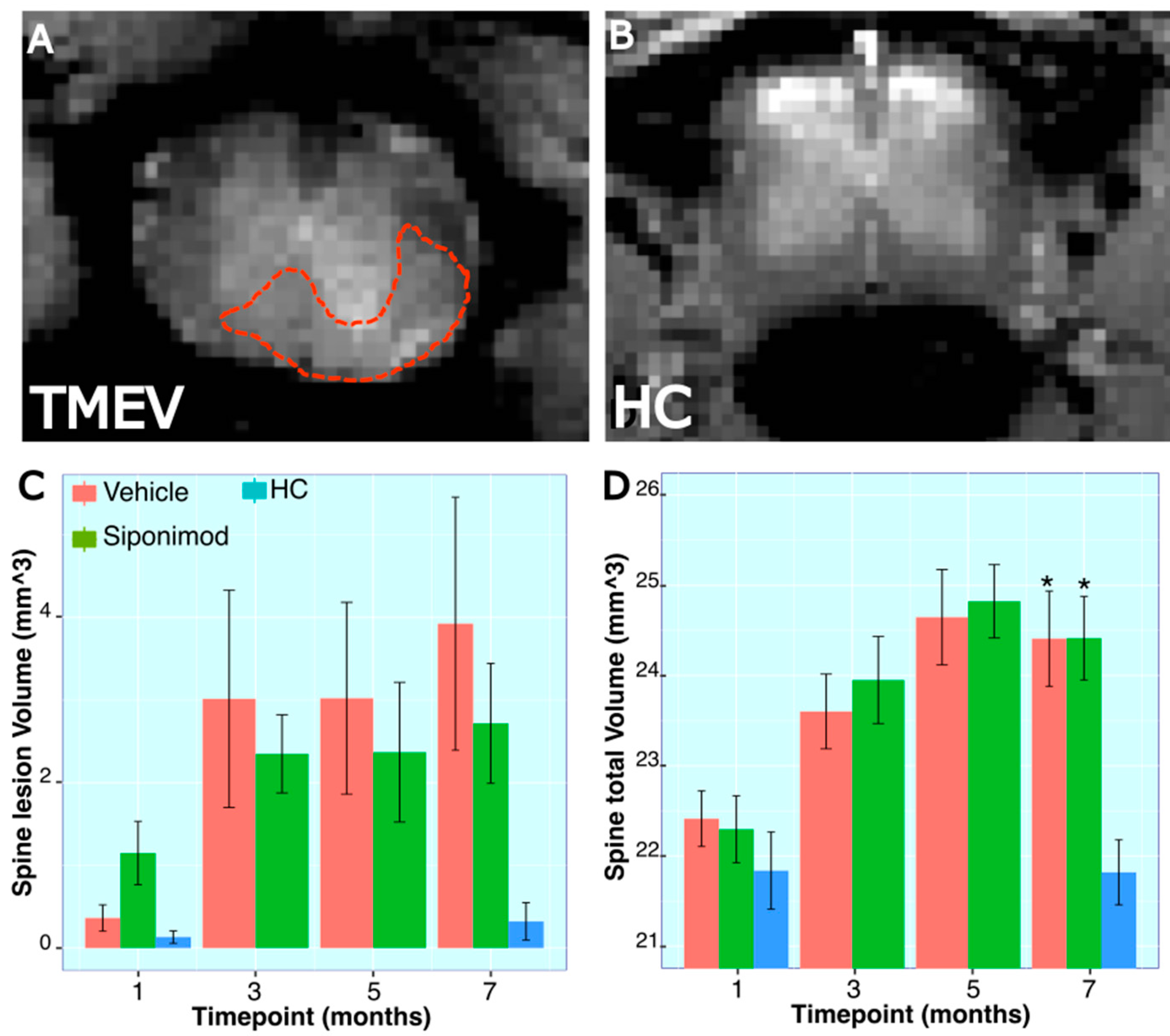
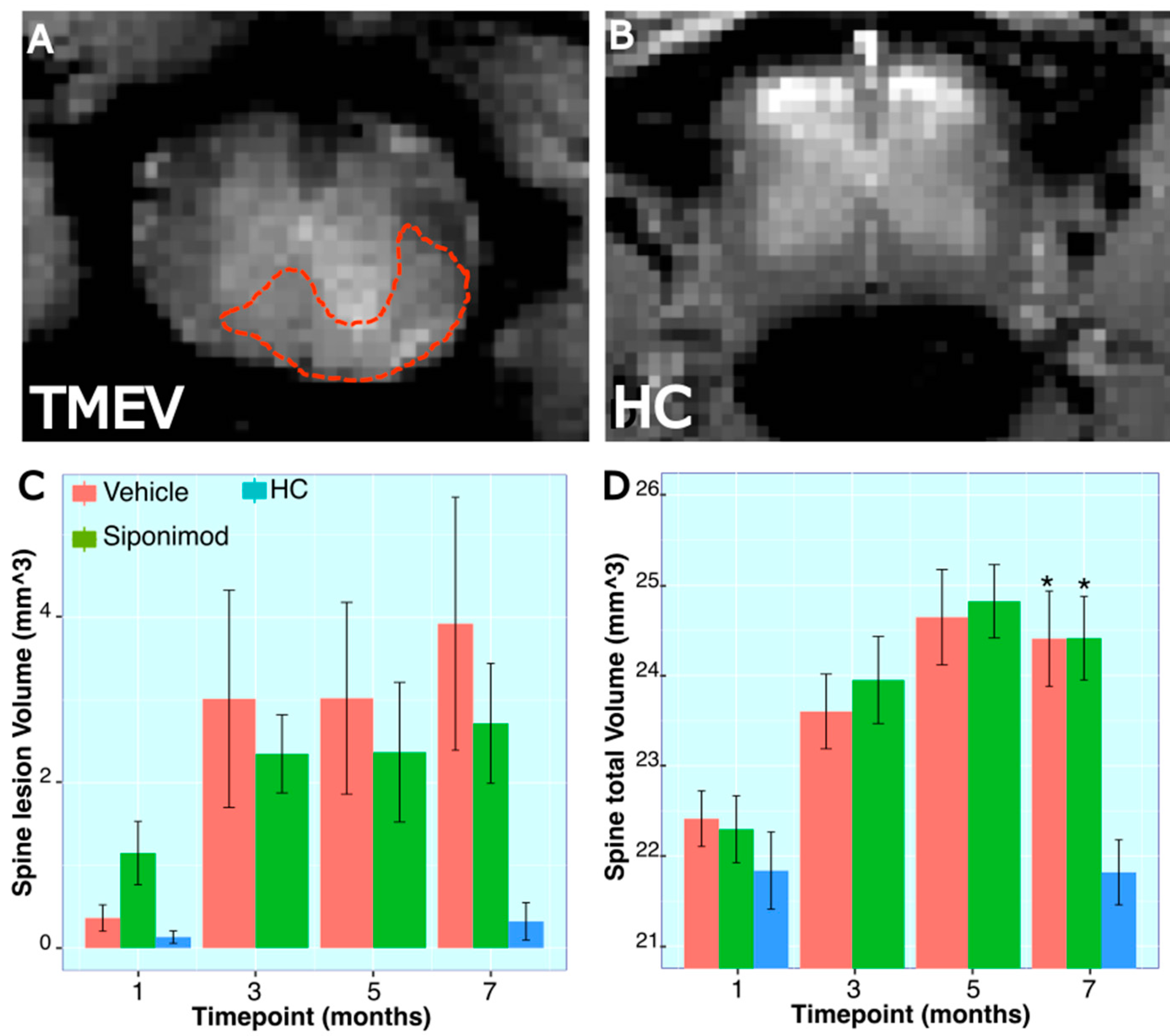
2.12. Longitudinal Changes in Spine Volume and Lesion Measures throughout the Disease’s Course
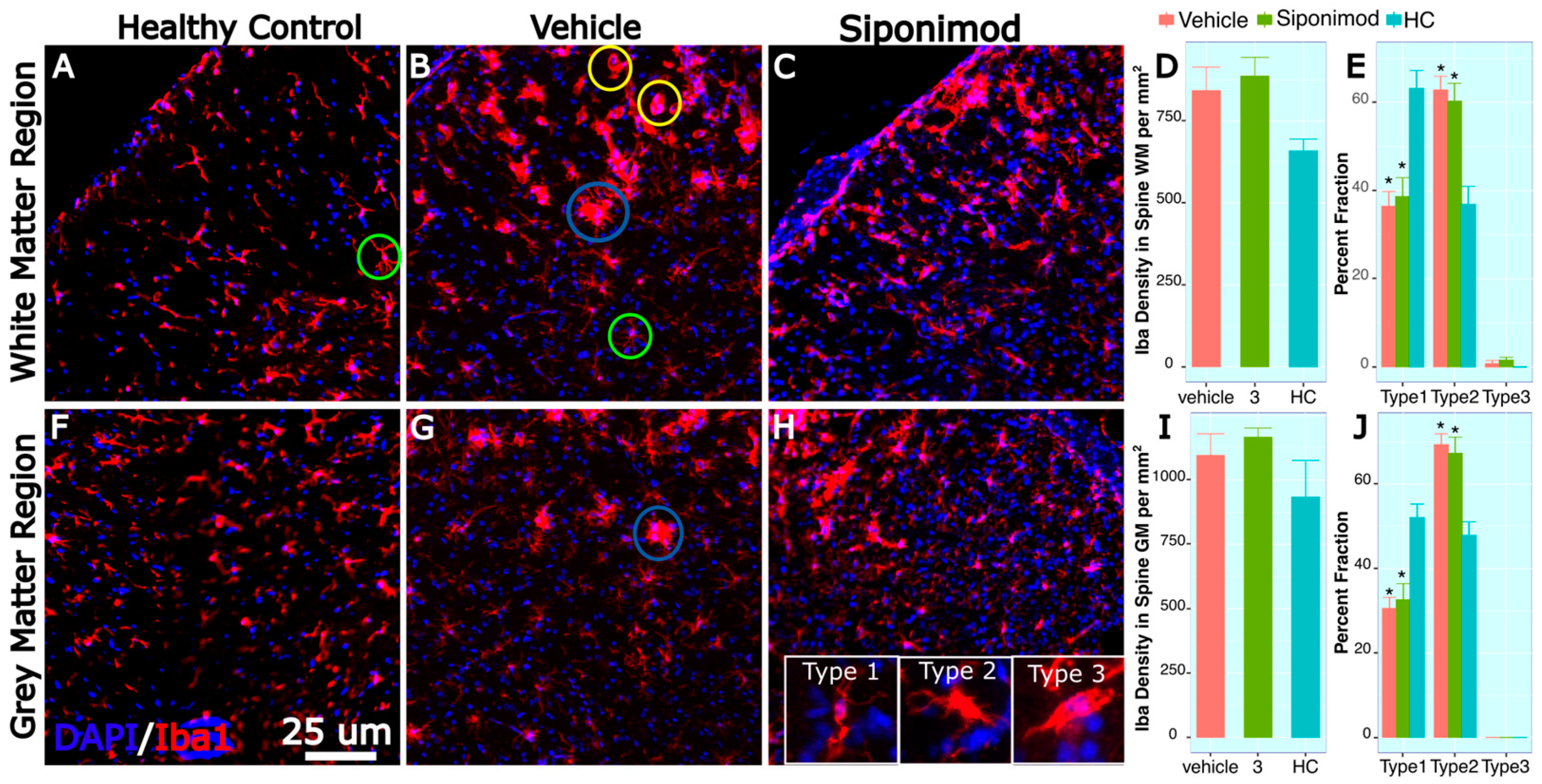
2.13. Microglia-Labeled Cell Density and Phenotype Changes in Response to Treatment
3. Discussion
3.1. SpT TMEV Animals Presented an Increased Disease Severity
3.2. SpT Has Limited Impact on Brain Region-Specific Volume Changes
3.3. Effect of Sp Treatment on TMEV-Induced Spine Volume Changes
3.4. Spine Histology
4. Materials and Methods
4.1. Subjects and Study Design
4.2. Induction of TMEV Infection
4.3. Treatment
4.4. Clinical Scoring
4.5. Rotarod
4.6. MRI Scanning
4.7. Brain Volume Determination
4.8. Spinal Cord Lesion and Volumetric Evaluation
4.9. Histological Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ganapathy Subramanian, R.; Zivadinov, R.; Bergsland, N.; Dwyer, M.G.; Weinstock-Guttman, B.; Jakimovski, D. Multiple sclerosis optic neuritis and trans-synaptic pathology on cortical thinning in people with multiple sclerosis. J. Neurol. 2023, 270, 3758–3769. [Google Scholar] [CrossRef]
- Jakimovski, D.; Vaughn, C.B.; Eckert, S.; Zivadinov, R.; Weinstock-Guttman, B. Long-term drug treatment in multiple sclerosis: Safety success and concerns. Expert Opin. Drug Saf. 2020, 19, 1121–1142. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Bleu, T.; Huang, W.; Hallmark, O.G.; Coughlin, S.R.; Goetzl, E.J. Identification of cDNAs encoding two G protein-coupled receptors for lysosphingolipids. FEBS Lett. 1997, 417, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.S.; Hinterding, K.; Cooke, N.G.; et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef]
- Van Doorn, R.; Van Horssen, J.; Verzijl, D.; Witte, M.; Ronken, E.; Van Het Hof, B.; Lakeman, K.; Dijkstra, C.D.; Van Der Valk, P.; Reijerkerk, A.; et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia 2010, 58, 1465–1476. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- McGavern, D.B.; Murray, P.D.; Rodriguez, M. Quantitation of spinal cord demyelination, remyelination, atrophy, and axonal loss in a model of progressive neurologic injury. J. Neurosci. Res. 1999, 58, 492–504. [Google Scholar] [CrossRef]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflamm. 2016, 13, 207. [Google Scholar] [CrossRef]
- McGinley, M.; Fox, R.J. Prospects of siponimod in secondary progressive multiple sclerosis. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418788013. [Google Scholar] [CrossRef]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Oleszak, E.L.; Chang, J.R.; Friedman, H.; Katsetos, C.D.; Platsoucas, C.D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004, 17, 174–207. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Sveinsson, M.; Sudyn, M.; Babek, N.; Siebert, D.; Bertolino, N.; Modica, C.M.; Preda, M.; Schweser, F.; Zivadinov, R. Teriflunomide’s Effect on Glia in Experimental Demyelinating Disease: A Neuroimaging and Histologic Study. J. Neuroimaging 2019, 29, 52–61. [Google Scholar] [CrossRef]
- Paz Soldan, M.M.; Raman, M.R.; Gamez, J.D.; Lohrey, A.K.; Chen, Y.; Pirko, I.; Johnson, A.J. Correlation of Brain Atrophy, Disability, and Spinal Cord Atrophy in a Murine Model of Multiple Sclerosis. J. Neuroimaging 2015, 25, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Modica, C.M.; Schweser, F.; Sudyn, M.L.; Bertolino, N.; Preda, M.; Polak, P.; Siebert, D.M.; Krawiecki, J.C.; Sveinsson, M.; Hagemeier, J.; et al. Effect of teriflunomide on cortex-basal ganglia-thalamus (CxBGTh) circuit glutamatergic dysregulation in the Theiler’s Murine Encephalomyelitis Virus mouse model of multiple sclerosis. PLoS ONE 2017, 12, e0182729. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.L.; Jin, F.; Pirko, I.; Johnson, A.J. Theiler’s murine encephalomyelitis virus as an experimental model system to study the mechanism of blood-brain barrier disruption. J. Neurovirol. 2014, 20, 107–112. [Google Scholar] [CrossRef]
- Pirko, I.; Johnson, A.; Chen, Y.; Lindquist, D.; Lohrey, A.; Ying, J.; Dunn, R.S. Brain atrophy correlates with functional outcome in a murine model of multiple sclerosis. Neuroimage 2011, 54, 802–806. [Google Scholar] [CrossRef]
- Althobity, A.A.; Khan, N.; Sandrock, C.J.; Woodruff, T.M.; Cowin, G.J.; Brereton, I.M.; Kurniawan, N.D. Multi-parametric MR for detection of pathological changes in CNS of mouse model of multiple sclerosis in vivo. NMR Biomed. 2023, e4964. [Google Scholar] [CrossRef]
- Lipton, H.L.; Melvold, R. Genetic analysis of susceptibility to Theiler’s virus-induced demyelinating disease in mice. J. Immunol. 1984, 132, 1821–1825. [Google Scholar] [CrossRef]
- Peterson, J.; Waltenbaugh, C.; Miller, S. IgG subclass responses to Theiler’s murine encephalomyelitis virus infection and immunization suggest a dominant role for Th1 cells in susceptible mouse strains. Immunology 1992, 75, 652. [Google Scholar]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J. Neuroimmune Pharmacol. 2010, 5, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Liang, S.; Schweser, F.; Dhanraj, R.; Schubart, A.; Preda, M.; Sveinsson, M.; Ramasamy, D.P.; Dwyer, M.G.; Weckbecker, G.; et al. Subcutaneous anti-CD20 antibody treatment delays gray matter atrophy in human myelin oligodendrocyte glycoprotein-induced EAE mice. Exp. Neurol. 2021, 335, 113488. [Google Scholar] [CrossRef]
- Modica, C.; Schweser, F.; Bertolino, N.; Rudra, T.; Sudyn, M.; Dwyer, M.; Zivadinov, R. Effect of Teriflunomide (Aubagio®) on cortico-basal ganglionic-thalamo-cortical gray matter connectivity in Mouse Model of Multiple Sclerosis. Neurology 2016, 86 (Suppl. S16), P5.312. [Google Scholar]
- Zivadinov, R.; Schweser, F.; Dwyer, M.G.; Pol, S. Detection of Monocyte/Macrophage and Microglia Activation in the TMEV Model of Chronic Demyelination Using USPIO-Enhanced Ultrahigh-Field Imaging. J. Neuroimaging 2020, 30, 769–778. [Google Scholar] [CrossRef]
- Smith, S.M.; Jenkinson, M.; Woolrich, M.W.; Beckmann, C.F.; Behrens, T.E.; Johansen-Berg, H.; Bannister, P.R.; De Luca, M.; Drobnjak, I.; Flitney, D.E.; et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 2004, 23 (Suppl. S1), S208–S219. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Miyamoto, O.; Shibuya, S.; Okada, M.; Igawa, H.; Janjua, N.A.; Norimatsu, H.; Itano, T. Different expression of macrophages and microglia in rat spinal cord contusion injury model at morphological and regional levels. Acta Med. Okayama 2005, 59, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Schweser, F.; Bertolino, N.; Preda, M.; Sveinsson, M.; Sudyn, M.; Babek, N.; Zivadinov, R. Characterization of leptomeningeal inflammation in rodent experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis. Exp. Neurol. 2019, 314, 82–90. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pol, S.; Dhanraj, R.; Taher, A.; Crever, M.; Charbonneau, T.; Schweser, F.; Dwyer, M.; Zivadinov, R. Effect of Siponimod on Brain and Spinal Cord Imaging Markers of Neurodegeneration in the Theiler’s Murine Encephalomyelitis Virus Model of Demyelination. Int. J. Mol. Sci. 2023, 24, 12990. https://doi.org/10.3390/ijms241612990
Pol S, Dhanraj R, Taher A, Crever M, Charbonneau T, Schweser F, Dwyer M, Zivadinov R. Effect of Siponimod on Brain and Spinal Cord Imaging Markers of Neurodegeneration in the Theiler’s Murine Encephalomyelitis Virus Model of Demyelination. International Journal of Molecular Sciences. 2023; 24(16):12990. https://doi.org/10.3390/ijms241612990
Chicago/Turabian StylePol, Suyog, Ravendra Dhanraj, Anissa Taher, Mateo Crever, Taylor Charbonneau, Ferdinand Schweser, Michael Dwyer, and Robert Zivadinov. 2023. "Effect of Siponimod on Brain and Spinal Cord Imaging Markers of Neurodegeneration in the Theiler’s Murine Encephalomyelitis Virus Model of Demyelination" International Journal of Molecular Sciences 24, no. 16: 12990. https://doi.org/10.3390/ijms241612990
APA StylePol, S., Dhanraj, R., Taher, A., Crever, M., Charbonneau, T., Schweser, F., Dwyer, M., & Zivadinov, R. (2023). Effect of Siponimod on Brain and Spinal Cord Imaging Markers of Neurodegeneration in the Theiler’s Murine Encephalomyelitis Virus Model of Demyelination. International Journal of Molecular Sciences, 24(16), 12990. https://doi.org/10.3390/ijms241612990