
Potential Role of Antibodies against Aquaporin-1 in Patients with Central Nervous System Demyelination

 , ,
, ,  ,
, 
Abstract
1. Introduction
1.1. Aquaporin Function, Structure, and Tissue Distribution
1.2. AQPs in Disease
1.3. Autoimmunity to AQPs
AQP4 Serum Antibodies in NMOSD Phenotype
2. AQP1
2.1. The Structural Biology of AQP1 Water Channel
2.2. The Role of AQP1 in the Human Body
2.3. Autoimmunity to AQP1 in Non-CNS Diseases
3. AQP1 Autoimmunity in NMOSD Phenotype
3.1. Assays for the Detection of Antibodies against AQP1
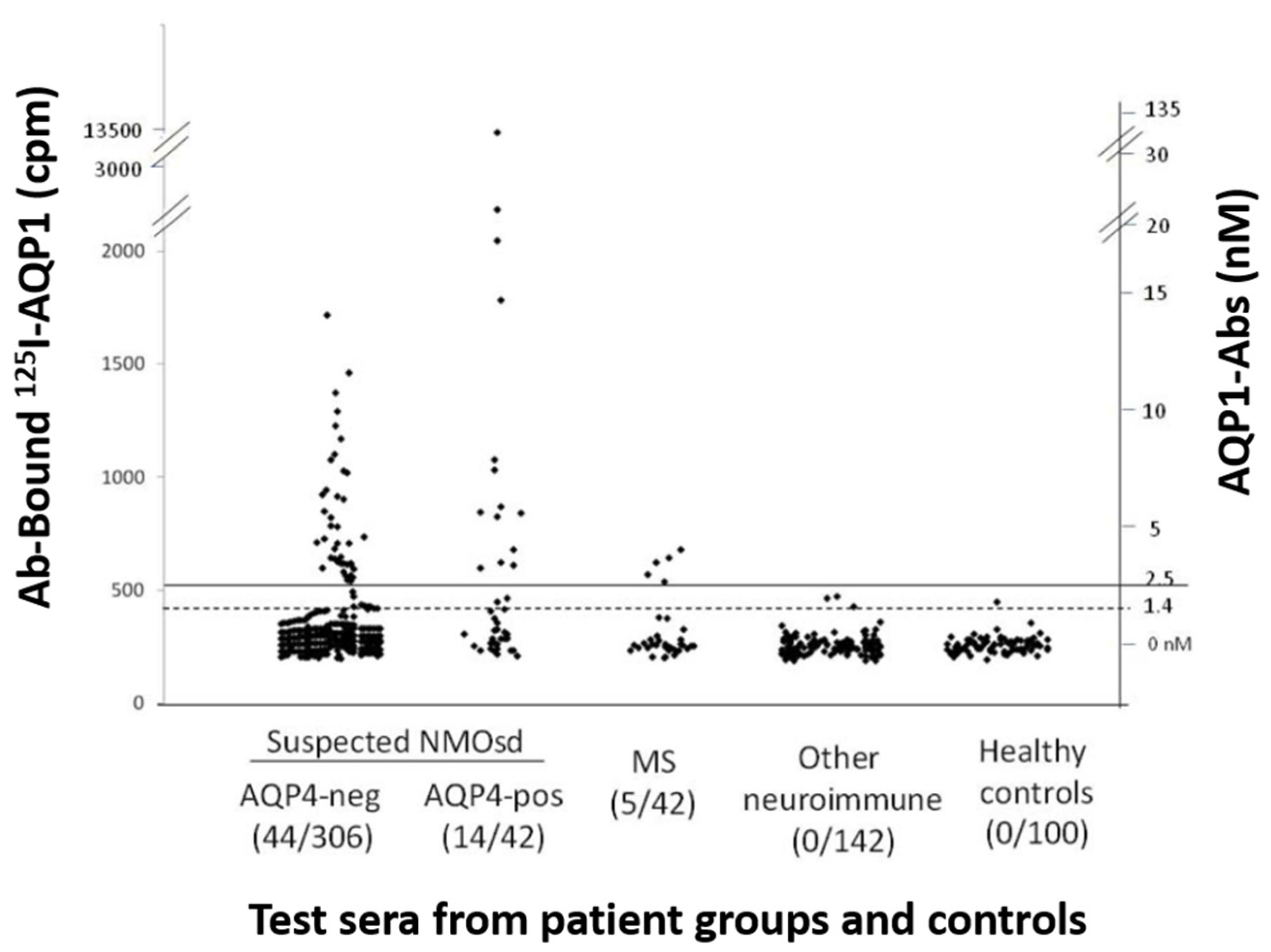
3.1.1. Assays for Antibody Binding to Cell-Free AQP1
- Radioimmunoprecipitation assay (RIPA)
- b.
- Western blotting with SDS-denatured AQP1 polypeptides
- c.
- ELISA with intact AQP1
- d.
- ELISA with AQP1 synthetic peptides (extracellular vs. intracellular location of the epitopes)
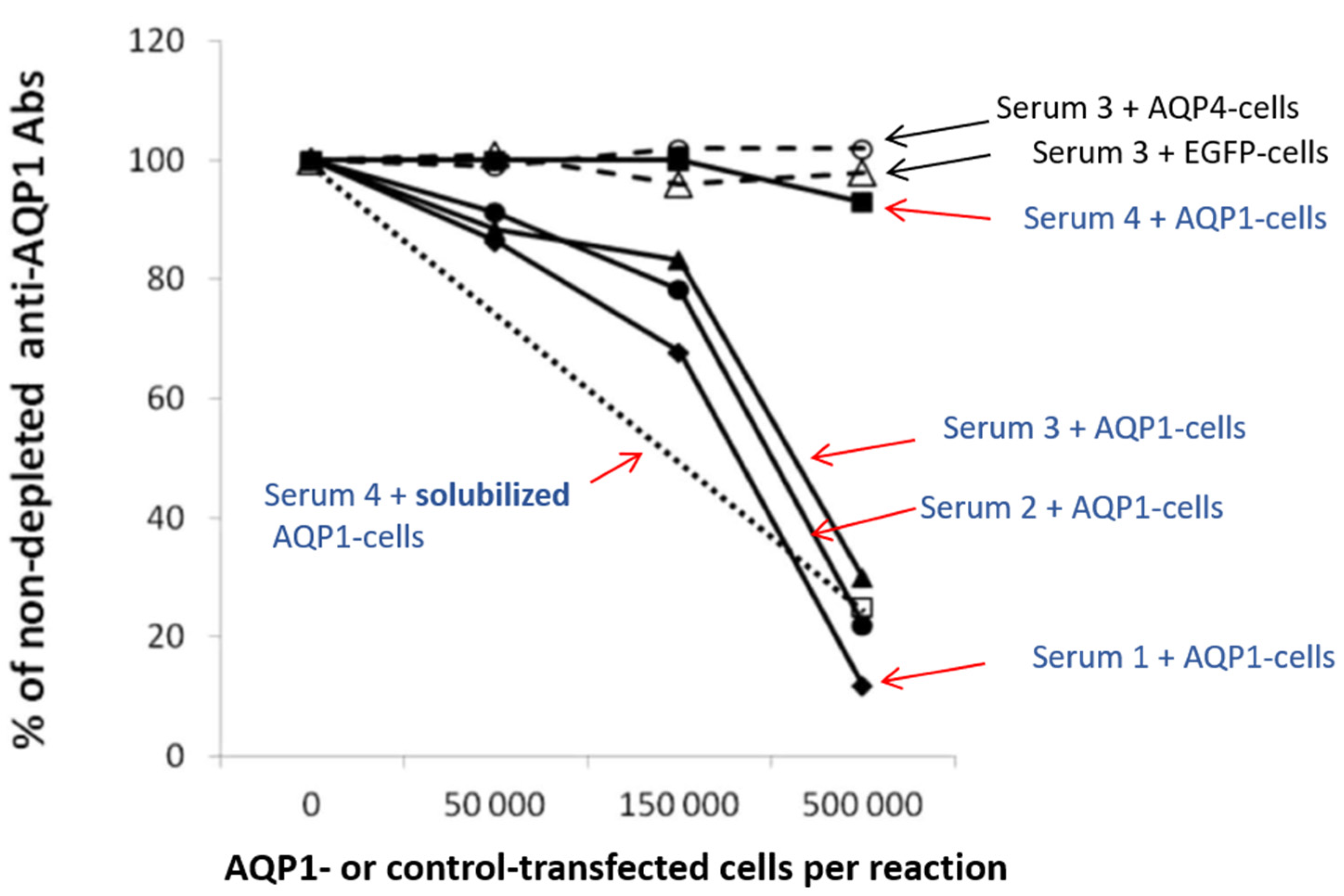
3.1.2. Assays for the Detection of the Autoantibody Binding to the Cell-Embedded AQP1
- Direct CBA
- b.
- Indirect CBA
3.1.3. Combination of Assays for the Detection of AQP1 Antibodies in NMOSD Phenotype
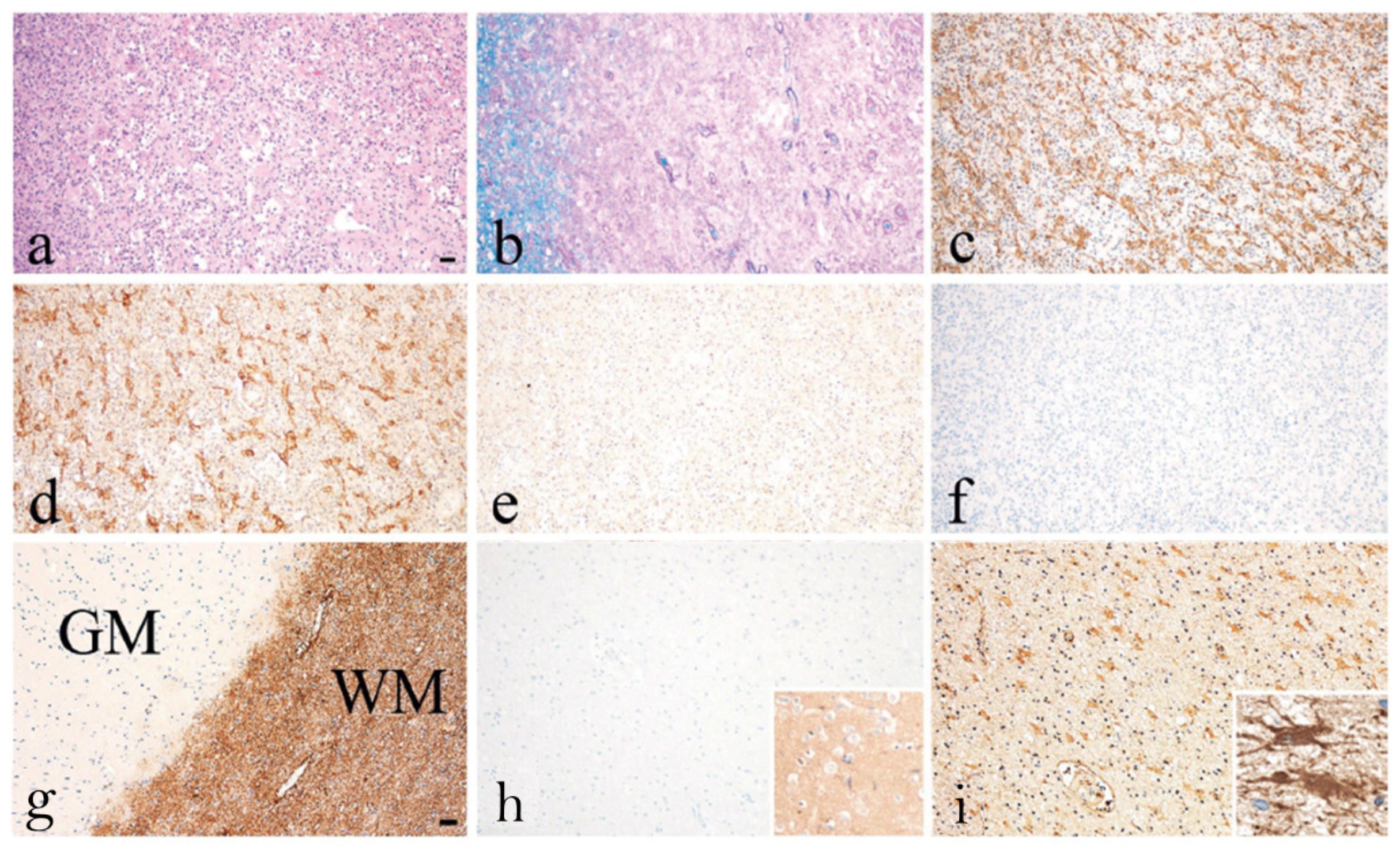
3.2. Are AQP1-Abs in NMOSD Pathogenic?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhan, J.; Cai, Q.; Xu, F.; Chai, R.; Lam, K.; Luan, Z.; Zhou, G.; Tsang, S.; Kipp, M.; et al. The Water Transport System in Astrocytes-Aquaporins. Cells 2022, 11, 2564. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xiao, M.; Li, S.; Yang, B. Aquaporins in Nervous System. Adv. Exp. Med. Biol. 2017, 969, 81–103. [Google Scholar] [CrossRef]
- Litman, T.; Sogaard, R.; Zeuthen, T. Ammonia and urea permeability of mammalian aquaporins. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 327–358. [Google Scholar] [CrossRef]
- Azad, A.K.; Raihan, T.; Ahmed, J.; Hakim, A.; Emon, T.H.; Chowdhury, P.A. Human Aquaporins: Functional Diversity and Potential Roles in Infectious and Non-infectious Diseases. Front. Genet. 2021, 12, 654865. [Google Scholar] [CrossRef] [PubMed]
- Filippidis, A.S.; Carozza, R.B.; Rekate, H.L. Aquaporins in Brain Edema and Neuropathological Conditions. Int. J. Mol. Sci. 2016, 18, 55. [Google Scholar] [CrossRef]
- Itoh, T.; Rai, T.; Kuwahara, M.; Ko, S.B.; Uchida, S.; Sasaki, S.; Ishibashi, K. Identification of a novel aquaporin, AQP12, expressed in pancreatic acinar cells. Biochem. Biophys. Res. Commun. 2005, 330, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Smith, B.L.; Christensen, E.I.; Agre, P. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc. Natl. Acad. Sci. USA 1993, 90, 7275–7279. [Google Scholar] [CrossRef]
- Oshio, K.; Watanabe, H.; Song, Y.; Verkman, A.S.; Manley, G.T. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2005, 19, 76–78. [Google Scholar] [CrossRef]
- Arcienega, I.I.; Brunet, J.F.; Bloch, J.; Badaut, J. Cell locations for AQP1, AQP4 and 9 in the non-human primate brain. Neuroscience 2010, 167, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Bichet, D.G.; Sasaki, S.; Kuwahara, M.; Arthus, M.F.; Lonergan, M.; Lin, Y.F. Two novel aquaporin-2 mutations responsible for congenital nephrogenic diabetes insipidus in Chinese families. J. Clin. Endocrinol. Metab. 2002, 87, 2694–2700. [Google Scholar] [CrossRef]
- Kavanagh, C.; Uy, N.S. Nephrogenic Diabetes Insipidus. Pediatr. Clin. N. Am. 2019, 66, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Sasaki, S. Updates and Perspectives on Aquaporin-2 and Water Balance Disorders. Int. J. Mol. Sci. 2021, 22, 12950. [Google Scholar] [CrossRef] [PubMed]
- Tradtrantip, L.; Jin, B.J.; Yao, X.; Anderson, M.O.; Verkman, A.S. Aquaporin-Targeted Therapeutics: State-of-the-Field. Adv. Exp. Med. Biol. 2017, 969, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Valsami, S.; Tzanetakos, D.; Stergiou, C.; Dandoulaki, M.; Barbarousi, D.; Psimenou, E.; Velonakis, G.; Stefanis, L.; Kilidireas, K. Autoimmune hemolytic anemia, demyelinating relapse, and AQP1 antibodies after alemtuzumab infusion. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Landegren, N.; Pourmousa Lindberg, M.; Skov, J.; Hallgren, A.; Eriksson, D.; Lisberg Toft-Bertelsen, T.; MacAulay, N.; Hagforsen, E.; Raisanen-Sokolowski, A.; Saha, H.; et al. Autoantibodies Targeting a Collecting Duct-Specific Water Channel in Tubulointerstitial Nephritis. J. Am. Soc. Nephrol. 2016, 27, 3220–3228. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Stergiou, C.; Daoussis, D.; Zisimopoulou, P.; Andonopoulos, A.P.; Zolota, V.; Tzartos, S.J. Antibodies to aquaporins are frequent in patients with primary Sjogren’s syndrome. Rheumatology 2017, 56, 2114–2122. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Aktas, O.; Ayzenberg, I.; Bellmann-Strobl, J.; Berthele, A.; Giglhuber, K.; Haussler, V.; Havla, J.; Hellwig, K.; Hummert, M.W.; et al. Update on the diagnosis and treatment of neuromyelits optica spectrum disorders (NMOSD)—Revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part I: Diagnosis and differential diagnosis. J. Neurol. 2023, 270, 3341–3368. [Google Scholar] [CrossRef]
- Guo, Y.; Lennon, V.A.; Parisi, J.E.; Popescu, B.; Vasquez, C.; Pittock, S.J.; Howe, C.L.; Lucchinetti, C.F. Spectrum of sublytic astrocytopathy in neuromyelitis optica. Brain 2022, 145, 1379–1390. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Veruki, M.L.; Torp, R.; Haug, F.M.; Laake, J.H.; Nielsen, S.; Agre, P.; Ottersen, O.P. Aquaporin-4 water channel protein in the rat retina and optic nerve: Polarized expression in Muller cells and fibrous astrocytes. J. Neurosci. 1998, 18, 2506–2519. [Google Scholar] [CrossRef]
- Trillo-Contreras, J.L.; Ramirez-Lorca, R.; Villadiego, J.; Echevarria, M. Cellular Distribution of Brain Aquaporins and Their Contribution to Cerebrospinal Fluid Homeostasis and Hydrocephalus. Biomolecules 2022, 12, 530. [Google Scholar] [CrossRef] [PubMed]
- Oshio, K.; Binder, D.K.; Yang, B.; Schecter, S.; Verkman, A.S.; Manley, G.T. Expression of aquaporin water channels in mouse spinal cord. Neuroscience 2004, 127, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Lasbennes, F.; Magistretti, P.J.; Regli, L. Aquaporins in brain: Distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 2002, 22, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Rosu, G.C.; Pirici, I.; Grigorie, A.A.; Istrate-Ofiteru, A.M.; Iovan, L.; Tudorica, V.; Pirici, D. Distribution of Aquaporins 1 and 4 in the Central Nervous System. Curr. Health Sci. J. 2019, 45, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Hogancamp, W.F.; O’Brien, P.C.; Weinshenker, B.G. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999, 53, 1107–1114. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Zekeridou, A.; Lennon, V.A. Aquaporin-4 autoimmunity. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e110. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sanchez, C.; Wingerchuk, D.M. Emerging Targeted Therapies for Neuromyelitis Optica Spectrum Disorders. BioDrugs 2021, 35, 7–17. [Google Scholar] [CrossRef]
- Satoh, J.; Tabunoki, H.; Yamamura, T.; Arima, K.; Konno, H. Human astrocytes express aquaporin-1 and aquaporin-4 in vitro and in vivo. Neuropathology 2007, 27, 245–256. [Google Scholar] [CrossRef]
- Weinshenker, B.G.; Wingerchuk, D.M.; Pittock, S.J.; Lucchinetti, C.F.; Lennon, V.A. NMO-IgG: A specific biomarker for neuromyelitis optica. Dis. Markers 2006, 22, 197–206. [Google Scholar] [CrossRef]
- Misu, T.; Hoftberger, R.; Fujihara, K.; Wimmer, I.; Takai, Y.; Nishiyama, S.; Nakashima, I.; Konno, H.; Bradl, M.; Garzuly, F.; et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013, 125, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Walz, T.; Hirai, T.; Murata, K.; Heymann, J.B.; Mitsuoka, K.; Fujiyoshi, Y.; Smith, B.L.; Agre, P.; Engel, A. The three-dimensional structure of aquaporin-1. Nature 1997, 387, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ricciardelli, C.; Yool, A.J. Targeting Aquaporins in Novel Therapies for Male and Female Breast and Reproductive Cancers. Cells 2021, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yool, A.J.; Schulten, K.; Tajkhorshid, E. Mechanism of gating and ion conductivity of a possible tetrameric pore in aquaporin-1. Structure 2006, 14, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Seyahian, E.A.; Cacciagiu, L.; Damiano, A.E.; Zotta, E. AQP1 expression in the proximal tubule of diabetic rat kidney. Heliyon 2020, 6, e03192. [Google Scholar] [CrossRef]
- Morelle, J.; Marechal, C.; Yu, Z.; Debaix, H.; Corre, T.; Lambie, M.; Verduijn, M.; Dekker, F.; Bovy, P.; Evenepoel, P.; et al. AQP1 Promoter Variant, Water Transport, and Outcomes in Peritoneal Dialysis. N. Engl. J. Med. 2021, 385, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Sugie, J.; Intaglietta, M.; Sung, L.A. Water transport and homeostasis as a major function of erythrocytes. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1098–H1107. [Google Scholar] [CrossRef]
- Endo, M.; Jain, R.K.; Witwer, B.; Brown, D. Water channel (aquaporin 1) expression and distribution in mammary carcinomas and glioblastomas. Microvasc. Res. 1999, 58, 89–98. [Google Scholar] [CrossRef]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Maltaneri, R.E.; Schiappacasse, A.; Chamorro, M.E.; Nesse, A.B.; Vittori, D.C. Aquaporin-1 plays a key role in erythropoietin-induced endothelial cell migration. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118569. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Takahashi, N.; Kurata, N.; Yamaguchi, A.; Matsui, H.; Kato, S.; Takeuchi, K. Involvement of aquaporin-1 in gastric epithelial cell migration during wound repair. Biochem. Biophys. Res. Commun. 2009, 386, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Verkman, A.S. Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J. Am. Soc. Nephrol. 2006, 17, 39–45. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Saadoun, S.; Verkman, A.S. Aquaporins and cell migration. Pflug. Arch. 2008, 456, 693–700. [Google Scholar] [CrossRef]
- Verkman, A.S.; Mitra, A.K. Structure and function of aquaporin water channels. Am. J. Physiol. Ren. Physiol. 2000, 278, F13–F28. [Google Scholar] [CrossRef] [PubMed]
- Trillo-Contreras, J.L.; Toledo-Aral, J.J.; Echevarria, M.; Villadiego, J. AQP1 and AQP4 Contribution to Cerebrospinal Fluid Homeostasis. Cells 2019, 8, 197. [Google Scholar] [CrossRef]
- Turkoglu, R.; Lassmann, H.; Aker, F.V.; Tzartos, J.; Tzartos, S.; Tuzun, E. Recurrent tumefactive demyelinating lesions: A pathological study. Clin. Neuropathol. 2017, 36, 195–198. [Google Scholar] [CrossRef]
- Shields, S.D.; Mazario, J.; Skinner, K.; Basbaum, A.I. Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain 2007, 131, 8–20. [Google Scholar] [CrossRef]
- Halverson, G.R.; Peyrard, T. A review of the Colton blood group system. Immunohematology 2010, 26, 22–26. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20795314 (accessed on 13 May 2023). [CrossRef]
- Covin, R.B.; Evans, K.S.; Olshock, R.; Thompson, H.W. Acute hemolytic transfusion reaction caused by anti-Coa. Immunohematology 2001, 17, 45–49. Available online: https://www.ncbi.nlm.nih.gov/pubmed/15373591 (accessed on 11 May 2023). [CrossRef] [PubMed]
- Alam, J.; Koh, J.H.; Kim, N.; Kwok, S.K.; Park, S.H.; Song, Y.W.; Park, K.; Choi, Y. Detection of autoantibodies against aquaporin-5 in the sera of patients with primary Sjogren’s syndrome. Immunol. Res. 2016, 64, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Choi, Y.S.; Koh, J.H.; Kwok, S.K.; Park, S.H.; Song, Y.W.; Park, K.; Choi, Y. Detection of Autoantibodies against Aquaporin-1 in the Sera of Patients with Primary Sjogren’s Syndrome. Immune Netw. 2017, 17, 103–109. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Stergiou, C.; Kilidireas, K.; Zisimopoulou, P.; Thomaidis, T.; Tzartos, S.J. Anti-aquaporin-1 autoantibodies in patients with neuromyelitis optica spectrum disorders. PLoS ONE 2013, 8, e74773. [Google Scholar] [CrossRef]
- Schanda, K.; Waters, P.; Holzer, H.; Aboulenein-Djamshidian, F.; Leite, M.I.; Palace, J.; Vukusic, S.; Marignier, R.; Berger, T.; Reindl, M. Antibodies to aquaporin-1 are not present in neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e160. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Gomar, I.; Diaz Sanchez, M.; Ucles Sanchez, A.J.; Casado Chocan, J.L.; Suarez-Luna, N.; Ramirez-Lorca, R.; Villadiego, J.; Toledo-Aral, J.J.; Echevarria, M. Comparative Analysis for the Presence of IgG Anti-Aquaporin-1 in Patients with NMO-Spectrum Disorders. Int. J. Mol. Sci. 2016, 17, 1195. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zheng, Y.; Shan, F.; Chen, M.; Fan, Y.; Zhang, B.; Gao, C.; Gao, Q.; Yang, N. Development of a cell-based assay for the detection of anti-aquaporin 1 antibodies in neuromyelitis optica spectrum disorders. J. Neuroimmunol. 2014, 273, 103–110. [Google Scholar] [CrossRef]
- Jasiak-Zatonska, M.; Michalak, S.; Osztynowicz, K.; Kozubski, W.; Kalinowska-Lyszczarz, A. Relationship between blood-brain permeability and antibodies against aquaporins in neuromyelitis optica spectrum disorders and multiple sclerosis patients. Neurol. Neurochir. Pol. 2022, 56, 308–317. [Google Scholar] [CrossRef]
- Vincent, A.; Waters, P.; Leite, M.I.; Jacobson, L.; Koneczny, I.; Cossins, J.; Beeson, D. Antibodies identified by cell-based assays in myasthenia gravis and associated diseases. Ann. N. Y. Acad. Sci. 2012, 1274, 92–98. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Iadarola, M.J.; Keller, J.M.; Warner, B.M. Autoantibodies Targeting Intracellular and Extracellular Proteins in Autoimmunity. Front. Immunol. 2021, 12, 548469. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of Autoantibody-Induced Pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Stergiou, C.; Zisimopoulou, P.; Kilidireas, C.; Tzartos, S.J. Antibodies to Surface Aquaporin-1 Are Present in NMO but Low AQP1 Expression Prohibits Use of CBA; Neurol. Neuroim. Neuroinfl 2016. Available online: https://nn.neurology.org/content/antibodies-surface-aquaporin-1-are-present-nmo-low-aqp1-expression-prohibits-use-cba (accessed on 27 June 2023).
- Siritho, S.; Nakashima, I.; Takahashi, T.; Fujihara, K.; Prayoonwiwat, N. AQP4 antibody-positive Thai cases: Clinical features and diagnostic problems. Neurology 2011, 77, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Lennon, V.A. Aquaporin-4 autoantibodies in a paraneoplastic context. Arch. Neurol. 2008, 65, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.A.; Romere, C.M.; Nguyen, L.; Saksena, A.; Abdullah, S.J.; Diaz, A.E. Severe Coombs Positive Autoimmune Hemolytic Anemia after Alemtuzumab Infusion for Relapsing Remitting Multiple Sclerosis. What Can We Learn? Blood 2018, 132, 2331. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010, 133, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Dolman, D.; Drndarski, S.; Abbott, N.J.; Rattray, M. Induction of aquaporin 1 but not aquaporin 4 messenger RNA in rat primary brain microvessel endothelial cells in culture. J. Neurochem. 2005, 93, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tan, M.; Gu, M.; Marshall, C.; Ding, J.; Hu, G.; Xiao, M. Cellular localization of aquaporin-1 in the human and mouse trigeminal systems. PLoS ONE 2012, 7, e46379. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Mde, C.; Hirt, L.; Bogousslavsky, J.; Regli, L.; Badaut, J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J. Neurosci. Res. 2006, 83, 1231–1240. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AQP | Function | Tissue Distribution |
|---|---|---|
| QP0 | Also known as MIP, major intrinsic protein forms water channels in lens fiber cells and contributes to lens transparency | Lens fiber cells |
| AQP1 | Water transport | Kidney (proximal tubule, thin descending limb of Henle’s loop), Brain (white matter astrocytes, choroid plexus epithelial cells), spinal cord (dorsal horns), optic nerve (corneal endothelium, keratocytes and the ciliary epithelium), inner ear (fibrocytes of the spiral ligament), red blood cells (AQP1 with Colton blood group antigens), lung (alveolar epithelium), and vascular endothelium |
| AQP2 | Water reabsorption | Kidney (principal cells of the collecting ducts) |
| AQP3 | Skin hydration and water transport | Kidney (proximal tubule), skin (basal and supra-basal layers), gastrointestinal tract, and other tissues |
| AQP4 | Water movement across the blood–brain barrier and plays a role in brain edema and water homeostasis | Brain (astrocyte end-feet at blood–brain barrier), optic nerve (retinal glia), spinal cord, skeletal muscle, and other tissues |
| AQP5 | Water transport | Salivary, lacrimal, and other exocrine glands, respiratory tract submucosal glands |
| AQP6 | Possible role in acid–base homeostasis in the kidney. | Kidney (collecting ducts, intercalated cells) |
| AQP7 | Glycerol transport | Adipose tissue, liver, kidney (proximal tubule), and other tissues |
| AQP8 | Transport of water, urea, and other small solutes | Kidney (proximal tubule, thin descending limb of Henle’s loop), liver, and other tissues |
| AQP9 | Transport of water, urea, and other small solutes | Liver (hepatocytes), kidney (proximal tubule), testis, and other tissues |
| AQP10 | Water and solute transport in the gastrointestinal tract and kidney | Tissue distribution: Intestine (colon, ileum, duodenum), kidney (proximal tubule), liver, and other tissues |
| AQP11 | Glycerol channel activity and water channel activity, transport hydrogen peroxide | In several organs such as liver, kidney, and brain |
| AQP12 | Digestive enzyme secretion such as maturation and exocytosis of secretory granules | Pancreatic acinar cells |
| Patient a | Sex | Epitopes b | Specific Binding on Live AQP1-Cells (“Indirect CBA”) c | MRI & Clinical Data (& Neoplasms) |
|---|---|---|---|---|
| 1 | F | Cytopl | ΝΤ | LETM and optic neuritis (NMO) |
| 2 | M | NT | Few antibodies (5%) | LETM and optic neuritis (NMO) |
| 3 | F | Extr-C | Most antibodies | LETM and optic neuritis (NMO) |
| 4 | F | Extr-A | Most antibodies | LETM and optic neuritis (NMO) |
| 5 | F | Extr-A | Most antibodies | LETM and optic neuritis (NMO) |
| 6 | F | Cytopl | NT | LETM |
| 7 | F | Cytopl | Few antibodies (16%) | LETM |
| 8 | F | Extr-E | Some antibodies (30%) | LETM |
| 9 | M | Extr-A | Most antibodies | LETM |
| 10 | M | Extr-A | Most antibodies | LETM |
| 11 | F | Extr-A | Most antibodies | LETM |
| 12 | F | Extr-A | Few antibodies (16%) | LETM |
| 13 | F | Extr-A | Most antibodies | LETM |
| 14 | F | Extr-A | Most antibodies | LETM |
| 15 | M | Extr & Cytopl | Few antibodies (19%) | LETM & Hodgkin lymphoma |
| 16 | M | Extr-A | Most antibodies | LETM & brain lesions (fulfilled Barkhof criteria) |
| 17 | M | Extr-A | Most antibodies | Transverse myelitis & kidney neoplasm |
| 18 | F | Extr-E | Most antibodies | MS & predominant spinal cord lesions |
| 19 | F | Extr-A | Most antibodies | MS & predominant spinal cord lesions & breast cancer |
| 20 | F | Cytopl | No antibodies | MS |
| 21 | F | Cytopl | No antibodies | MS |
| 22 | F | Cytopl | No antibodies | MS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pechlivanidou, M.; Xenou, K.; Tzanetakos, D.; Koutsos, E.; Stergiou, C.; Andreadou, E.; Voumvourakis, K.; Giannopoulos, S.; Kilidireas, C.; Tüzün, E.; et al. Potential Role of Antibodies against Aquaporin-1 in Patients with Central Nervous System Demyelination. Int. J. Mol. Sci. 2023, 24, 12982. https://doi.org/10.3390/ijms241612982
Pechlivanidou M, Xenou K, Tzanetakos D, Koutsos E, Stergiou C, Andreadou E, Voumvourakis K, Giannopoulos S, Kilidireas C, Tüzün E, et al. Potential Role of Antibodies against Aquaporin-1 in Patients with Central Nervous System Demyelination. International Journal of Molecular Sciences. 2023; 24(16):12982. https://doi.org/10.3390/ijms241612982
Chicago/Turabian StylePechlivanidou, Maria, Konstantina Xenou, Dimitrios Tzanetakos, Emmanuel Koutsos, Christos Stergiou, Elisabeth Andreadou, Konstantinos Voumvourakis, Sotirios Giannopoulos, Constantinos Kilidireas, Erdem Tüzün, and et al. 2023. "Potential Role of Antibodies against Aquaporin-1 in Patients with Central Nervous System Demyelination" International Journal of Molecular Sciences 24, no. 16: 12982. https://doi.org/10.3390/ijms241612982
APA StylePechlivanidou, M., Xenou, K., Tzanetakos, D., Koutsos, E., Stergiou, C., Andreadou, E., Voumvourakis, K., Giannopoulos, S., Kilidireas, C., Tüzün, E., Tsivgoulis, G., Tzartos, S., & Tzartos, J. (2023). Potential Role of Antibodies against Aquaporin-1 in Patients with Central Nervous System Demyelination. International Journal of Molecular Sciences, 24(16), 12982. https://doi.org/10.3390/ijms241612982