
Structure-Based Modeling of Sigma 1 Receptor Interactions with Ligands and Cholesterol and Implications for Its Biological Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
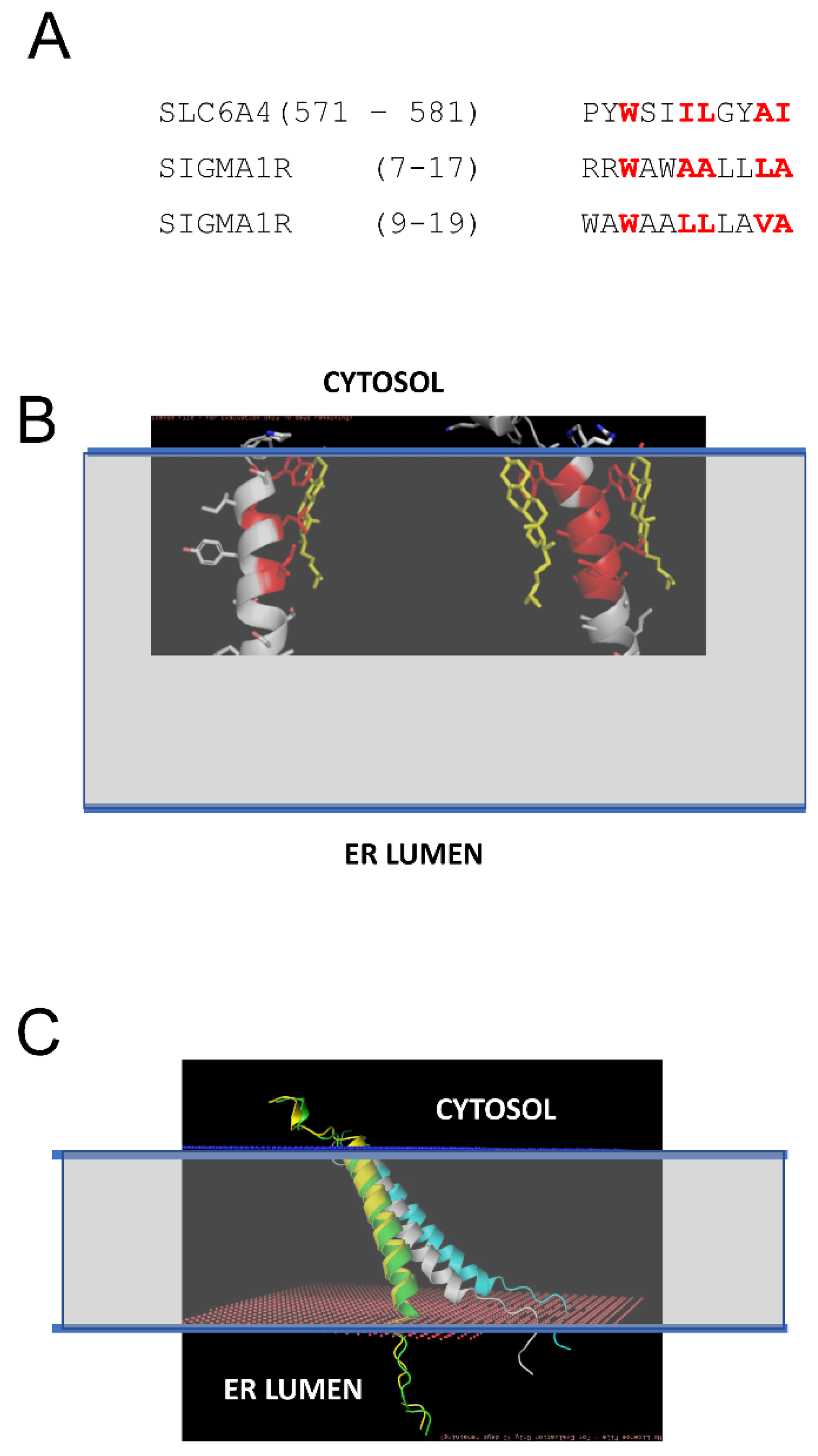
2.1. Co-Existence of Two Conformations of Sigma 1 Receptor BIND Domain
2.2. Effects of Association with Cholesterol on Sigma 1 Receptor H1 α-Helix Conformation
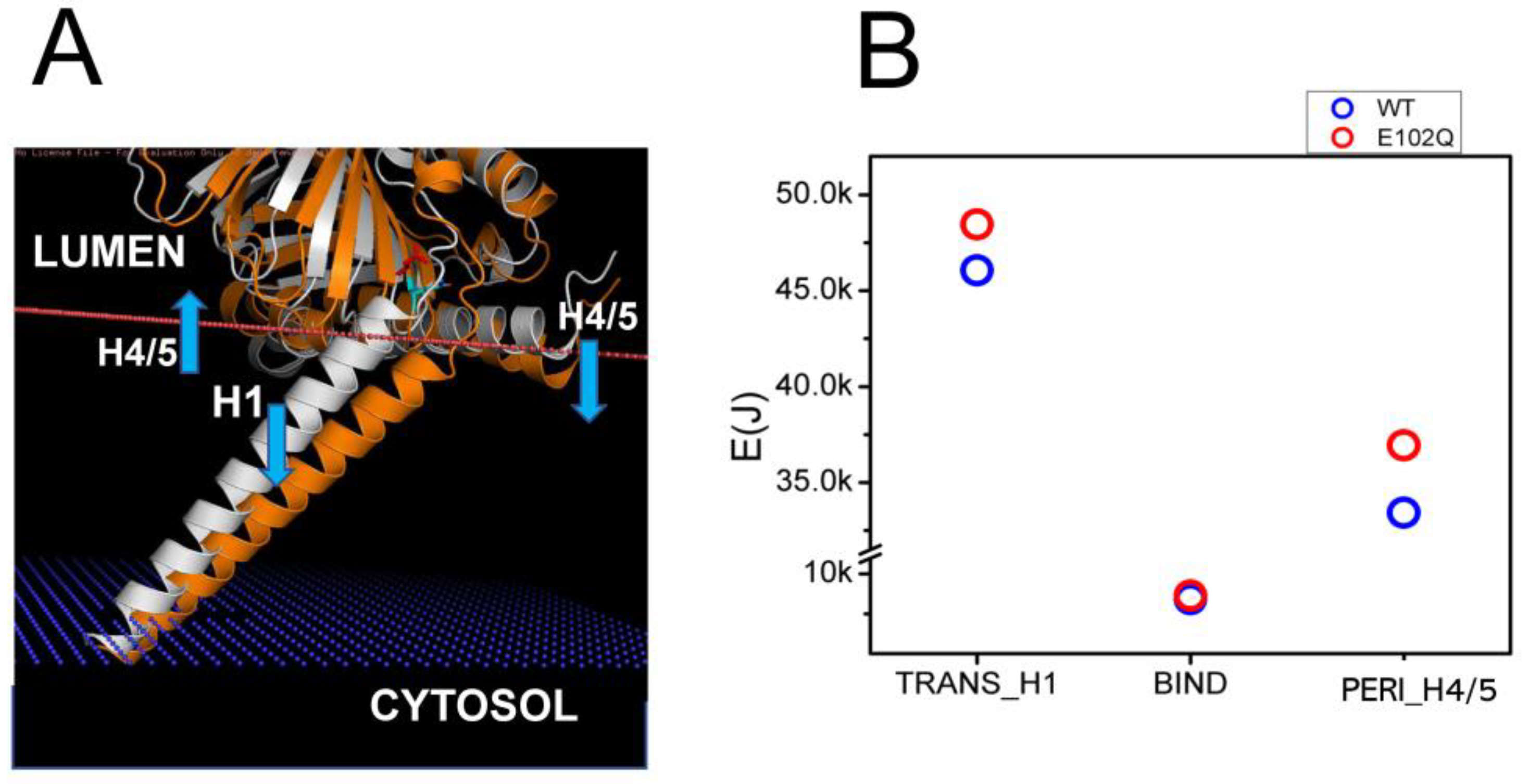
2.3. Effects of Pathogenic Mutations on Sigma 1 Receptor Association with the Membrane
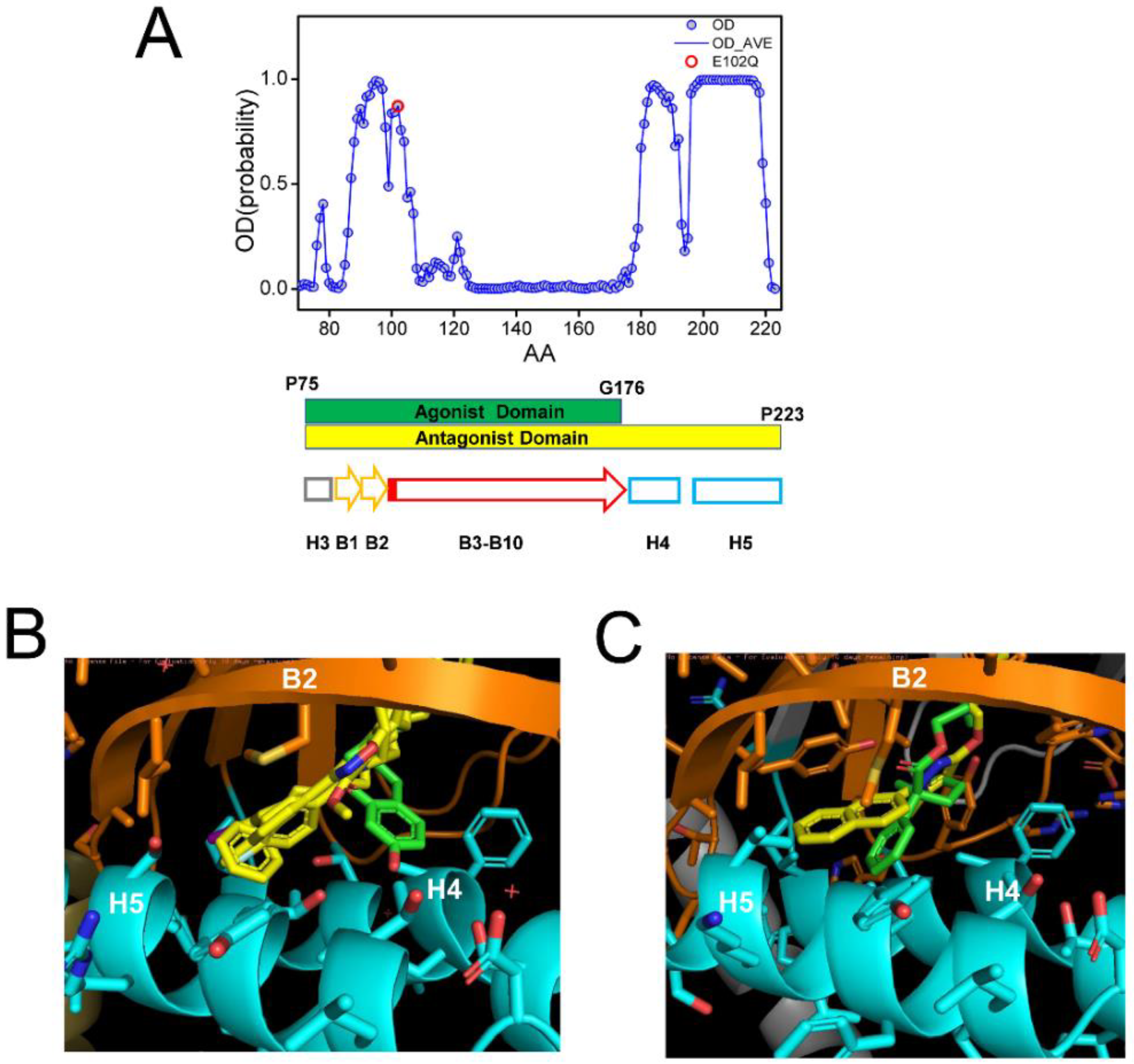
2.4. Effects of Ligand Association on Sigma 1 Receptor Conformation
3. Discussion
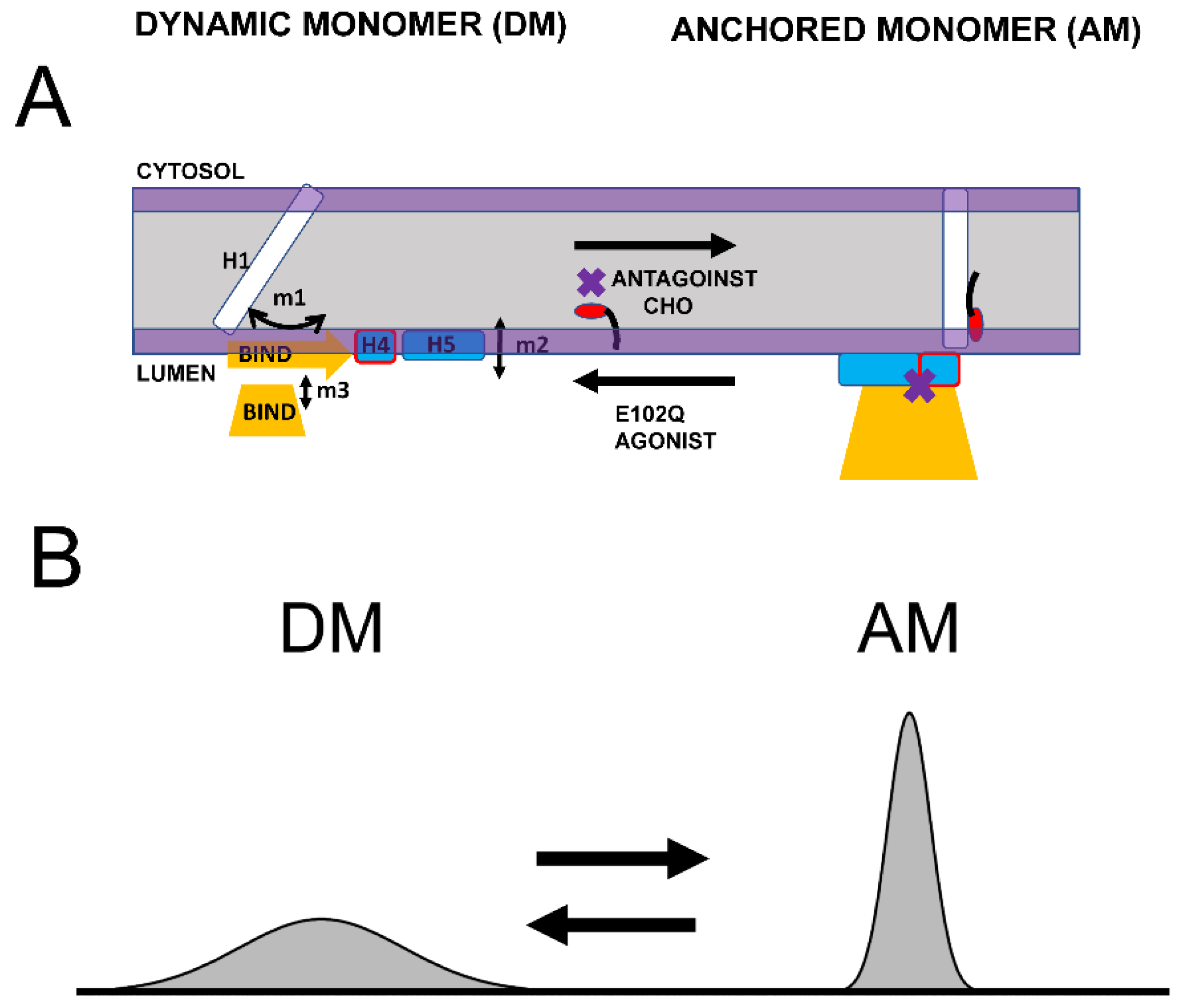
3.1. Dynamic Equilibrium of Two Conformations of Sigma 1 Receptor
3.2. Implications for Sigma 1 Receptor Biological Function
4. Materials and Methods
4.1. Secondary Structure Analysis
4.2. The Calculation of the Membrane-Association Energy of S1R
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayashi, T. The Sigma-1 Receptor in Cellular Stress Signaling. Front. Neurosci. 2019, 13, 733. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of sigma Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Alonso, G.; Phan, V.; Guillemain, I.; Saunier, M.; Legrand, A.; Anoal, M.; Maurice, T. Immunocytochemical localization of the sigma(1) receptor in the adult rat central nervous system. Neuroscience 2000, 97, 155–170. [Google Scholar] [CrossRef]
- Kitaichi, K.; Chabot, J.G.; Moebius, F.F.; Flandorfer, A.; Glossmann, H.; Quirion, R. Expression of the purported sigma(1) (sigma(1)) receptor in the mammalian brain and its possible relevance in deficits induced by antagonism of the NMDA receptor complex as revealed using an antisense strategy. J. Chem. Neuroanat. 2000, 20, 375–387. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Guo, L.W.; Epstein, M.L.; Ruoho, A.E. Role of the Sigma-1 receptor in Amyotrophic Lateral Sclerosis (ALS). J. Pharmacol. Sci. 2015, 127, 10–16. [Google Scholar] [CrossRef]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Li, X.; Hu, Z.; Liu, L.; Xie, Y.; Zhan, Y.; Zi, X.; Wang, J.; Wu, L.; Xia, K.; Tang, B.; et al. A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 2015, 84, 2430–2437. [Google Scholar] [CrossRef]
- Almendra, L.; Laranjeira, F.; Fernandez-Marmiesse, A.; Negrao, L. SIGMAR1 gene mutation causing Distal Hereditary Motor Neuropathy in a Portuguese family. Acta Myol. 2018, 37, 2–4. [Google Scholar]
- Ververis, A.; Dajani, R.; Koutsou, P.; Aloqaily, A.; Nelson-Williams, C.; Loring, E.; Arafat, A.; Mubaidin, A.F.; Horany, K.; Bader, M.B.; et al. Distal hereditary motor neuronopathy of the Jerash type is caused by a novel SIGMAR1 c.500A>T missense mutation. J. Med. Genet. 2020, 57, 178–186. [Google Scholar] [CrossRef]
- Gregianin, E.; Pallafacchina, G.; Zanin, S.; Crippa, V.; Rusmini, P.; Poletti, A.; Fang, M.; Li, Z.; Diano, L.; Petrucci, A.; et al. Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum. Mol. Genet. 2016, 25, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Horga, A.; Tomaselli, P.J.; Gonzalez, M.A.; Laura, M.; Muntoni, F.; Manzur, A.Y.; Hanna, M.G.; Blake, J.C.; Houlden, H.; Zuchner, S.; et al. SIGMAR1 mutation associated with autosomal recessive Silver-like syndrome. Neurology 2016, 87, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Ujike, H.; Tanaka, Y.; Sakai, A.; Yamamoto, M.; Fujisawa, Y.; Kanzaki, A.; Kuroda, S. A variant of the sigma receptor type-1 gene is a protective factor for Alzheimer disease. Am. J. Geriatr. Psychiatry 2005, 13, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T. Bi-phasic dose response in the preclinical and clinical developments of sigma-1 receptor ligands for the treatment of neurodegenerative disorders. Expert. Opin. Drug Discov. 2020, 16, 373–389. [Google Scholar] [CrossRef]
- Herrando-Grabulosa, M.; Gaja-Capdevila, N.; Vela, J.M.; Navarro, X. Sigma 1 receptor as a therapeutic target for amyotrophic lateral sclerosis. Br. J. Pharmacol. 2020, 178, 1336–1352. [Google Scholar] [CrossRef]
- Zhemkov, V.; Geva, M.; Hayden, M.R.; Bezprozvanny, I. Sigma-1 Receptor (S1R) Interaction with Cholesterol: Mechanisms of S1R Activation and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4082. [Google Scholar] [CrossRef]
- Couly, S.; Yasui, Y.; Su, T.P. SIGMAR1 Confers Innate Resilience against Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 7767. [Google Scholar] [CrossRef]
- Kourrich, S.; Hayashi, T.; Chuang, J.Y.; Tsai, S.Y.; Su, T.P.; Bonci, A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 2013, 152, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Zhemkov, V.; Liou, J.; Bezprozvanny, I. Sigma 1 Receptor, Cholesterol and Endoplasmic Reticulum Contact Sites. Contact 2021, 4, 25152564211026505. [Google Scholar] [CrossRef]
- Zhemkov, V.; Ditlev, J.A.; Lee, W.R.; Wilson, M.; Liou, J.; Rosen, M.K.; Bezprozvanny, I. The role of sigma 1 receptor in organization of endoplasmic reticulum signaling microdomains. eLife 2021, 10, e65192. [Google Scholar] [CrossRef]
- Palmer, C.P.; Mahen, R.; Schnell, E.; Djamgoz, M.B.; Aydar, E. Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 2007, 67, 11166–11175. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Fujimoto, M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 2010, 77, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 14949–14954. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human sigma1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Betz, R.M.; Dror, R.O.; Kruse, A.C. Structural basis for sigma1 receptor ligand recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Xiao, Y.; Ji, Y.; Sun, Z.; Zhou, X. An open-like conformation of the sigma-1 receptor reveals its ligand entry pathway. Nat. Commun. 2022, 13, 1267. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Yang, H.; Epstein, M.L.; Ruoho, A.E.; Yang, J.; Guo, L.W. APEX2-enhanced electron microscopy distinguishes sigma-1 receptor localization in the nucleoplasmic reticulum. Oncotarget 2017, 8, 51317–51330. [Google Scholar] [CrossRef]
- Ortega-Roldan, J.L.; Ossa, F.; Amin, N.T.; Schnell, J.R. Solution NMR studies reveal the location of the second transmembrane domain of the human sigma-1 receptor. FEBS Lett. 2015, 589, 659–665. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef]
- Fantini, J.; Di Scala, C.; Baier, C.J.; Barrantes, F.J. Molecular mechanisms of protein-cholesterol interactions in plasma membranes: Functional distinction between topological (tilted) and consensus (CARC/CRAC) domains. Chem. Phys. Lipids 2016, 199, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Di Scala, C.; Evans, L.S.; Williamson, P.T.; Barrantes, F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 2016, 6, 21907. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Baier, C.J.; Evans, L.S.; Williamson, P.T.F.; Fantini, J.; Barrantes, F.J. Relevance of CARC and CRAC Cholesterol-Recognition Motifs in the Nicotinic Acetylcholine Receptor and Other Membrane-Bound Receptors. Curr. Top. Membr. 2017, 80, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kenworthy, A.K.; Sanders, C.R. Cholesterol as a co-solvent and a ligand for membrane proteins. Protein Sci. 2014, 23, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E. Enhanced fold recognition using efficient short fragment clustering. J. Mol. Biochem. 2012, 1, 76–85. [Google Scholar]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB(1). Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Abramyan, A.M.; Yano, H.; Xu, M.; Liu, L.; Naing, S.; Fant, A.D.; Shi, L. The Glu102 mutation disrupts higher-order oligomerization of the sigma 1 receptor. Comput. Struct. Biotechnol. J. 2020, 18, 199–206. [Google Scholar] [CrossRef]
- Hong, W.C. Distinct Regulation of sigma 1 Receptor Multimerization by Its Agonists and Antagonists in Transfected Cells and Rat Liver Membranes. J. Pharmacol. Exp. Ther. 2020, 373, 290–301. [Google Scholar] [CrossRef]
- Mishra, A.K.; Mavlyutov, T.; Singh, D.R.; Biener, G.; Yang, J.; Oliver, J.A.; Ruoho, A.; Raicu, V. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 2015, 466, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Bonifazi, A.; Xu, M.; Guthrie, D.A.; Schneck, S.N.; Abramyan, A.M.; Fant, A.D.; Hong, W.C.; Newman, A.H.; Shi, L. Pharmacological profiling of sigma 1 receptor ligands by novel receptor homomer assays. Neuropharmacology 2018, 133, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Gromek, K.A.; Suchy, F.P.; Meddaugh, H.R.; Wrobel, R.L.; LaPointe, L.M.; Chu, U.B.; Primm, J.G.; Ruoho, A.E.; Senes, A.; Fox, B.G. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 2014, 289, 20333–20344. [Google Scholar] [CrossRef] [PubMed]
- Rossino, G.; Orellana, I.; Caballero, J.; Schepmann, D.; Wunsch, B.; Rui, M.; Rossi, D.; Gonzalez-Avendano, M.; Collina, S.; Vergara-Jaque, A. New Insights into the Opening of the Occluded Ligand-Binding Pocket of Sigma1 Receptor: Binding of a Novel Bivalent RC-33 Derivative. J. Chem. Inf. Model. 2020, 60, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Hajipour, A.R.; Fontanilla, D.; Ramachandran, S.; Chu, U.B.; Mavlyutov, T.; Ruoho, A.E. Identification of regions of the sigma-1 receptor ligand binding site using a novel photoprobe. Mol. Pharmacol. 2007, 72, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Liu, L.; Naing, S.; Shi, L. The Effects of Terminal Tagging on Homomeric Interactions of the Sigma 1 Receptor. Front. Neurosci. 2019, 13, 1356. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Nugent, T.; Jones, D.T. Membrane protein orientation and refinement using a knowledge-based statistical potential. BMC Bioinform. 2013, 14, 276. [Google Scholar] [CrossRef]
- Kufareva, I.; Lenoir, M.; Dancea, F.; Sridhar, P.; Raush, E.; Bissig, C.; Gruenberg, J.; Abagyan, R.; Overduin, M. Discovery of novel membrane binding structures and functions. Biochem. Cell Biol. 2014, 92, 555–563. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Bezprozvanny, I. Structure-Based Modeling of Sigma 1 Receptor Interactions with Ligands and Cholesterol and Implications for Its Biological Function. Int. J. Mol. Sci. 2023, 24, 12980. https://doi.org/10.3390/ijms241612980
Kim M, Bezprozvanny I. Structure-Based Modeling of Sigma 1 Receptor Interactions with Ligands and Cholesterol and Implications for Its Biological Function. International Journal of Molecular Sciences. 2023; 24(16):12980. https://doi.org/10.3390/ijms241612980
Chicago/Turabian StyleKim, Meewhi, and Ilya Bezprozvanny. 2023. "Structure-Based Modeling of Sigma 1 Receptor Interactions with Ligands and Cholesterol and Implications for Its Biological Function" International Journal of Molecular Sciences 24, no. 16: 12980. https://doi.org/10.3390/ijms241612980
APA StyleKim, M., & Bezprozvanny, I. (2023). Structure-Based Modeling of Sigma 1 Receptor Interactions with Ligands and Cholesterol and Implications for Its Biological Function. International Journal of Molecular Sciences, 24(16), 12980. https://doi.org/10.3390/ijms241612980