

Parp Inhibitors and Radiotherapy: A New Combination for Prostate Cancer (Systematic Review)

 , , and
, , and
Abstract
:1. Introduction
2. Methods
2.1. Search Strategy
2.2. Article Selection
3. Results
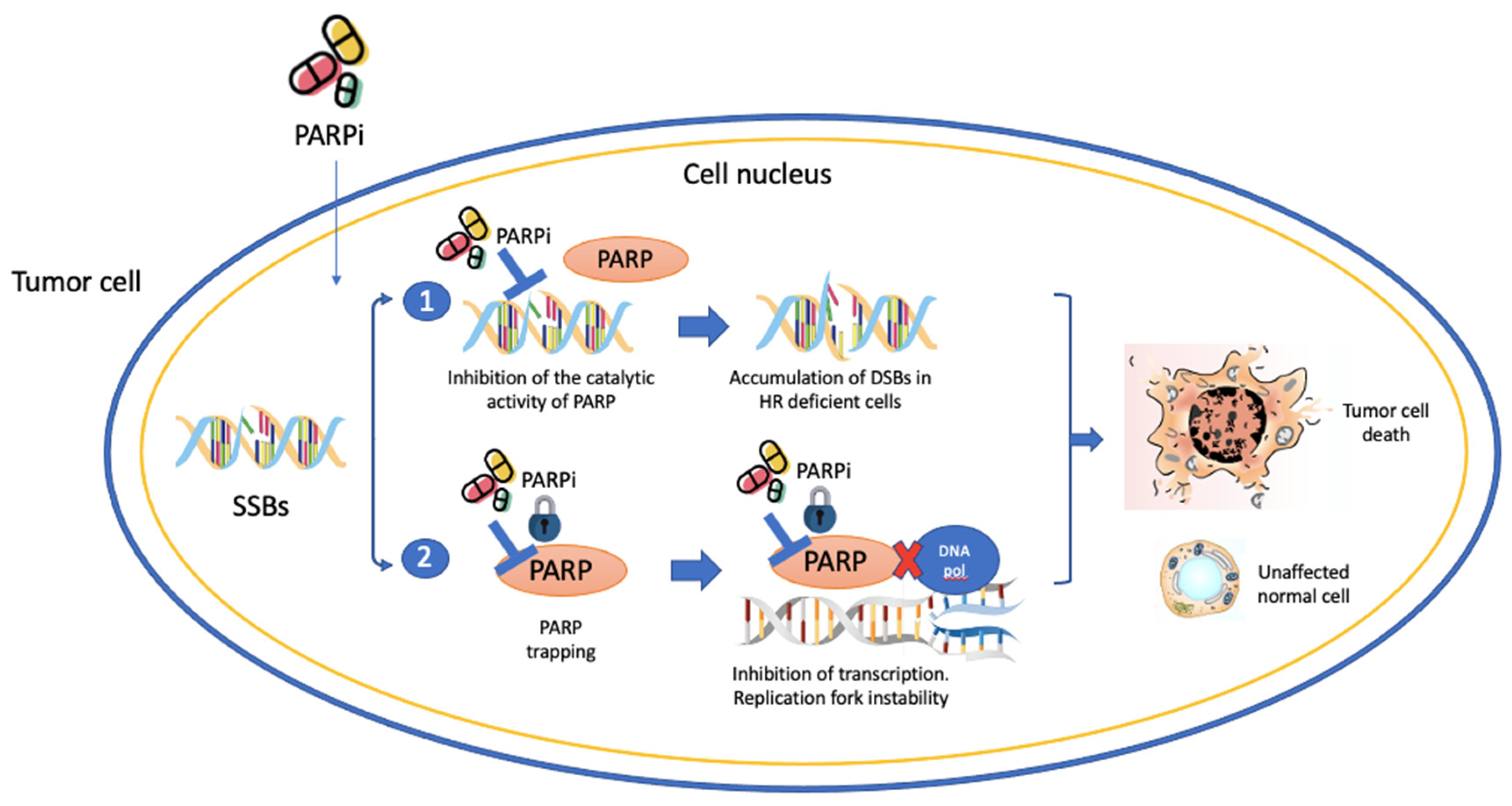
3.1. PARP Inhibitors in Prostate Cancer
3.2. PARPi as Radiosensitizers in Prostate Cancer
3.2.1. The Rationale to Combine PARPi and Radiotherapy
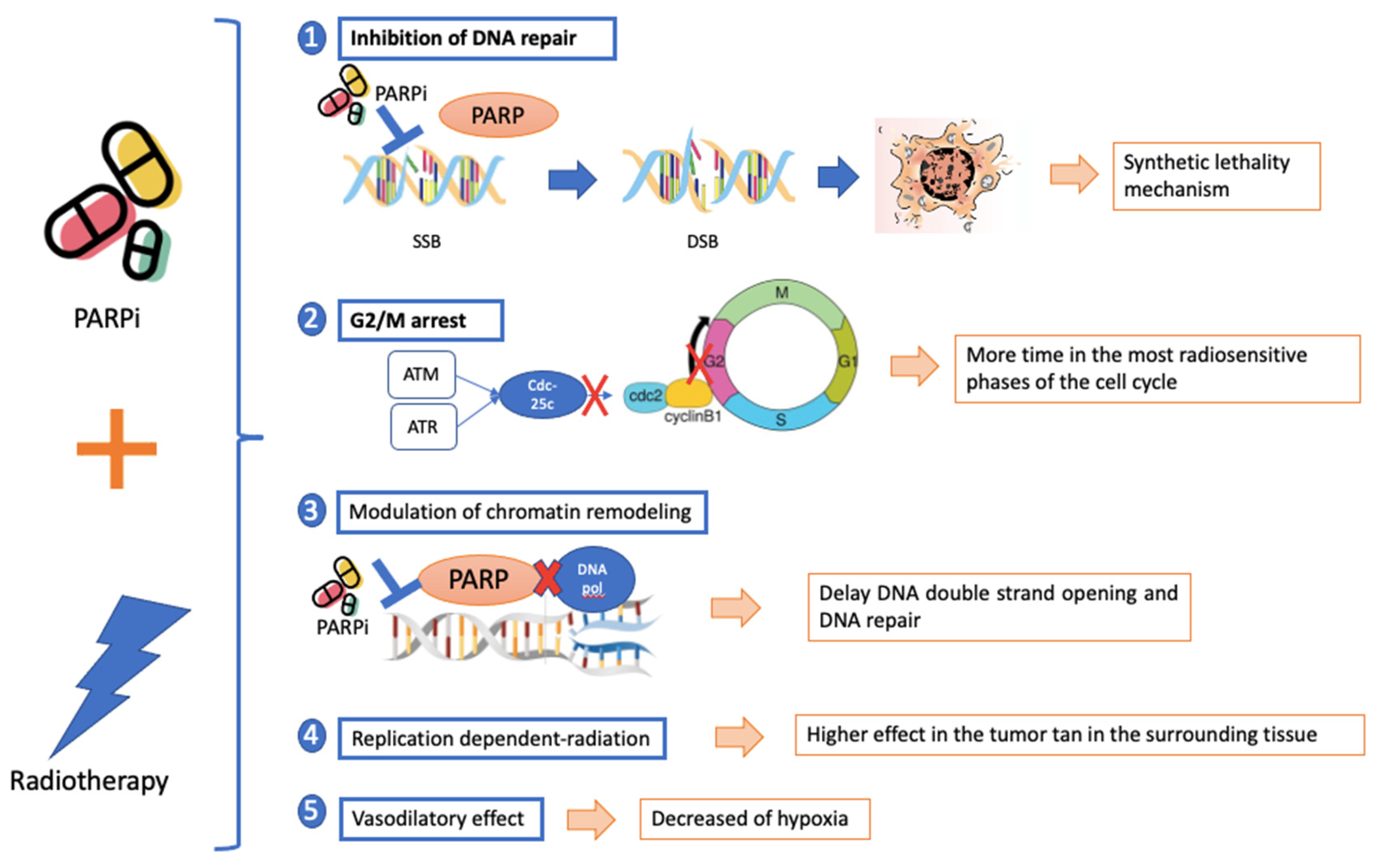
3.2.2. Mechanism of Radiosensitization of PARPi
3.2.3. The Combination of PARPi and Radiotherapy in Prostate Cancer: Preclinical Studies
3.2.4. The Combination of PARPi and Radiotherapy in Prostate Cancer: Clinical Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Castro, E.; Fizazi, K.; Heidenreich, A.; Ost, P.; Procopio, G.; Tombal, B.; Gillessen, S.; on behalf of the ESMO Guidelines Committee. Prostate cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Mary-Ellen Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and prostate cancer: To infinity and Beyond BRCA. Oncologist 2021, 26, e115–e129. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Powell, C.; Mikropoulos, C.; Kaye, S.B.; Nutting, C.M.; Bhide, S.A.; Newbold, K.; Harrington, K.J. Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. Cancer Treat. Rev. 2010, 36, 566–575. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Loap, P.; Loirat, D.; Berger, F.; Rodrigues, M.; Bazire, L.; Pierga, J.-Y.; Vincent-Salomon, A.; Laki, F.; Boudali, L.; Raizonville, L.; et al. Concurrent Olaparib and Radiotherapy in Patients with Triple-Negative Breast Cancer: The Phase 1 Olaparib and Radiation Therapy for Triple-Negative Breast Cancer Trial. JAMA Oncol. 2022, 8, 1802–1808. [Google Scholar] [CrossRef]
- Deng, S.; Vlatkovic, T.; Li, M.; Zhan, T.; Veldwijk, M.R.; Herskind, C. Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer. Cancers 2022, 14, 4874. [Google Scholar] [CrossRef]
- Yang, S.-H.; Kuo, T.-C.; Wu, H.; Guo, J.-C.; Hsu, C.; Hsu, C.-H.; Tien, Y.-W.; Yeh, K.-H.; Cheng, A.-L.; Kuo, S.-H. Perspectives on the combination of radiotherapy and targeted therapy with DNA repair inhibitors in the treatment of pancreatic cancer. World J. Gastroenterol. 2016, 22, 7275–7288. [Google Scholar] [CrossRef] [PubMed]
- De Haan, R.; Van Den Heuvel, M.M.; Van Diessen, J.; Peulen, H.M.U.; Van Werkhoven, E.; De Langen, A.J. Phase I and Pharmacologic Study of Olaparib in Combination with High-dose Radiotherapy with and without Concurrent Cisplatin for Non–Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 1256–1266. [Google Scholar] [CrossRef]
- Mumtaz, S.; Nahushkumar Rana, J.; Ha Choi, E.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A. Radiation Track Structure: How the Spatial Distribution of Energy Deposition Drives Biological Response. Clin. Oncol. 2020, 32, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Lyu, F.; Shang, S.-Y.; Gao, X.-S.; Ma, M.-W.; Xie, M.; Ren, X.-Y.; Liu, M.-Z.; Chen, J.-Y.; Li, S.-S.; Huang, L. Uncovering the Secrets of Prostate Cancer’s Radiotherapy Resistance: Advances in Mechanism Research. Biomedicines 2023, 11, 1628. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.; Hill, M.A.; Parsons, J.L. The Cellular Response to Complex DNA Damage Induced by Ionising Radiation. Int. J. Mol. Sci. 2023, 24, 4920. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. Energy deposition stochastics and track structure: What about the target? Radiat. Prot. Dosim. 2006, 122, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Shi, J.; Bian, C.; Yu, X. Poly(ADP-ribose) mediates the BRCA2-dependent early DNA damage response. Cell Rep. 2015, 13, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Shi, J.; Chen, S.-H.; Bian, C.; Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Res. 2015, 43, 10782–10794. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Dejsuphong, D.; Fukushima, T.; Morrison, C.; Sonoda, E.; Schreiber, V.; Zhao, G.Y.; Saberi, A.; Masutani, M.; Adachi, N.; et al. Parp-1 protects homologous recombination from interference by Ku and ligase IV in vertebrate cells. EMBO J. 2006, 25, 1305–1314. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. Arequirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [PubMed]
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003, 23, 3974–3981. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Yu, X. Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Sci. Adv. 2019, 5, eaav4340. [Google Scholar] [CrossRef] [PubMed]
- Ame, J.C.; Fouquerel, E.; Gauthier, L.R.; Biard, D.; Boussin, F.D.; Dantzer, F.; de Murcia, G.; Schreiber, V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J. Cell Sci. 2009, 122, 1990–2002. [Google Scholar] [CrossRef]
- Jump, D.B.; Butt, T.R.; Smulson, M. Nuclear protein modification and chromatin substructure. 3. Relationship between poly(adenosine diphosphate) ribosylation and different functional forms of chromatin. Biochemistry 1979, 18, 983–990. [Google Scholar] [CrossRef]
- Anachkova, B.; Russev, G.; Poirier, G.G. DNA replication and poly(ADP-ribosyl)ation of chromatin. Cytobios 1989, 58, 19–28. [Google Scholar]
- Dantzer, F.; Nasheuer, H.P.; Vonesch, J.L.; de Murcia, G.; Ménissier-de Murcia, J. Functional association of poly(ADP-ribose) polymerase with DNA polymerase α-primase complex: A link between DNA strand break detection and DNA replication. Nucleic Acids Res. 1998, 26, 1891–1898. [Google Scholar] [CrossRef]
- Bryant, H.E.; Helleday, T. Inhibition of poly (ADP-ribose) polymerase activates ATMwhich is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006, 34, 1685–1691. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of PARP-1 does not interfere with the repair of DNA doublestrand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Takebayashi, S.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef]
- Min, W.; Bruhn, C.; Grigaravicius, P.; Zhou, Z.W.; Li, F.; Krüger, A.; Siddeek, B.; Greulich, K.O.; Popp, O.; Meisezahl, C.; et al. Poly (ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 2013, 4, 2993. [Google Scholar] [CrossRef]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.; Samara, R.; Espinoza, L.A.; Hassa, P.O.; Hottiger, M.O.; Smulson, M.E. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene 2003, 22, 8460–8471. [Google Scholar] [CrossRef]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-ribose) polymerase: Mechanisms and therapeutic potential. Int. J. Radiat. Oncol. 2008, 72, 1188–1197. [Google Scholar] [CrossRef]
- Cumpston, M.; Li, T.; Page, M.; Chandler, J.; Welch, V.; Higgins, J.P.; Thomas, J. Updated guidance for trusted systematic reviews: A new edition of the Cochrane Handbook for Systematic Reviews of Interventions. Cochrane Database Syst. Rev. 2019, 2019, ED000142. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Int. J. Surg. 2010, 8, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; PRISMA-P Group. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: Elaboration and explanation. BMJ 2015, 350, g7647. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specifickilling of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Francica, P.; Rottenberg, S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Moka, N.; Hamstra, D.A.; Ryan, C.J. Co-Inhibition of androgen receptor and PARP as a novel treatment paradigm in prostate cancer-where are we now? Cancers 2022, 14, 801. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Congregado, B.; Rivero, I.; Osman, I.; Saez, C.; Medina López, R. PARP inhibitors: A new horizon for patients with prostate cancer. Biomedicines 2022, 10, 1416. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Smith, M.R.; Sandhu, S.K.; Kelly, W.K.; Scher, H.I. Pre-specified interim analysis of GALAHAD: A phase 2 study of niraparib in patients (pts) with metastatic castrationresistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects. Ann. Oncol. 2019, 30, v884–v885. [Google Scholar] [CrossRef]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef] [PubMed]
- Thiery-Vuillemin, A.; Saad, F.; Armstrong, A.J.; Oya, M.; Vianna, K.C.M.; Özgüroğlu, M.; Gedye, C.; Buchschacher, G.L.; Lee, J.Y.; Emmenegger, U.; et al. Tolerability of abiraterone (abi) combined with olaparib (ola) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): Further results from the phase III PROpel trial. J. Clin. Oncol. 2022, 40, 5019. [Google Scholar] [CrossRef]
- Chi, K.N.; Rathkopf, D.E.; Smith, M.R.; Efstathiou, E.; Attard, G.; Olmos, D.; Lee, J.Y.; Small, E.J.; Gomes, A.J.; Roubaud, G.; et al. Phase 3 MAGNITUDE study: First results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) withmetastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J. Clin. Oncol. 2022, 40, 12. [Google Scholar] [CrossRef]
- Brown, J.M.; Carlson, D.J.; Brenner, D.J. The tumor radiobiology of SRS and SBRT: Are more than the 5 Rs involved? Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 254–262. [Google Scholar] [CrossRef]
- Borgmann, K.; Kocher, S.; Kriegs, M.; Mansour, W.Y.; Parplys, A.C.; Rieckmann, T.; Rothkamm, K. DNA Repair. In Molecular Radio-Oncology: Recent Results in Cancer Research; Springer: Berlin/Heidelberg, Germany, 2016; Volume 198, pp. 1–24. [Google Scholar]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef]
- Rothkamm, K.; Krüger, I.; Thompson, L.H.; Lübrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Kitagawa, R.; Kastan, M.B. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Barnard, S.; Moquet, J.; Ellender, M.; Rana, Z.; Burdak-Rothkamm, S. DNA damage foci: Meaning and significance. Environ. Mol. Mutagen. 2015, 56, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Höhne, K.; von Neubeck, C.; Thames, H.D.; Yaromina, A.; Dahm-Daphi, J.; Baumann, M.; Krause, M. Residual γH2AX;H2AX foci predict local tumour control after radiotherapy. Radiother. Oncol. 2013, 108, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Menegakis, A.; De Colle, C.; Yaromina, A.; Hennenlotter, J.; Stenzl, A.; Scharpf, M.; Fend, F.; Noell, S.; Tatagiba, M.; Brucker, S.; et al. Residual gammaH2AX foci after ex vivo irradiation of patient samples with known tumour-type specific differences in radio-responsiveness. Radiother. Oncol. 2015, 116, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; McCrudden, C.M.; Hirst, D.G.; O’Connor, M.J.; Wedge, S.R.; Stratford, I.J. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol. Cancer Ther. 2011, 10, 1949–1958. [Google Scholar] [CrossRef]
- Dulaney, C.; Marcrom, S.; Stanley, J.; Yang, E.S. Poly(ADP-ribose) polymerase activity and inhibition in cancer. Semin. Cell Dev. Biol. 2017, 63, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Mansour, W.Y.; Schumacher, S.; Rosskopf, R.; Rhein, T.; Schmidt-Petersen, F.; Gatzemeier, F.; Haag, F.; Borgmann, K.; Willers, H.; Dahm-Daphi, J. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res. 2008, 36, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Magrini, S.M.; Buglione, M.; Corvò, R.; Pirtoli, L.; Paiar, F.; Ponticelli, P.; Petrucci, A.; Bacigalupo, A.; Crociani, M.; Lastrucci, L.; et al. Cetuximab and radiotherapy versus cisplatin and radiotherapy for locally advanced head and neck cancer: A randomized phase II trial. J. Clin. Oncol. 2016, 34, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, P.; Chevalier, F.; Austry, J.B.; Waissi, W.; Burckel, H.; Noël, G. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: A systematic review of pre-clinical and clinical human studies. Oncotarget 2017, 8, 69105–69124. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Noël, G.; Godon, C.; Fernet, M.; Giocanti, N.; Mégnin-Chanet, F.; Favaudon, V. Radiosensitization by the poly(ADP-ribose) polymerase inhibitor 4-amino-1,8-naphthalimide is specific of the S phase of the cell cycle and involves arrest of DNA synthesis. Mol. Cancer Ther. 2006, 5, 564–574. [Google Scholar] [CrossRef]
- Ali, M.; Telfer, B.A.; McCrudden, C.; O’Rourke, M.; Thomas, H.D.; Kamjoo, M.; Kyle, S.; Robson, T.; Shaw, C.; Hirst, D.G.; et al. Vasoactivity of AG014699, a clinically active small molecule inhibitor of poly(ADP-ribose) polymerase: A contributory factor to chemopotentiation in vivo? Clin. Cancer Res. 2009, 15, 6106–6112. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- Han, S.; Brenner, J.C.; Sabolch, A.; Jackson, W.; Speers, C.; Wilder-Romans, K. Targeted Radiosensitization of ETS Fusion–Positive Prostate Cancer. Neoplasia 2013, 15, 1207–1217. [Google Scholar] [CrossRef]
- Gani, C.; Coackley, C.; Kumareswaran, R.; Schütze, C.; Krause, M.; Zafarana, G.; Bristow, R.G. In vivo studies of the PARP inhibitor, AZD-2281, in combination with fractionated radiotherapy: An exploration of the therapeutic ratio. Radiother. Oncol. 2015, 116, 486–494. [Google Scholar] [CrossRef]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 3947. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, A.L.; Tangutoori, S.; Baldwin, P.; Qiao, J.; Gharagouzloo, C.; Seitzer, N.; Clohessy, J.G.; Makrigiorgos, G.M.; Cormack, R.; Pandolfi, P.P.; et al. Nanoformulation of Olaparib amplifies PARP inhibition and sensitizes PTEN/TP53-deficient prostate cancer to radiation. Mol. Cancer Ther. 2017, 16, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Tennstedt, P.; Simon, R.; Volquardsen, J.; Borgmann, K.; Bokemeyer, C. BCL2-overexpressing prostate cancer cells rely on PARP1-dependent end-joining and are sensitive to combined PARP inhibitor and radiation therapy. Cancer Lett. 2018, 423, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Köcher, S.; Beyer, B.; Lange, T.; Nordquist, L.; Volquardsen, J.; Burdak-Rothkamm, S. A functional ex vivo assay to detect PARP1-EJ repair and radiosensitization by PARP-inhibitor in prostate cancer. Int. J. Cancer 2019, 144, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Fan, H.; Quan, Z.; Wu, X. Ionizing Radiation Combined with PARP1 Inhibitor Reduces Radioresistance in Prostate Cancer with RB1/TP53 Loss. Cancer Investig. 2021, 39, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Bolla, M.; Van Tienhoven, G.; Warde, P.; Dubois, J.B.; Mirimanoff, R.O.; Storme, G.; Bernier, J.; Kuten, A.; Sternberg, C.; Billiet, I.; et al. External irradiation with or without long-term androgen suppression for prostate cancer with high metastatic risk: 10-year results of an EORTC randomised study. Lancet Oncol. 2010, 11, 1066. [Google Scholar] [CrossRef]
- Denham, J.W.; Steigler, A.; Lamb, D.S.; Joseph, D.; Turner, S.; Matthews, J.; Atkinson, C.; North, J.; Christie, D.; Spry, N.A.; et al. Short-term neoadjuvant androgen deprivation and radiotherapy for locally advanced prostate cancer: 10-year data from the TROG 96.01 randomised trial. Lancet Oncol. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Mottet, N.; Bellmunt, J.; Briers, E.; Bolla, M.; Bourke, L.; Cornford, P.; De Santis, M.; Henry, A.M.; Joniau, S.; Lam, T.B.; et al. EAU–ESTRO–ESUR–SIOG Guidelines on Prostate Cancer; EAU Guidelines Office: Arnhem, The Netherlands, 2023. [Google Scholar]
- Makhov, P.; Fazliyeva, R.; Tufano, A.; Uzzo, R.G.; Kolenko, V.M. Examining the Effect of PARP-1 Inhibitors on Transcriptional Activity of Androgen Receptor in Prostate Cancer Cells. Methods Mol. Biol. 2023, 2609, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Zumsteg, Z.; Karrison, T.; Michaelson, M.; Tran, P.; Kudchadker, R.; Feng, F. 689TiP NRG Oncology’s GU007 (NADIR): A randomized phase II trial of niraparib with standard combination androgen deprivation therapy (ADT) and radiotherapy (RT) in high-risk prostate cancer (PC) (with initial phase I). Ann. Oncol. 2020, 31, 546. [Google Scholar] [CrossRef]
- Parker, C.C.; James, N.D.; Brawley, C.D.; Clarke, N.W.; Ali, A.; Amos, C.L.; Attard, G.; Chowdhury, S.; Cook, A.; Cross, W.; et al. Radiotherapy to the prostate for men with metastatic prostate cancer in the UK and Switzerland: Long-term results from the STAMPEDE randomised controlled trial. PLoS Med. 2022, 19, e1003998. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Hoyle, A.; Haran, Á.M.; Brawley, C.D.; Cook, A.; Amos, C.; Calvert, J.; Douis, H.; Mason, M.D.; Dearnaley, D.; et al. Association of Bone Metastatic Burden With Survival Benefit From Prostate Radiotherapy in Patients with Newly Diagnosed Metastatic Prostate Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2021, 7, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Ost, P.; Reynders, D.; Decaestecker, K.; Fonteyne, V.; Lumen, X.; De Bruycker, A.; Lambert, B.; Delrue, L.; Bultijnck, R.; Claeys, T.; et al. Surveillance or Metastasis-Directed Therapy for Oligometastatic Prostate Cancer Recurrence: A Prospective, Randomized, Multicenter Phase II Trial. J. Clin. Oncol. 2018, 36, 446–453. [Google Scholar] [CrossRef]
- Phillips, R.; Shi, W.Y.; Deek, M.; Radwan, N.; Lim, S.J.; Antonarakis, E.S.; Rowe, S.P.; Ross, A.E.; Gorin, M.A.; Deville, C.; et al. Outcomes of Observation vs Stereotactic Ablative Radiation for Oligometastatic Prostate Cancer: The ORIOLE Phase 2 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 650–659. [Google Scholar] [CrossRef]
- Deek, M.P.; Van der Eecken, K.; Sutera, P.; Deek, R.A.; Fonteyne, V.; Mendes, A.A.; Decaestecker, K.; Kiess, A.P.; Lumen, N.; Phillips, R.; et al. Long-Term Outcomes and Genetic Predictors of Response to Metastasis-Directed Therapy Versus Observation in Oligometastatic Prostate Cancer: Analysis of STOMP and ORIOLE Trials. J. Clin. Oncol. 2022, 40, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, M.; Chung, P.; Parker, C.; Bristow, R.; Toi, A.; Panzarella, T.; Warde, P.; Catton, C.; Menard, C.; Bayley, A.; et al. Androgen Withdrawal in Patients Reduces Prostate Cancer Hypoxia. Implications for Disease Progression and Radiation Response. Cancer Res. 2007, 67, 6022–6025. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A Hormone-DNA Repair Circuit Governs the Response to Genotoxic Insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanijv, K.; Gelali, E.; Massie, C.E. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Drug/Approval Situation | Setting | HRR Mutations | Outcomes |
|---|---|---|---|---|
| PROfound phase III, randomized [63] | Olaparib vs. AA/Enzalutamide Approved by FDA and EMA | mCRPC who progress after AA or Enzalutamide | Selected | Longer rPFS (5.8 months vs. 3.5 months, p < 0.001) Higher ORR (22% vs. 4%; odds ratio 5.93, 95% CI: 2.01–25.40) |
| TRITON-2, phase II single arm [64] | Rucaparib Approved by FDA | mCRPC who progress after 1–2 novel antiandrogens and paclitaxel | Selected | ORR of 43.5% and a PSA response rate of 54.8% |
| GALAHAD, phase II single arm [65] | Niraparib Under evaluation by FDA | mCRPC who progress after paclitaxel and AR targeted therapy | Selected | ORR of 41%, CRR of 63%, median rPFS and OS of 8.2 and 12.6 respectively |
| TALAPRO-1, phase II single arm [66] | Talazoparib Under evaluation by FDA | mCRPC after paclitaxel therapy | Selected | ORR of 29.8%, and 46% BRCA1/2 mutations |
| PROpel, phase III randomized [67] | Olaparib + AA vs. placebo + AA | First line mCRPC | Unselected | Improvement in rPFS (HR 0.66 [95% CI 0.54–0.81]) in Olaparib + AA arm |
| MAGNITUDE, phase III randomized [68] | Niraparib + AA vs. placebo + AA | First line mCRPC | Selected | Preliminary results: Improved on the rPFS (relative risk 0.53, 95% CI: 0.36–0.79, p = 0.0014) and a reduced the risk of disease progression/death (47% vs. 27%) |
| Author, Year | In Vitro: Cell Line and Intervention | In Vivo: Animal Model and Intervention | Ex Vivo: Model | Outcomes |
|---|---|---|---|---|
| Han et al., 2013 [92] | PC3 and DU145 PCa cell lines with ERG overexpression Intervention: 1.0 μM Olaparib 1h pre Radiation (2 Gy/min) | Xenograft models: subcutaneous mice model of PC3-control and PC3-ERG Intervention: ABT-888 (100 mg/kg) twice daily, or radiation alone (2 Gy for 5 days), or in combination | In vitro: olaparib radiosensitized ERG-positive cells by a factor of 1.52 (±0.03) relative to ERG-negative cells In vivo: ERG overexpression confers radiation resistance through increased efficiency of DNA repair following radiation that can be reversed through inhibition of PARP1. | |
| Gani et al., 2015 [93] | 22Rv1 PCa cells Intervention: RT 2 Gys + 1.0 μM PARP inhibitor (AZD-2281) | 22Rv1 prostate xenografts: Old male CD1 nude mice were injected s.c. with 2.0 × 106 cells Intervention: (a) vehicle alone; (b) AZD-2281 100 mg/kg. daily for 3 days; (c) 8 Gy plus vehicle i.p. for 3 days; (d) 8 Gy plus AZD-2281 100 mg/kg for 3 days; (e) 5x2 Gy plus 7 days vehicle i.p.; (f) 5x2 Gy plus 7 days of AZD-2281 100 mg/kg | Combining AZD-2281 with fractionated RT led to a significant delay in tumor growth and increased clonogenic cell death without increasing gut toxicity | |
| Mansour et al., 2017 [94] | Cell lines DU145, BPH1 Intervention: Irradiation of 0.8 Gy/min + 1 μM olaparib for 4–6 h | Tissue spots from 3261 radical prostatectomies (RP) | PTEN status in PCa may be a potential predictor of both RT and PARPi response, alone or in combination | |
| Van de Ven et al., 2018 [95] | Three PTEN-deficient PCa cell lines: human PC3 (ATCC), human LNCaP (ATCC), and mouse FKO1 Intervention: 1μM Olaparib or NanoOlaparib for 24 hr, irradiated (0–10 Gy) | Subcutaneous mouse model of radiation resistant PCa generated from FKO1 Intervention: NanoOlaparib (40 mg/kg) IV twice weekly 12 weeks + irradiation one dose of 10 Gy | In vitro: radiosensitivity increased after NanoOlaparib. Changes in γ-H2AX In vivo: Triple the mouse OS compared to RT alone. 50% complete response after 13 weeks. MRI studies revealed that NanoOlaparib enhances the intratumural accumulation of systemically administered nanoparticles (ferumoxytol) | |
| Oing et al., 2018 [96] | Cell lines DU145, BPH1 Intervention: Irradiation of 0.8 Gy/min + 1 μM olaparib | Samples from RP | Olaparib could enhance radiosensitivity of cells overexpressing BCL2 | |
| Köcher et al., 2019 [97] | Freshly collected tumor samples from PCa | an ex vivo assay could be used to detect radiosensitivity in tumor biopsies helping to personalize treatments | ||
| Fan et al., 2021 [98] | Cell lines: LNCaP, 22RV1, PC3, and DU145 Drugs: Enzalutamide, KU55933, Olaparib (AZD2281) RT: 2–8 Gy | Loss RB1 increased vulnerability to the DNA damage pathway. RT + PARP1 increased radiosensitivity. |
| Study | Design | Estimated Enrollment | Setting | Intervention | Primary Endpoint |
|---|---|---|---|---|---|
| NADIR (NCT04037254) [103] | Phase II, randomized clinical trial | 180 | High risk localized or locally advanced PCa (no prior treatment) with or without HRR mutations | Niraparib + RT + ADT vs. niraparib alone vs. RT + ADT | Maintenance of disease-free state |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivero Belenchón, I.; Congregado Ruiz, C.B.; Saez, C.; Osman García, I.; Medina López, R.A. Parp Inhibitors and Radiotherapy: A New Combination for Prostate Cancer (Systematic Review). Int. J. Mol. Sci. 2023, 24, 12978. https://doi.org/10.3390/ijms241612978
Rivero Belenchón I, Congregado Ruiz CB, Saez C, Osman García I, Medina López RA. Parp Inhibitors and Radiotherapy: A New Combination for Prostate Cancer (Systematic Review). International Journal of Molecular Sciences. 2023; 24(16):12978. https://doi.org/10.3390/ijms241612978
Chicago/Turabian StyleRivero Belenchón, Inés, Carmen Belen Congregado Ruiz, Carmen Saez, Ignacio Osman García, and Rafael Antonio Medina López. 2023. "Parp Inhibitors and Radiotherapy: A New Combination for Prostate Cancer (Systematic Review)" International Journal of Molecular Sciences 24, no. 16: 12978. https://doi.org/10.3390/ijms241612978
APA StyleRivero Belenchón, I., Congregado Ruiz, C. B., Saez, C., Osman García, I., & Medina López, R. A. (2023). Parp Inhibitors and Radiotherapy: A New Combination for Prostate Cancer (Systematic Review). International Journal of Molecular Sciences, 24(16), 12978. https://doi.org/10.3390/ijms241612978