
Exploration into Natural Variation Genes Associated with Determinate and Capitulum-like Inflorescence in Brassica napus


Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization of the Mutant (MT) Plants
2.2. Genetic Analysis of the MT Phenotype
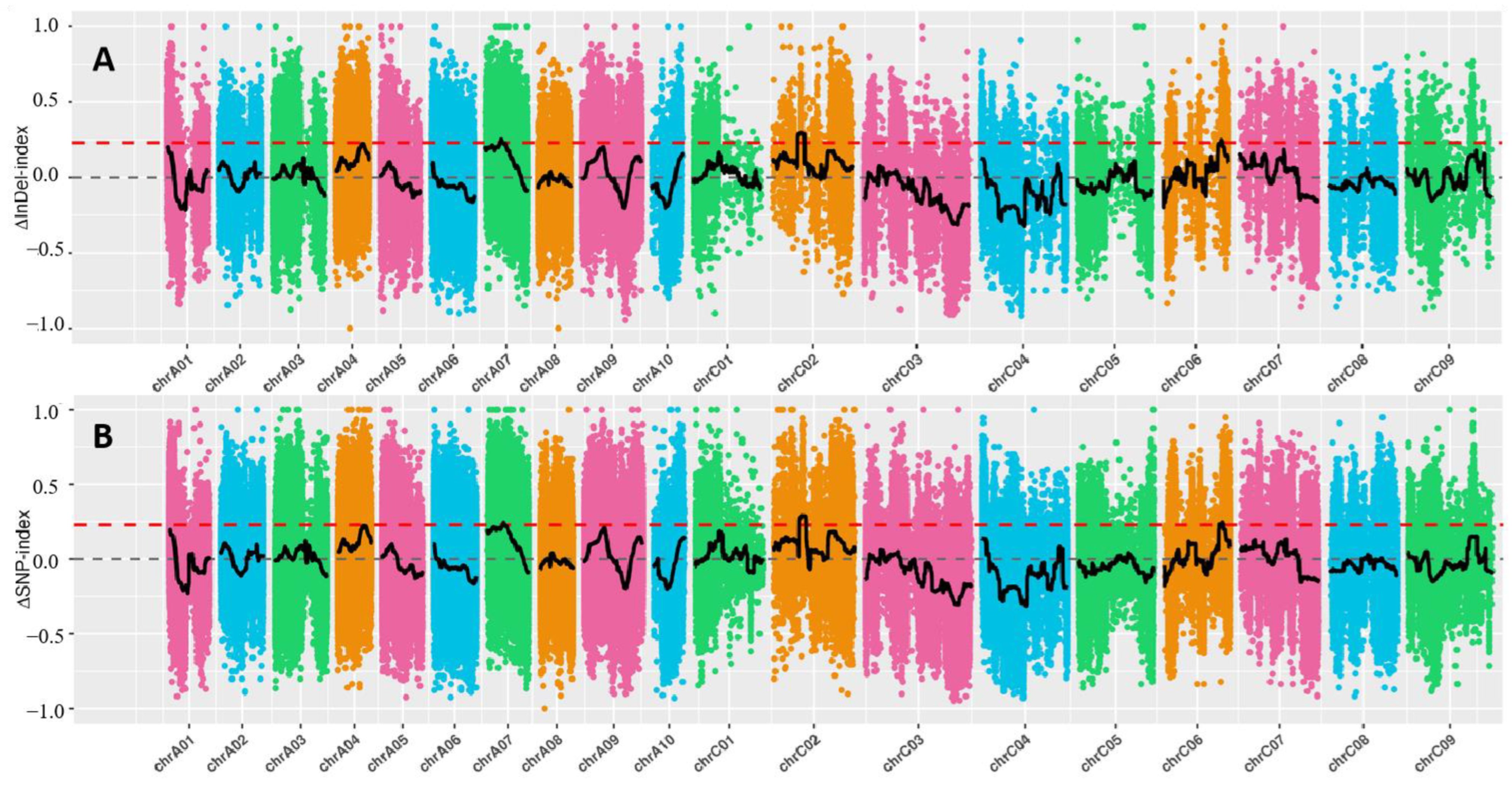
2.3. QTL Mapping by BSA-Seq Technique
2.4. Gene Expression Profile Analysis
2.5. Candidate Genes for Regulating the Determinate Inflorescence
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Field Experiment
4.2. Investigation and Analysis of the Agronomic Traits of MT and WT Plants
4.3. BSA-Seq Analysis
4.4. RNA Extraction, Preparation, Sequencing, and Data Analysis
4.5. Identification and Annotation of Differentially Expressed Genes (DEGs)
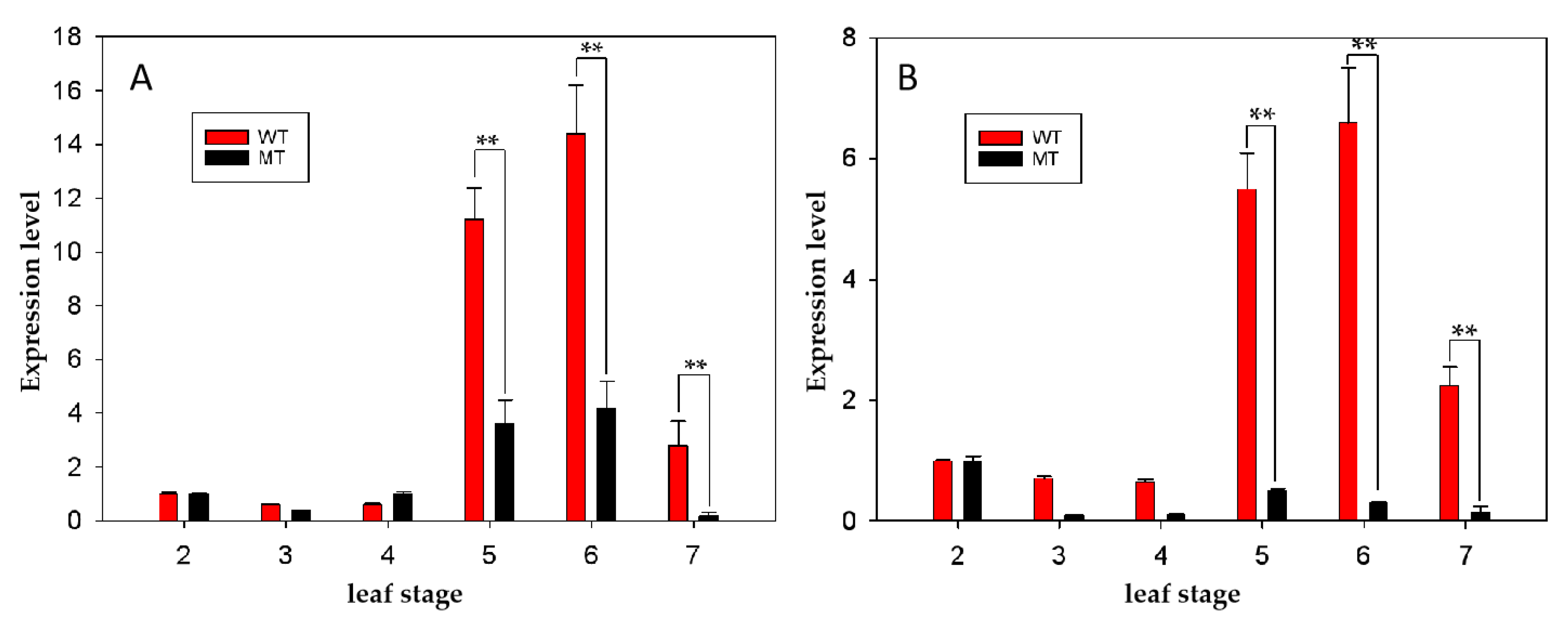
4.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beckman, C. Vegetable oils: Competition in a changing market. Bi-Wkly. Bull. Agric. Agri-Food Can. 2005, 11. [Google Scholar]
- Fu, T. Breeding and Utilization of Rapeseed Hybrid; Hubei Science Technology Press: Wuhan, China, 2000; pp. 167–169. [Google Scholar]
- Prusinkiewicz, P.; Erasmus, Y.; Lane, B.; Harder, L.D.; Coen, E. Evolution and development of inflorescence architectures. Science 2007, 316, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Plackett, A.R.G.; Powers, S.J.; Phillips, A.L.; Wilson, Z.A.; Hedden, P.; Thomas, S.G. The early inflorescence of Arabidopsis thaliana demonstrates positional effects in floral organ growth and meristem patterning. Plant Reprod. 2018, 31, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Shannon, S.; Meeks-Wagner, D.R. A Mutation in the Arabidopsis TFL1 Gene Affects Inflorescence Meristem Development. Plant Cell 1991, 3, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Honma, T.; Goto, K. CENTRORADIALIS/TERMINAL FLOWER 1 gene homolog is conserved inN. tabacum, a determinate inflorescence plant. J. Plant Res. 1998, 111, 289–294. [Google Scholar] [CrossRef]
- Zhang, H.; Miao, H.; Li, C.; Wei, L.; Duan, Y.; Ma, Q.; Kong, J.; Xu, F.; Chang, S. Ultra-dense SNP genetic map construction and identification of SiDt gene controlling the determinate growth habit in Sesamum indicum L. Sci. Rep. 2016, 6, 31556. [Google Scholar] [CrossRef]
- Kaur, H.; Banga, S.S. Discovery and mapping of Brassica juncea Sdt1 gene associated with determinate plant growth habit. Theor. Appl. Genet. 2015, 128, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yao, Y.; Xiao, L.; Zhao, Z.; Guo, S.; Fu, Z.; Du, D. Fine mapping of the Brassica napus Bnsdt1 gene associated with determinate growth habit. Theor. Appl. Genet. 2018, 131, 193–208. [Google Scholar] [CrossRef]
- Parcy, F.; Nilsson, O.; Busch, M.A.; Lee, I.; Weigel, D. A genetic framework for floral patterning. Nature 1998, 395, 561. [Google Scholar] [CrossRef]
- Saddic, L.A.; Huvermann, B.; Bezhani, S.; Su, Y.; Winter, C.M.; Kwon, C.S.; Collum, R.P.; Wagner, D. The LEAFY target LMI1 is a meristem identity regulator and acts together with LEAFY to regulate expression of CAULIFLOWER. Development 2006, 133, 1673–1682. [Google Scholar] [CrossRef]
- Lee, J.; Oh, M.; Park, H.; Lee, I. SOC1 translocated to the nucleus by interaction with AGL24 directly regulates LEAFY. Plant J. 2008, 55, 832–843. [Google Scholar] [CrossRef]
- Liu, C.; Teo, Z.W.N.; Bi, Y.; Song, S.; Xi, W.; Yang, X.; Yin, Z.; Yu, H. A conserved genetic pathway determines inflorescence architecture in Arabidopsis and rice. Dev. Cell 2013, 24, 612–622. [Google Scholar] [CrossRef]
- Gregis, V.; Andrés, F.; Sessa, A.; Guerra, R.F.; Simonini, S.; Mateos, J.L.; Torti, S.; Zambelli, F.; Prazzoli, G.M.; Bjerkan, K.N.; et al. Identification of pathways directly regulated by SHORT VEGETATIVE PHASE during vegetative and reproductive development in Arabidopsis. Genome Biol. 2013, 14, R56. [Google Scholar] [CrossRef]
- Wils, C.R.; Kaufmann, K. Gene-regulatory networks controlling inflorescence and flower development in Arabidopsis thaliana. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.; Guli, C.L.; Yu, X.-H.; Smyth, D.R. terminal flower: A gene affecting inflorescence development in Arabidopsis thaliana. Plant J. 1992, 2, 103–116. [Google Scholar] [CrossRef]
- Ohshima, S.; Murata, M.; Sakamoto, W.; Ogura, Y.; Motoyoshi, F. Cloning and molecular analysis of the Arabidopsis gene Terminal Flower 1. Mol. Gen. Genet. MGG 1997, 254, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, T.; Duan, M.; Song, J.; Li, X. De novo Transcriptome Analysis of Sinapis alba in Revealing the Glucosinolate and Phytochelatin Pathways. Front. Plant Sci. 2016, 7, 259. [Google Scholar] [CrossRef]
- Ma, Q.; Liu, X.; Franks, R.G.; Xiang, Q.-Y.J. Alterations of CorTFL1 and CorAP1 expression correlate with major evolutionary shifts of inflorescence architecture in Cornus (Cornaceae)—A proposed model for variation of closed inflorescence forms. New Phytol. 2017, 216, 519–535. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, H.; Shen, Y.; Wang, J. Transcriptomic analysis of rice (Oryza sativa) endosperm using the RNA-Seq technique. Plant Mol. Biol. 2013, 81, 363–378. [Google Scholar] [CrossRef]
- Jones, S.I.; Vodkin, L.O. Using RNA-Seq to Profile Soybean Seed Development from Fertilization to Maturity. PLoS ONE 2013, 8, e59270. [Google Scholar] [CrossRef] [PubMed]
- Loraine, A.E.; McCormick, S.; Estrada, A.; Patel, K.; Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol. 2013, 162, 1092–1109. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Dong, N.; Wang, Y.; Li, G.; Wang, L.; Guo, X.; Li, J.; Wen, Z.; Wei, W. RNA-seq transcriptome analysis of the immature seeds of two Brassica napus lines with extremely different thousand-seed weight to identify the candidate genes related to seed weight. PLoS ONE 2018, 13, e0191297. [Google Scholar] [CrossRef]
- Wenger, J.W.; Schwartz, K.; Sherlock, G. Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS Genet. 2010, 6, e1000942. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S. QTL–seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Guo, Z.; Cai, L.; Chen, Z.; Wang, R.; Zhang, L.; Guan, S.; Zhang, S.; Ma, W.; Liu, C.; Pan, G. Identification of candidate genes controlling chilling tolerance of rice in the cold region at the booting stage by BSA-Seq and RNA-Seq. R. Soc. Open Sci. 2020, 7, 201081. [Google Scholar] [CrossRef]
- Ye, S.; Yan, L.; Ma, X.; Chen, Y.; Wu, L.; Ma, T.; Zhao, L.; Yi, B.; Ma, C.; Tu, J. Combined BSA-seq based mapping and RNA-seq profiling reveal candidate genes associated with plant architecture in Brassica napus. Int. J. Mol. Sci. 2022, 23, 2472. [Google Scholar] [CrossRef]
- Klein, H.; Xiao, Y.; Conklin, P.A.; Govindarajulu, R.; Kelly, J.A.; Scanlon, M.J.; Whipple, C.J.; Bartlett, M. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3 Genes. Genomes Genet. 2018, 8, 3583–3592. [Google Scholar]
- Luo, M.; Lu, B.; Shi, Y.; Zhao, Y.; Liu, J.; Zhang, C.; Wang, Y.; Liu, H.; Shi, Y.; Fan, Y. Genetic basis of the oil biosynthesis in ultra-high-oil maize grains with an oil content exceeding 20%. Front. Plant Sci. 2023, 14, 1168216. [Google Scholar]
- Yu, K.; He, Y.; Li, Y.; Li, Z.; Zhang, J.; Wang, X.; Tian, E. Quantitative Trait Locus Mapping Combined with RNA Sequencing Reveals the Molecular Basis of Seed Germination in Oilseed Rape. Biomolecules 2021, 11, 1780. [Google Scholar] [CrossRef]
- Khattak, A.N.; Wang, T.; Yu, K.; Yang, R.; Wan, W.; Ye, B.; Tian, E. Exploring the basis of 2-propenyl and 3-butenyl glucosinolate synthesis by QTL mapping and RNA-sequencing in Brassica juncea. PLoS ONE 2019, 14, e0220597. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Zhang, Y.; Wang, L.; Gao, Y.; Li, D.; Yu, J.; Zhou, R.; Zhang, X. Genetic dissection and fine mapping of a novel dt gene associated with determinate growth habit in sesame. BMC Genet. 2018, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Bernard, R.L. Two Genes Affecting Stem Termination in Soybeans1. Crop Sci. 1972, 12, 235–239. [Google Scholar] [CrossRef]
- Li, B.; Tang, M.; Nelson, A.; Caligagan, H.; Zhou, X.; Clark-Wiest, C.; Ngo, R.; Brady, S.M.; Kliebenstein, D.J. Network-Guided Discovery of Extensive Epistasis between Transcription Factors Involved in Aliphatic Glucosinolate Biosynthesis. Plant Cell 2018, 30, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H. MutMap+: Genetic mapping and mutant identification without crossing in rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Huang, Z.; Li, Z.; Zhang, S.; Liu, X.; Gu, D. De novo transcriptome sequencing of Cryptotermes domesticus and comparative analysis of gene expression in response to different wood species. Gene 2016, 575 Pt 3, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Items | WT (Mean) | MT (Mean) |
|---|---|---|
| Raw Reads Number | 48,705,785 | 46,319,327 |
| Raw Bases Number | 7,305,867,800 | 6,947,899,100 |
| Clean Reads Number (%) | 47,184,448 (96.88) | 44,944,151 (96.96) |
| Clean Bases Number | 7,077,667,200 | 6,741,622,600 |
| Low-Quality Reads Number (%) | 344,567 (0.71) | 360,269 (0.78) |
| Mapped Reads (%) | 41,135,862 (87.18) | 39,824,899 (88.60) |
| Unmapped Reads | 6,048,586 | 5,119,252 |
| Multi-Map Reads (%) | 6,988,386 (14.81) | 6,857,621 (15.26) |
| Ns Reads Number (%) | 3115 (0.01) | 3694 (0.01) |
| Adapter Polluted Reads Number (%) | 1,173,656 (2.41) | 1,011,214 (2.18) |
| Raw Q30 Bases Rate (%) | 93.84 | 93.67 |
| Clean Q30 Bases Rate (%) | 94.14 | 94.01 |
| Exon (%) | 15,111,070 (94.36) | 14,619,066 (94.53) |
| Intron (%) | 249,023 (1.56) | 221,680 (1.43) |
| Intergenic (%) | 649,815 (4.07) | 623,039 (4.04) |
| Novel Transcripts | 160,798 | 129,446 |
| Gene ID in Darmor-Bzh | Arabidopsis ID | E-Value | Gene Name | Gene Function |
|---|---|---|---|---|
| BnaC02g02980D | AT5G03680 | 0 | PETAL LOSS | Organ initiation and orientation |
| BnaC02g02940D | AT2G18960 | 4 × 10102 | OPEN STOMATA 2 | Regulation of stomatal movement |
| BnaC02g02910D | AT2G46030 | 3 × 1085 | UBIQUITIN-CONJUGATING ENZYME 6 | Ubiquitin-dependent protein catabolic process |
| BnaC02g02900D | AT5G03840 | 0 | TERMINAL FLOWER 1 | Controls inflorescence meristem identity |
| BnaC02g02880D | AT5G64140 | 1 × 1055 | RIBOSOMAL PROTEIN S28 | Ribosomal small subunit assembly and translation |
| BnaC02g02870D | AT5G03860 | 0 | MALATE SYNTHASE | Encodes a protein with malate synthase activity |
| BnaC02g02830D | AT5G03940 | 0 | 54 CHLOROPLAST PROTEIN | Protein import into chloroplast thylakoid membrane |
| BnaC02g02820D | ||||
| BnaC06g25530D | AT1G68990 | 0 | MALE GAMETOPHYTE DEFECTIVE 3 | Transcription of mitochondrial genes |
| BnaC06g25500D | AT1G69120 | 0 | APETALA1, AP1 | Inflorescence meristem identity; specifies floral meristem and sepal identity |
| BnaC06g25460D | AT3G08900 | 2 × 1048 | REVERSIBLY GLYCOSYLATED POLYPEPTIDE 3 | UDP-L-arabinose metabolic process |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, W.; Zhao, H.; Yu, K.; Xiang, Y.; Dai, W.; Du, C.; Tian, E. Exploration into Natural Variation Genes Associated with Determinate and Capitulum-like Inflorescence in Brassica napus. Int. J. Mol. Sci. 2023, 24, 12902. https://doi.org/10.3390/ijms241612902
Wan W, Zhao H, Yu K, Xiang Y, Dai W, Du C, Tian E. Exploration into Natural Variation Genes Associated with Determinate and Capitulum-like Inflorescence in Brassica napus. International Journal of Molecular Sciences. 2023; 24(16):12902. https://doi.org/10.3390/ijms241612902
Chicago/Turabian StyleWan, Wei, Haifei Zhao, Kunjiang Yu, Yang Xiang, Wendong Dai, Caifu Du, and Entang Tian. 2023. "Exploration into Natural Variation Genes Associated with Determinate and Capitulum-like Inflorescence in Brassica napus" International Journal of Molecular Sciences 24, no. 16: 12902. https://doi.org/10.3390/ijms241612902
APA StyleWan, W., Zhao, H., Yu, K., Xiang, Y., Dai, W., Du, C., & Tian, E. (2023). Exploration into Natural Variation Genes Associated with Determinate and Capitulum-like Inflorescence in Brassica napus. International Journal of Molecular Sciences, 24(16), 12902. https://doi.org/10.3390/ijms241612902