

Glucagon and Its Receptors in the Mammalian Heart

Abstract
1. Introduction
2. Glucagon Receptor
3. Glucagon Receptor Regulation
4. Glucagon Receptor Agonists and Antagonists
5. Pharmacokinetics of Glucagon
6. Formation and Degradation of Glucagon
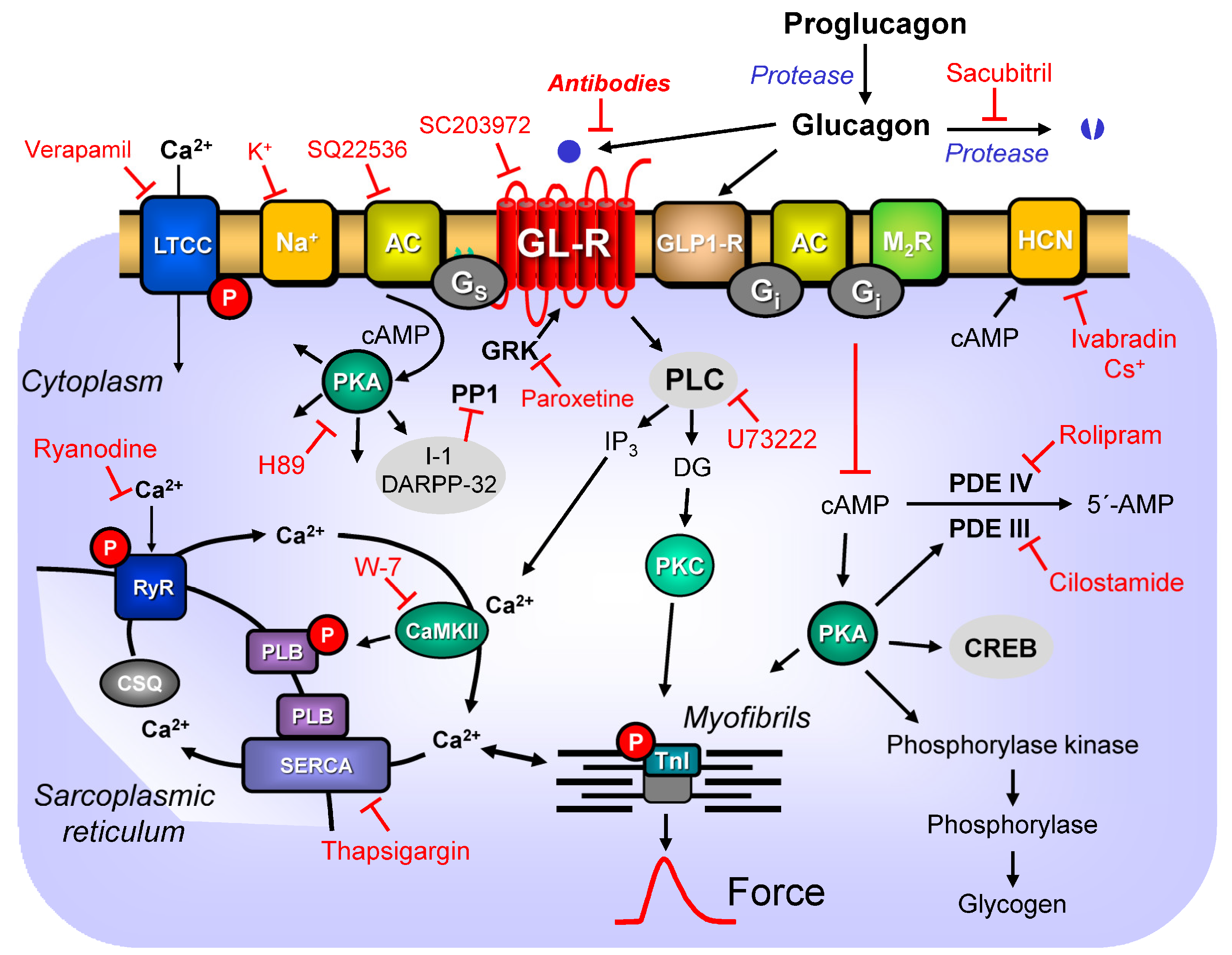
7. Signal Transduction
8. Inotropic and Chronotropic Effects of Glucagon in Animal Hearts
9. Electrophysiological Effects of Glucagon in Mammalian Hearts
10. Species
11. Region
12. Inotropic and Chronotropic Effects of Glucagon in Isolated Human Hearts
13. Clinical Relevance
13.1. Glucagon in Heart Failure
13.2. Glucagon in Cardiac Ischemia
13.3. Glucagon to Treat Intoxication
13.4. Glucagon and the Diabetic Heart
13.5. Glucagon and Arrhythmias
13.6. Glucagon Receptor Mutations and Functions
14. Glucagon Outside the Heart
15. Summary
16. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AC | adenylyl cyclase |
| AV-artery | special artery that transports blood into the AV-node |
| AV-block | failure of conduction of depolarization to pass through the AV-node |
| AV-node | specialized muscle fibers in the atrioventricular node of the heart |
| cAMP | 3′,5′ cyclic adenosine monophosphate |
| cGMP | 3′,5′ cyclic guanosine monophosphate |
| Ca2+ | divalent calcium ion(s) |
| DG | 1,2-diacylglycerol |
| EC50 | half maximally stimulatory concentration of a drug |
| EPAC | cAMP-binding exchange protein |
| ERK | extracellular signal-activated kinase |
| Fura2 | a drug that shows Ca2+-dependent changes in its fluorescence |
| GLP-1 | glucagon-like-peptide-1 |
| GLP-1-R | glucagon-like-peptide-1-receptor |
| Gi | G-protein that inhibits the activity of AC |
| Gq | G-protein that stimulates PLC |
| Gs | G-protein that increases the activity of AC |
| GR | glucagon receptor |
| G-protein | GTP-binding protein |
| GTP | guanosine triphosphate |
| h | hours |
| H89 | an inhibitor of the activity of PKA |
| IBMX | 3-isobutyl-1-methyl-xanthine, an inhibitor of the activity of PDEs |
| IC50 | half maximally inhibitory concentration of a drug |
| IP3 | inositol-1,2,5 trisphosphate |
| kDa | kilo Dalton (molecular weight of a protein) |
| KO | knockout, animal with genetic deletion of a gene |
| LTCC | L-type calcium ion channel |
| mg | milli (10−3) gram |
| µg | micro (10−6) gram |
| MAP kinase | mitogen activated protein kinase |
| min | minute(s) |
| mL | milli liter |
| mM | milli (10−3) molar concentration of drug |
| µM | micro (10−6) molar concentration of drug |
| Mn2+ | divalent form of magnesium |
| mRNA | messenger ribonucleotides |
| nd | not determined |
| nM | nano (10−9) molar concentration of drug |
| PCE | positive chronotropic effect (increase the number of heartbeats per minute) |
| PDE | phosphodiesterase |
| PIE | positive inotropic effect (increase in force of contraction) |
| pH | potentia hydrogenii: negative decadic concentration of free protons in an aqueous solution |
| PKA | cAMP-dependent protein kinase |
| PLB | phospholamban |
| PLC | phospholipase C |
| PPARα | perixosome proliferation-activated receptor alpha |
| pM | pico (10−12) molar concentration of drug |
| RNAse | ribonucleotide degrading enzyme |
| SERCA | sarcoplasmic reticular Ca2+-ATP(adenosine triphosphate)ase |
| SGLT2 | sodium glucose transporter 2 |
| TnI | inhibitory subunit of troponin |
| X-ray | Roentgen rays |
References
- Ahrén, B. Glucagon—Early breakthroughs and recent discoveries. Peptides 2015, 67, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, E.W.; Rall, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Farah, A.; Tuttle, R. Studies on the pharmacology of glucagon. J. Pharmacol. Exp. Ther. 1960, 129, 49–55. [Google Scholar] [PubMed]
- Lucchesi, B.R. Cardiac actions of glucagon. Circ. Res. 1968, 22, 777–787. [Google Scholar] [CrossRef]
- Glick, G.; Parmley, W.W.; Wechsler, A.S.; Sonnenblick, E.H. Glucagon. Its enhancement of cardiac performance in the cat and dog and persistence of its inotropic action despite beta-receptor blockade with propranolol. Circ. Res. 1968, 22, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Baiio, L.L.; Yusta, B.; Mulvihill, E.E.; Cao, X.; Streutker, C.J.; Butany, J.; Cappola, T.P.; Margulies, K.B.; Drucker, D.J. GLP 1 receptor expression within the human heart. Endocrinology 2018, 159, 1570–1584. [Google Scholar]
- Munroe, D.G.; Gupta, A.K.; Kooshesh, F.; Vyas, T.B.; Rizkalla, G.; Wang, H.; Demchyshyn, L.; Yang, Z.J.; Kamboj, R.K.; Chen, H. Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 1999, 96, 1569–1573. [Google Scholar] [CrossRef]
- Jelinek, L.J.; Lok, S.; Rosenberg, G.B.; Smith, R.A.; Grant, F.J.; Biggs, S.; Bensch, P.A.; Kuijper, J.L.; Sheppard, P.O.; Sprecher, C.A.; et al. Expression cloning and signaling properties of the rat glucagon receptor. Science 1993, 259, 1614–1616. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, D.J.; Occi, J.L.; Hey, P.J.; Strader, C.D.; Graziano, M.P. Cloning and expression of a human glucagon receptor. Biochem. Biophys. Res. Commun. 1994, 198, 328–334. [Google Scholar] [CrossRef]
- Koth, C.M.; Murray, J.M.; Mukund, S.; Madjidi, A.; Minn, A.; Clarke, H.J.; Wong, T.; Chiang, V.; Luis, E.; Estevez, A.; et al. Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 14393–14398. [Google Scholar] [CrossRef]
- Yang, D.H.; Zhou, C.H.; Liu, Q.; Wang, M.W. Landmark studies on the glucagon subfamily of GPCRs: From small molecule modulators to a crystal structure. Acta Pharmacol. Sin. 2015, 36, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, D.; de Graaf, C.; Moeller, A.; West, G.M.; Dharmarajan, V.; Wang, C.; Siu, F.Y.; Song, G.; Reedtz-Runge, S. Conformational states of the full-length glucagon receptor. Nat. Commun. 2015, 6, 7859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiao, A.; Yang, L.; Van Eps, N.; Frederiksen, K.S.; Yang, D.; Dai, A.; Cai, X.; Zhang, H.; Yi, C.; et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 2018, 553, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Krishna Kumar, K.; O’Brien, E.S.; Habrian, C.H.; Latorraca, N.R.; Wang, H.; Tuneew, I.; Montabana, E.; Marqusee, S.; Hilger, D.; Isacoff, E.Y.; et al. Negative allosteric modulation of the glucagon receptor by RAMP2. Cell 2023, 186, 1465–1477.e18. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Tastenoy, M.; Vertongen, P.; Robberecht, P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol. Cell. Endocrinol. 1994, 105, 131–137. [Google Scholar] [CrossRef]
- Hansen, L.H.; Abrahamsen, N.; Nishimura, E. Glucagon receptor mRNA distribution in rat tissues. Peptides 1995, 16, 1163–1166. [Google Scholar] [CrossRef]
- Ali, S.; Ussher, J.R.; Baggio, L.L.; Kabir, M.G.; Charron, M.J.; Ilkayeva, O.; Newgard, C.B.; Drucker, D.J. Cardiomyocyte glucagon receptor signaling modulates outcomes in mice with experimental myocardial infarction. Mol. Metab. 2014, 4, 132–143. [Google Scholar] [CrossRef]
- Baggio, L.L.; Ussher, J.R.; McLean, B.A.; Cao, X.; Kabir, M.G.; Mulvihill, E.E.; Mighiu, A.S.; Zhang, H.; Ludwig, A.; Seeley, R.J.; et al. The autonomic nervous system and cardiac GLP-1 receptors control heart rate in mice. Mol. Metab. 2017, 6, 1339–1349. [Google Scholar] [CrossRef]
- Watanabe, M.; Hirose, Y.; Sugimoto, M.; Nakanishi, M.; Watanabe, H.; Shimada, M. The distribution of tissue insulin receptors in the mouse by whole-body autoradiography. J. Recept. Res. 1992, 12, 13–37. [Google Scholar] [CrossRef]
- Lok, S.; Kuijper, J.L.; Jelinek, L.J.; Kramer, J.M.; Whitmore, T.E.; Sprecher, C.A.; Mathewes, S.; Grant, F.J.; Biggs, S.H.; Rosenberg, G.B.; et al. The human glucagon receptor encoding gene: Structure, cDNA sequence and chromosomal localization. Gene 1994, 140, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, P.; Wheeler, M.B.; Irwin, D.M. Evolution of receptors for proglucagon-derived peptides: Isolation of frog glucagon receptors. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 128, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Domene, R.; Orenes-Piñero, E.; Arribas-Leal, J.M.; Canovas-Lopez, S.; Hernández-Cascales, J. Evidence for a lack of inotropic and chronotropic effects of glucagon and glucagon receptors in the human heart. Cardiovasc. Diabetol. 2023, 22, 128. [Google Scholar] [CrossRef] [PubMed]
- Bomholt, A.B.; Johansen, C.D.; Christensen, J.B.; Kjeldsen, S.A.S.; Galsgaard, K.D.; Winther-Sørensen, M.; Serizawa, R.; Hornum, M.; Porrini, E.; Pedersen, J.; et al. Evaluation of commercially available glucagon receptor antibodies and glucagon receptor expression. Commun. Biol. 2022, 5, 1278. [Google Scholar] [CrossRef] [PubMed]
- Nobel-Allen, N.; Kirsch, M.; Lucchesi, B.R. Glucagon: Its enhancement of cardiac performance in the cat with chronic heart failure. J. Pharmacol. Exp. Ther. 1973, 187, 475–481. [Google Scholar] [PubMed]
- Sattler, R.W.; van Zwieten, P.A. The positive inotropic action of glucagon on the cat heart in situ. Klin. Wochenschr. 1972, 50, 531–533. [Google Scholar] [CrossRef]
- Winokur, S.; Nobel-Allen, N.L.; Lucchesi, B.R. The positive inotropic effect of glucagon in the chronically failed right ventricle as demonstrated in the isolated cat heart. Eur. J. Pharmacol. 1975, 32, 349–356. [Google Scholar] [CrossRef]
- Gold, H.K.; Prindle, K.H.; Levey, G.S.; Epstein, S.E. Effects of experimental heart failure on the capacity of glucagon to augment myocardial contractility and activate adenyl cyclase. J. Clin. Investig. 1970, 49, 999–1006. [Google Scholar] [CrossRef][Green Version]
- Marcus, M.L.; Skelton, C.L.; Prindle, K.H., Jr.; Epstein, S.E. Potentiation of the inotropic effects of glucagon by theophylline. J. Pharmacol. Exp. Ther. 1971, 179, 331–337. [Google Scholar]
- Regan, T.J.; Lehan, P.H.; Henneman, D.H.; Behar, A.; Hellems, H.K. Myocardial metabolic and contractile response to glucagon and Epinephrine. J. Lab. Clin. Med. 1964, 63, 638–647. [Google Scholar]
- Endoh, M. Correlation of cyclic AMP and cyclic GMP levels with changes in contractile force of dog ventricular myocardium during cholinergic antagonism of positive inotropic actions of histamine, glucagon, theophylline and papaverine. JPN J. Pharmacol. 1979, 29, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Antonaccio, M.J.; Lucchesi, B.R. The interaction of glucagon with theophylline, PGE1, isoproteerenol, ouabain, and CaC1 on the dog isolated papillary muscle. Life Sci. 1970, 9, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Nayler, W.G.; McInnes, I.; Chipperfield, D.; Carson, V.; Daile, P. The effect of glucagon on calcium exchangeability, coronary blood flow, myocardial function and high energy phosphate stores. J. Pharmacol. Exp. Ther. 1970, 171, 265–275. [Google Scholar] [PubMed]
- Lipski, J.I.; Kaminsky, D.; Donoso, E.; Friedberg, C.K. Electrophysiological effects of glucagon on the normal canine heart. Am. J. Physiol. 1972, 222, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Saegusa, K.; Ogiwara, Y.; Chiba, S. Different effectiveness of glucagon on the pacemaker activity and contractility in intact dog hearts and in isolated perfused right atria. JPN Heart J. 1986, 27, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Positive chronotropic and inotropic effects of glucagon on the canine isolated atrium. Tohoku J. Exp. Med. 1975, 115, 61–65. [Google Scholar] [CrossRef][Green Version]
- Kimura, T.; Kokubun, M.; Hashimoto, K. Primary effect of glucagon on positive chronotropism. Jpn. J. Pharmacol. 1974, 24, 279–283. [Google Scholar] [CrossRef]
- Smitherman, T.C.; Osborn, R.C., Jr.; Atkins, J.M. Cardiac dose response relationship for intravenously infused glucagon in normal intact dogs and men. Am. Heart J. 1978, 96, 363–371. [Google Scholar] [CrossRef]
- Bache, R.J.; McHale, P.A.; Curry, C.L.; Alexander, J.A.; Greenfield, J.C., Jr. Coronary and systemic hemodynamic effects of glucagon in the intact unanesthetized dog. J. Appl. Physiol. 1970, 29, 769–774. [Google Scholar] [CrossRef]
- Moir, T.W.; Naylor, W.G. Coronary vascular effects of glucagon in the isolated dog heart. Circ. Res. 1970, 26, 29–34. [Google Scholar] [CrossRef]
- Iwanij, V.; Hur, K.C. Development of physiological responsiveness to glucagon during embryogenesis of avian heart. Dev. Biol. 1987, 122, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Méry, P.F.; Brechler, V.; Pavoine, C.; Pecker, F.; Fischmeister, R. Glucagon stimulates the cardiac Ca2+ current by activation of adenylyl cyclase and inhibition of phosphodiesterase. Nature 1990, 345, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.F.; MacLeod, K.M.; McNeill, J.H. Glucagon-induced densensitization: Correlation between cyclic AMP levels and contractile force. Eur. J. Pharmacol. 1982, 79, 147–150. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, K.M.; Rodgers, R.L.; McNeill, J.H. Characterization of glucagon-induced changes in rate, contractility and cyclic AMP levels in isolated cardiac preparations of the rat and guinea pig. J. Pharmacol. Exp. Ther. 1981, 217, 798–804. [Google Scholar]
- Rodgers, R.L.; MacLeod, K.M.; McNeill, J.H. Responses of rat and guinea pig hearts to glucagon. Lack of evidence for a dissociation between changes in myocardial cyclic 3′5′-adenosine monophosphate and contractility. Circ. Res. 1981, 49, 216–225. [Google Scholar] [CrossRef]
- Hadházy, P. Actions of glucagon and dibutyryl cyclic 3′,5′-AMP on chronotropic responses to vagal stimulation and acetylcholine. Pharmacology 1973, 9, 285–293. [Google Scholar] [CrossRef]
- Green, R.L. Paradoxical effects of calcium-glucagon interaction on cardiac muscle contractility of isolated guinea pig atria. Pharmacology 1977, 15, 519–528. [Google Scholar] [CrossRef]
- Spilker, B. Comparison of the inotropic response to glucagon, ouabain and noradrenaline. Br. J. Pharmacol. 1970, 40, 382–395. [Google Scholar] [CrossRef]
- Klein, S.W.; Morch, J.E.; Mahon, W.A. Cardiovascular effects of glucagon in man. Can. Med. Assoc. J. 1968, 98, 1161–1164. [Google Scholar]
- Strauer, B.E. The influence of glucagon on myocardial mechanics of papillary muscles obtained from patients with chronic congestive heart failure. Naunyn Schmiedebergs Arch. Pharmakol. 1971, 270, 90–93. [Google Scholar] [CrossRef]
- Linhart, J.W.; Barold, S.S.; Cohen, L.S.; Hildner, F.J.; Samet, P. Cardiovascular effects of glucagon in man. Am. J. Cardiol. 1968, 22, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.F., Jr. Glucagon and the cardiovascular system. Ann. Intern. Med. 1969, 71, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Manchester, J.H.; Parmley, W.W.; Matloff, J.M.; Leidtke, A.J.; LaRaia, P.J.; Herman, M.V.; Sonnenblock, E.H.; Gorlin, R. Effects of glucagon on myocardial oxygen consumption and coronary blood flow in man and in dog. Circulation 1970, 41, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wildenthal, K.; Allen, D.O.; Karlsson, J.; Wakeland, J.R.; Clark, C.M., Jr. Responsiveness to glucagon in fetal hearts. Species variability and apparent disparities between changes in beating, adenylate cyclase activation, and cyclic AMP concentration. J. Clin. Investig. 1976, 57, 551–558. [Google Scholar] [CrossRef]
- Boder, G.B.; Harley, R.J.; Johnson, I.S. Recording system for monitoring automaticity of heart cells in culture. Nature 1971, 231, 531–532. [Google Scholar] [CrossRef]
- Boder, G.B.; Johnson, I.S. Comparative effects of some cardioactive agents on automaticity of cultured heart cells. J. Mol. Cell. Cardiol. 1972, 4, 453–463. [Google Scholar] [CrossRef]
- Necco, A.; Dasdia, T.; Di Francesco, D.; Ferroni, A. Action of ouabain, oligomycin, and glucagon on cultured heart cells treated with adriamycin. Pharmacol. Res. Commun. 1976, 8, 105–109. [Google Scholar] [CrossRef]
- Greeff, K. Einfluss von Pharmaka auf die Kontraktilität des Herzens [Effect of drugs on the contractility of the heart]. Verh. Dtsch. Ges. Kreislaufforsch. 1976, 42, 80–92. (In German) [Google Scholar]
- Henry, P.D.; Dobson, J.G., Jr.; Sobel, B.E. Dissociations between changes in myocardial cyclic adenosine monophosphate and contractility. Circ. Res. 1975, 36, 392–400. [Google Scholar] [CrossRef]
- Goldstein, R.E.; Skelton, C.L.; Levey, G.S.; Glancy, D.L.; Beiser, G.D.; Epstein, S.E. Effects of chronic heart failure on the capacity of glucagon to enhance contractility and adenyl cyclase activity of human papillary muscles. Circulation 1971, 44, 638–648. [Google Scholar] [CrossRef]
- Prasad, K. Glucagon-induced changes in the action potential, contraction, and Na+-K+-ATPase of cardiac muscle. Cardiovasc. Res. 1975, 9, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cascales, J. Does glucagon have a positive inotropic effect in the human heart? Cardiovasc. Diabetol. 2018, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.M.; Bøgevig, S.; Holst, J.J.; Knop, F.K.; Christensen, M.B. Hemodynamic Effects of Glucagon: A Literature Review. J. Clin. Endocrinol. Metab. 2018, 103, 1804–1812. [Google Scholar] [CrossRef]
- Parmley, W.W.; Glick, G.; Sonnenblick, E.H. Cardiovascular effects of glucagon in man. N. Engl. J. Med. 1968, 279, 12–17. [Google Scholar] [CrossRef]
- Mukharji, A.; Drucker, D.J.; Charron, M.J.; Swoap, S.J. Oxyntomodulin increases intrinsic heart rate through the glucagon receptor. Physiol. Rep. 2013, 1, e00112. [Google Scholar] [CrossRef]
- Kiss, Z.; Tkachuk, V.A. Guanine-nucleotide-dependent inhibition of adenylate cyclase of rabbit heart by glucagon. Eur. J. Biochem. 1984, 142, 323–328. [Google Scholar] [CrossRef]
- Gonzalez-Muñoz, C.; Nieto-Cerón, S.; Cabezas-Herrera, J.; Hernández-Cascales, J. Glucagon increases contractility in ventricle but not in atrium of the rat heart. Eur. J. Pharmacol. 2008, 587, 243–247. [Google Scholar] [CrossRef]
- Harney, J.A.; Rodgers, R.L. Insulin-like stimulation of cardiac fuel metabolism by physiological levels of glucagon: Involvement of PI3K but not cAMP. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E155–E161. [Google Scholar] [CrossRef] [PubMed]
- Brunt, M.E.; McNeill, J.H. The effect of glucagon on rat cardiac cyclic AMP, phosphorylase a and force of contraction. Arch. Int. Pharmacodyn. Ther. 1978, 233, 42–52. [Google Scholar]
- Merino, B.; Quesada, I.; Hernández-Cascales, J. Glucagon Increases Beating Rate but Not Contractility in Rat Right Atrium. Comparison with Isoproterenol. PLoS ONE 2015, 10, e0132884. [Google Scholar] [CrossRef]
- Mayer, S.E.; Namm, D.H.; Rice, L. Effect of glucagon on cyclic 3′,5′-AMP, phosphorylase activity and contractility of heart muscle of the rat. Circ. Res. 1970, 26, 225–233. [Google Scholar] [CrossRef]
- Clark, C.M., Jr.; Beatty, B.; Allen, D.O. Evidence for delayed development of the glucagon receptor of adenylate cyclase in the fetal and neonatal rat heart. J. Clin. Investig. 1973, 52, 1018–1025. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graziano, M.P.; Hey, P.J.; Borkowski, D.; Chicchi, G.G.; Strader, C.D. Cloning and functional expression of a human glucagon-like peptide-1 receptor. Biochem. Biophys. Res. Commun. 1993, 196, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Levey, G.S.; Epstein, S.E. Activation of adenyl cyclase by glucagon in cat and human heart. Circ. Res. 1969, 24, 151–156. [Google Scholar] [CrossRef]
- Sauvadet, A.; Rohn, T.; Pecker, F.; Pavoine, C. Synergistic actions of glucagon and miniglucagon on Ca2+ mobilization in cardiac cells. Circ. Res. 1996, 78, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, P.; Robberecht, P.; Waelbroeck, M.; De Neef, P.; Camus, J.C.; Huu, A.N.; Roba, J.; Christophe, J. Topographical distribution of the secretin- and VIP-stimulated adenylate cyclase system in the heart of five animal species. Pflug. Arch. 1983, 397, 100–105. [Google Scholar] [CrossRef]
- Brechler, V.; Pavoine, C.; Hanf, R.; Garbarz, E.; Fischmeister, R.; Pecker, F. Inhibition by glucagon of the cGMP-inhibited low-Km cAMP phosphodiesterase in heart is mediated by a pertussis toxin-sensitive G-protein. J. Biol. Chem. 1992, 267, 15496–15501. [Google Scholar] [CrossRef]
- Kilts, J.D.; Gerhardt, M.A.; Richardson, M.D.; Sreeram, G.; Mackensen, G.B.; Grocott, H.P.; White, W.D.; Davis, R.D.; Newman, M.F.; Reves, J.G.; et al. Beta(2)-adrenergic and several other G protein-coupled receptors in human atrial membranes activate both G(s) and G(i). Circ. Res. 2000, 87, 705–709. [Google Scholar] [CrossRef]
- Menon, K.M.; Giese, S.; Jaffe, R.B. Hormone- and fluoride-sensitive adenylate cyclases in human fetal tissues. Biochim. Biophys. Acta 1973, 304, 203–209. [Google Scholar] [CrossRef][Green Version]
- Brown, H.D.; Chattopadhyay, S.K.; Matthews, W.S. Glucagon stimulation of adenyl cyclase activity of cardiac muscle. Naturwissenschaften 1968, 55, 181–182. [Google Scholar] [CrossRef]
- Christophe, J.; Waelbroeck, M.; Chatelain, P.; Robberecht, P. Heart receptors for VIP, PHI and secretin are able to activate adenylate cyclase and to mediate inotropic and chronotropic effects. Species variations and physiopathology. Peptides 1984, 5, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, P.; Damien, C.; Moroder, L.; Coy, D.H.; Wünsch, E.; Christophe, J. Receptor occupancy and adenylate cyclase activation in rat liver and heart membranes by 10 glucagon analogs modified in position 2, 3, 4, 25, 27 and/or 29. Regul. Pept. 1988, 21, 117–128. [Google Scholar] [CrossRef] [PubMed]
- England, P.J. Studies on the phosphorylation of the inhibitory subunit of troponin during modification of contraction in perfused rat heart. Biochem. J. 1976, 160, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Juan-Fita, M.J.; Vargas, M.L.; Kaumann, A.J.; Hernández Cascales, J. Rolipram reduces the inotropic tachyphylaxis of glucagon in rat ventricular myocardium. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 324–329. [Google Scholar] [CrossRef]
- Juan-Fita, M.J.; Vargas, M.L.; Hernández, J. The phosphodiesterase 3 inhibitor cilostamide enhances inotropic responses to glucagon but not to dobutamine in rat ventricular myocardium. Eur. J. Pharmacol. 2005, 512, 207–213. [Google Scholar] [CrossRef]
- Rochais, F.; Abi-Gerges, A.; Horner, K.; Lefebvre, F.; Cooper, D.M.; Conti, M.; Fischmeister, R.; Vandecasteele, G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ. Res. 2006, 98, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.C.; Grosso, D.S.; Romero, S.A.; Frangakis, C.J.; Byus, C.V.; Bressler, R. Ultrastructural studies of metabolically active isolated adult rat heart myocytes. J. Mol. Cell. Cardiol. 1978, 10, 449–459. [Google Scholar] [CrossRef]
- Watanabe, A.M.; Hathaway, D.R.; Besch, H.R., Jr.; Farmer, B.B.; Harris, R.A. alpha-Adrenergic reduction of cyclic adenosine monophosphate concentrations in rat myocardium. Circ. Res. 1977, 40, 596–602. [Google Scholar] [CrossRef]
- Cascieri, M.A.; Koch, G.E.; Ber, E.; Sadowski, S.J.; Louizides, D.; de Laszlo, S.E.; Hacker, C.; Hagmann, W.K.; MacCoss, M.; Chicchi, G.G.; et al. Characterization of a novel, non-peptidyl antagonist of the human glucagon receptor. J. Biol. Chem. 1999, 274, 8694–8697. [Google Scholar] [CrossRef]
- Murad, F.; Vaughan, M. Effect of glucagon on rat heart adenyl cyclase. Biochem. Pharmacol. 1969, 18, 1053–1059. [Google Scholar] [CrossRef]
- Shiao, L.L.; Cascieri, M.A.; Trumbauer, M.; Chen, H.; Sullivan, K.A. Generation of mice expressing the human glucagon receptor with a direct replacement vector. Transgenic Res. 1999, 8, 295–302. [Google Scholar] [CrossRef]
- Livingston, J.N.; Einarsson, K.; Backman, L.; Ewerth, S.; Arner, P. Glucagon receptor of human liver. Studies of its molecular weight and binding properties, and its ability to activate hepatic adenylyl cyclase of non-obese and obese subjects. J. Clin. Investig. 1985, 75, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.L. A reappraisal of the role of cyclic AMP in the physiological action of glucagon. Peptides 2023, 159, 170906. [Google Scholar] [CrossRef] [PubMed]
- Wallner, M.; Kolesnik, E.; Ablasser, K.; Khafaga, M.; Wakula, P.; Ljubojevic, S.; Thon-Gutschi, E.M.; Sourij, H.; Kapl, M.; Edmunds, N.J.; et al. Exenatide exerts a PKA-dependent positive inotropic effect in human atrial myocardium: GLP-1R mediated effects in human myocardium. J. Mol. Cell. Cardiol. 2015, 89 Pt B, 365–375. [Google Scholar] [CrossRef]
- Huh, K.Y.; Hwang, J.G.; Shin, W.; Baek, S.; Choi, J.; Lee, N.; Cho, Y.M.; Lee, H. A double-blind, placebo-controlled, single-ascending dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of HM15136, a novel long-acting glucagon analogue, in healthy subjects. Diabetes Obes. Metab. 2022, 24, 411–420. [Google Scholar] [CrossRef]
- Qureshi, S.A.; Rios Candelore, M.; Xie, D.; Yang, X.; Tota, L.M.; Ding, V.D.; Li, Z.; Bansal, A.; Miller, C.; Cohen, S.M.; et al. A novel glucagon receptor antagonist inhibits glucagon-mediated biological effects. Diabetes 2004, 53, 3267–3273. [Google Scholar] [CrossRef]
- O’Harte, F.P.; Franklin, Z.J.; Rafferty, E.P.; Irwin, N. Characterisation of structurally modified analogues of glucagon as potential glucagon receptor antagonists. Mol. Cell. Endocrinol. 2013, 381, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Chepurny, O.G.; Matsoukas, M.T.; Liapakis, G.; Leech, C.A.; Milliken, B.T.; Doyle, R.P.; Holz, G.G. Nonconventional glucagon and GLP-1 receptor agonist and antagonist interplay at the GLP-1 receptor revealed in high-throughput FRET assays for cAMP. J. Biol. Chem. 2019, 294, 3514–3531, Erratum in J. Biol. Chem. 2019, 294, 8714. [Google Scholar] [CrossRef]
- Irwin, N.; Franklin, Z.J.; O’Harte, F.P. desHis1Glu9-glucagon-[mPEG] and desHis1Glu9(Lys30PAL)-glucagon: Long-acting peptide-based PEGylated and acylated glucagon receptor antagonists with potential antidiabetic activity. Eur. J. Pharmacol. 2013, 709, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, H.; Xu, H.; Liu, Y.; Cai, M.; Shi, Y.; Zhang, J.; Wang, H. High glucose-induced glucagon resistance and membrane distribution of GCGR revealed by super-resolution imaging. iScience 2023, 26, 105967. [Google Scholar] [CrossRef]
- Kjeldsen, S.A.S.; Hansen, L.H.; Esser, N.; Mongovin, S.; Winther-Sørensen, M.; Galsgaard, K.D.; Hunt, J.E.; Kissow, H.; Ceutz, F.R.; Terzic, D.; et al. Neprilysin Inhibition Increases Glucagon Levels in Humans and Mice with Potential Effects on Amino Acid Metabolism. J. Endocr. Soc. 2021, 5, bvab084. [Google Scholar] [CrossRef]
- Liang, Y.; Osborne, M.C.; Monia, B.P.; Bhanot, S.; Gaarde, W.A.; Reed, C.; She, P.; Jetton, T.L.; Demarest, K.T. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 2004, 53, 410–417. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, M.G.; Geerts, B.F.; Morgan, E.S.; Brandt, T.A.; de Kam, M.L.; Romijn, J.A.; Cohen, A.F.; Bhanot, S.; Burggraaf, J. First proof of pharmacology in humans of a novel glucagon receptor antisense drug. J. Clin. Pharmacol. 2015, 55, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, N.; Lundgren, K.; Nishimura, E. Regulation of glucagon receptor mRNA in cultured primary rat hepatocytes by glucose and cAMP. J. Biol. Chem. 1995, 270, 15853–15857. [Google Scholar] [CrossRef]
- Robberecht, P.; Pochet, R.; Chatelain, P.; Verloes, A.; Camus, J.C.; de Neef, P.; Christophe, J. Decreased secretin and glucagon responsiveness of adenylate cyclase in cardiac membranes from hypothyroid rats. FEBS Lett. 1981, 132, 33–36. [Google Scholar] [CrossRef]
- Chatelain, P.; Robberecht, P.; De Neef, P.; Camus, J.C.; Christophe, J. Early decrease in secretin-, glucagon-, and isoproterenol-stimulated cardiac adenylate cyclase activity in rats treated with isoproterenol. Biochem. Pharmacol. 1982, 31, 347–352. [Google Scholar] [CrossRef]
- Taton, G.; Chatelain, P.; Delhaye, M.; Camus, J.C.; De Neef, P.; Waelbroeck, M.; Tatemoto, K.; Robberecht, P.; Christophe, J. Vasoactive intestinal peptide (VIP) and peptide having N-terminal histidine and C-terminal isoleucine amide (PHI) stimulate adenylate cyclase activity in human heart membranes. Peptides 1982, 3, 897–900. [Google Scholar] [CrossRef]
- Srikant, C.B.; Freeman, D.; McCorkle, K.; Unger, R.H. Binding and biologic activity of glucagon in liver cell membranes of chronically hyperglucagonemic rats. J. Biol. Chem. 1977, 252, 7434–7438. [Google Scholar] [CrossRef]
- Iyengar, R.; Mintz, P.W.; Swartz, T.L.; Birnbaumer, L. Divalent cation-induced desensitization of glucagon-stimulable adenylyl cyclase in rat liver plasma membrane. GTP-dependent stimulation by glucagon. J. Biol. Chem. 1980, 255, 11875–11882. [Google Scholar] [CrossRef]
- Krilov, L.; Nguyen, A.; Miyazaki, T.; Unson, C.G.; Williams, R.; Lee, N.H.; Ceryak, S.; Bouscarel, B. Dual mode of glucagon receptor internalization: Role of PKCα, GRKs and β-arrestins. Exp. Cell Res. 2011, 317, 2981–2994. [Google Scholar] [CrossRef]
- Heurich, R.O.; Buggy, J.J.; Vandenberg, M.T.; Rossomando, A.J. Glucagon induces a rapid and sustained phosphorylation of the human glucagon receptor in Chinese hamster ovary cells. Biochem. Biophys. Res. Commun. 1996, 220, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Chen, Y.; Shenoy, S.K. Agonist-activated glucagon receptors are deubiquitinated at early endosomes by two distinct deubiquitinases to facilitate Rab4a-dependent recycling. J. Biol. Chem. 2020, 295, 16630–16642. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Seaquist, E.R. Translational aspects of glucagon: Current use and future prospects. J. Endocrinol. 2023, 257, e220278. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Sokrat, B.; Capozzi, M.E.; El, K.; Bai, Y.; Jazic, A.; Han, B.; Krishnakumar, K.; D’Alessio, D.A.; Campbell, J.E.; et al. The Ubiquitination Status of the Glucagon Receptor determines Signal Bias. J. Biol. Chem. 2023, 299, 104690. [Google Scholar] [CrossRef]
- Schinner, S.; Dellas, C.; Schroder, M.; Heinlein, C.A.; Chang, C.; Fischer, J.; Knepel, W. Repression of glucagon gene transcription by peroxisome proliferator-activated receptor gamma through inhibition of Pax6 transcriptional activity. J. Biol. Chem. 2002, 277, 1941–1948. [Google Scholar] [CrossRef]
- Mortensen, O.H.; Dichmann, D.S.; Abrahamsen, N.; Grunnet, N.; Nishimura, E. Identification of a novel human glucagon receptor promoter: Regulation by cAMP and PGC-1alpha. Gene 2007, 393, 127–136. [Google Scholar] [CrossRef]
- Iizuka, K.; Tomita, R.; Takeda, J.; Horikawa, Y. Rat glucagon receptor mRNA is directly regulated by glucose through transactivation of the carbohydrate response element binding protein. Biochem. Biophys. Res. Commun. 2012, 417, 1107–1112. [Google Scholar] [CrossRef]
- Vila Petroff, M.G.; Egan, J.M.; Wang, X.; Sollott, S.J. Glucagon-like peptide-1 increases cAMP but fails to augment contraction in adult rat cardiac myocytes. Circ. Res. 2001, 89, 445–452. [Google Scholar] [CrossRef]
- Mayo, K.E.; Miller, L.J.; Bataille, D.; Dalle, S.; Göke, B.; Thorens, B.; Drucker, D.J. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev. 2003, 55, 167–194. [Google Scholar] [CrossRef]
- Hawkes, C.P.; De Leon, D.D.; Rickels, M.R. Novel Preparations of Glucagon for the Prevention and Treatment of Hypoglycemia. Curr. Diab. Rep. 2019, 19, 97. [Google Scholar] [CrossRef]
- Watanabe, M.; Tamayama, T.; Hayasaki, H.; Kuno, M.; Kuroda, E.; Watanabe, Y.; Shimada, M. Whole body radioautographic analysis of the in vivo distribution of glucagon receptors in mice. Acta Histochem. Cytochem. 1997, 30, 471–476. [Google Scholar] [CrossRef][Green Version]
- Abraham, M.A.; Lam, T.K.T. Glucagon action in the brain. Diabetologia 2016, 59, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Müller, L.; De Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D.; et al. Chemical Hybridization of Glucagon and Thyroid Hormone Optimizes Therapeutic Impact for Metabolic Disease. Cell 2016, 167, 843–857.e14. [Google Scholar] [CrossRef] [PubMed]
- Wewer Albrechtsen, N.J. Glucagon receptor signaling in metabolic diseases. Peptides 2018, 100, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Gu, W.; Yang, J.; Bi, V.; Shen, Y.; Lee, E.; Winters, K.A.; Komorowski, R.; Zhang, C.; Patel, J.J.; et al. Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J. Pharmacol. Exp. Ther. 2009, 329, 102–111. [Google Scholar] [CrossRef]
- Velikyan, I.; Haack, T.; Bossart, M.; Evers, A.; Laitinen, I.; Larsen, P.; Plettenburg, O.; Johansson, L.; Pierrou, S.; Wagner, M.; et al. First-in-class positron emission tomography tracer for the glucagon receptor. EJNMMI Res. 2019, 9, 17. [Google Scholar] [CrossRef]
- Rodgers, R.L. Glucagon, cyclic AMP, and hepatic glucose mobilization: A half-century of uncertainty. Physiol. Rep. 2022, 10, e15263. [Google Scholar] [CrossRef]
- Teigen, I.A.; Åm, M.K.; Carlsen, S.M.; Christiansen, S.C. Pharmacokinetics of Intraperitoneally Delivered Glucagon in Pigs: A Hypothesis of First Pass Metabolism. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 505–511. [Google Scholar] [CrossRef]
- Slama, G.; Bennis, D.; Zirinis, P. Modes of glucagon administration. In Lefebvre; Springer: Berlin/Heidelberg, Germany, 2012; pp. 159–169. [Google Scholar]
- Pontiroli, A.E.; Tagliabue, E. Therapeutic Use of Intranasal Glucagon: Resolution of Hypoglycemia. Int. J. Mol. Sci. 2019, 20, 3646. [Google Scholar] [CrossRef]
- Pontiroli, A.E.; Calderara, A.; Perfetti, M.G.; Bareggi, S.R. Pharmacokinetics of intranasal, intramuscular and intravenous glucagon in healthy subjects and diabetic patients. Eur. J. Clin. Pharmacol. 1993, 45, 555–558. [Google Scholar] [CrossRef]
- Hinds, C.E.; Owen, B.M.; Hope, D.C.D.; Pickford, P.; Jones, B.; Tan, T.M.; Minnion, J.S.; Bloom, S.R. A glucagon analogue decreases body weight in mice via signalling in the liver. Sci. Rep. 2021, 11, 22577. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Chernish, S.M.; Brunelle, R.L. Gastrointestinal radiography with glucagon. Gastrointest. Radiol. 1979, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mühlhauser, I.; Koch, J.; Berger, M. Pharmacokinetics and bioavailability of injected glucagon: Differences between intramuscular, subcutaneous, and intravenous administration. Diabetes Care 1985, 8, 39–42. [Google Scholar] [CrossRef]
- Blauw, H.; Wendl, I.; DeVries, J.H.; Heise, T.; Jax, T.; PCDIAB Consortium. Pharmacokinetics and pharmacodynamics of various glucagon dosages at different blood glucose levels. Diabetes Obes. Metab. 2016, 18, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Rickels, M.R.; DuBose, S.N.; Toschi, E.; Beck, R.W.; Verdejo, A.S.; Wolpert, H.; Cummins, M.J.; Newswanger, B.; Riddell, M.C.; T1D Exchange Mini-Dose Glucagon Exercise Study Group. Mini-Dose Glucagon as a Novel Approach to Prevent Exercise-Induced Hypoglycemia in Type 1 Diabetes. Diabetes Care 2018, 41, 1909–1916. [Google Scholar] [CrossRef]
- Müller, T.D.; Finan, B.; Clemmensen, C.; DiMarchi, R.D.; Tschöp, M.H. The New Biology and Pharmacology of Glucagon. Physiol. Rev. 2017, 97, 721–766. [Google Scholar] [CrossRef] [PubMed]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sørensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef]
- Zeigerer, A.; Sekar, R.; Kleinert, M.; Nason, S.; Habegger, K.M.; Müller, T.D. Glucagon’s Metabolic Action in Health and Disease. Compr. Physiol. 2021, 11, 1759–1783. [Google Scholar] [CrossRef]
- Beaubien, G.; Schäfer, M.K.; Weihe, E.; Dong, W.; Chrétien, M.; Seidah, N.G.; Day, R. The distinct gene expression of the pro-hormone convertases in the rat heart suggests potential substrates. Cell Tissue Res. 1995, 279, 539–549. [Google Scholar] [CrossRef]
- Furuta, M.; Yano, H.; Zhou, A.; Rouillé, Y.; Holst, J.J.; Carroll, R.; Ravazzola, M.; Orci, L.; Furuta, H.; Steiner, D.F. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc. Natl. Acad. Sci. USA 1997, 94, 6646–6651. [Google Scholar] [CrossRef]
- Jones, H.B.; Reens, J.; Brocklehurst, S.R.; Betts, C.J.; Bickerton, S.; Bigley, A.L.; Jenkins, R.P.; Whalley, N.M.; Morgan, D.; Smith, D.M. Islets of Langerhans from prohormone convertase-2 knockout mice show α-cell hyperplasia and tumorigenesis with elevated α-cell neogenesis. Int. J. Exp. Pathol. 2014, 95, 29–48. [Google Scholar] [CrossRef]
- Fitzpatrick, D.; Ikram, H.; Nicholls, M.G.; Espiner, E.A. Hemodynamic, hormonal and electrolyte responses to prenalterol infusion in heart failure. Circulation 1983, 67, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.P.; Riley, M.; Elborn, J.S.; Stanford, C.F.; Shaw, C.; McKillop, J.M.; Buchanan, K.D. Regulatory peptides in the plasma of patients with chronic cardiac failure at rest and during exercise. Eur. Heart J. 1992, 13, 1399–1404. [Google Scholar] [CrossRef]
- Melenovsky, V.; Benes, J.; Franekova, J.; Kovar, J.; Borlaug, B.A.; Segetova, M.; Tura, A.; Pelikanova, T. Glucose Homeostasis, Pancreatic Endocrine Function, and Outcomes in Advanced Heart Failure. J. Am. Heart Assoc. 2017, 6, e005290. [Google Scholar] [CrossRef]
- Esser, N.; Mongovin, S.M.; Parilla, J.; Barrow, B.M.; Mundinger, T.O.; Fountaine, B.S.; Larmore, M.J.; Castillo, J.J.; Akter, R.; Hull, R.L.; et al. Neprilysin inhibition improves intravenous but not oral glucose-mediated insulin secretion via GLP-1R signaling in mice with β-cell dysfunction. Am. J. Physiol. Endocrinol. Metab. 2022, 322, E307–E318. [Google Scholar] [CrossRef] [PubMed]
- Trebbien, R.; Klarskov, L.; Olesen, M.; Holst, J.J.; Carr, R.D.; Deacon, C.F. Neutral endopeptidase 24.11 is important for the degradation of both endogenous and exogenous glucagon in anesthetized pigs. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E431–E438. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Figini, M.; Emanueli, C.; Geppetti, P.; Grady, E.F.; Gerard, N.P.; Ansell, J.; Payan, D.G.; Gerard, C.; Bunnett, N. The control of microvascular permeability and blood pressure by neutral endopeptidase. Nat. Med. 1997, 3, 904–907. [Google Scholar] [CrossRef]
- Bozkurt, B.; Nair, A.P.; Misra, A.; Scott, C.Z.; Mahar, J.H.; Fedson, S. Neprilysin Inhibitors in Heart Failure: The Science, Mechanism of Action, Clinical Studies, and Unanswered Questions. JACC Basic Transl. Sci. 2022, 8, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Fontés, G.; Lajoix, A.D.; Bergeron, F.; Cadel, S.; Prat, A.; Foulon, T.; Gross, R.; Dalle, S.; Le-Nguyen, D.; Tribillac, F.; et al. Miniglucagon (MG)-generating endopeptidase, which processes glucagon into MG, is composed of N-arginine dibasic convertase and aminopeptidase B. Endocrinology 2005, 146, 702–712. [Google Scholar] [CrossRef]
- Howard, J.W.; Kay, R.G.; Tan, T.; Minnion, J.; Creaser, C.S. Identification of plasma protease derived metabolites of glucagon and their formation under typical laboratory sample handling conditions. Rapid. Commun. Mass. Spectrom. 2015, 29, 171–181. [Google Scholar] [CrossRef]
- Pavoine, C.; Brechler, V.; Kervran, A.; Blache, P.; Le-Nguyen, D.; Laurent, S.; Bataille, D.; Pecker, F. Miniglucagon [glucagon-(19-29)] is a component of the positive inotropic effect of glucagon. Am. J. Physiol. 1991, 260 Pt 1, C993–C999. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, J.A.; Lin, S.Y.; Martino, T.A.; Arab, S.; Liu, P.; Husain, M.; Sole, M.J.; Belsham, D.D. Diurnal profiling of neuroendocrine genes in murine heart, and shift in proopiomelanocortin gene expression with pressure-overload cardiac hypertrophy. J. Mol. Endocrinol. 2008, 41, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Han, S.; Li, X.; Li, Z.; Zhao, P.; Dai, A.; Chang, R.; Tai, L.; Tan, Q.; Chu, X.; et al. Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 2020, 367, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Harding, S.E. The effect of pertussis toxin on beta-adrenoceptor responses in isolated cardiac myocytes from noradrenaline-treated guinea-pigs and patients with cardiac failure. Br. J. Pharmacol. 1992, 106, 115–122. [Google Scholar] [CrossRef]
- Ngan, E.S.; Chow, L.S.; Tse, D.L.; Du, X.; Wei, Y.; Mojsov, S.; Chow, B.K. Functional studies of a glucagon receptor isolated from frog Rana tigrina rugulosa: Implications on the molecular evolution of glucagon receptors in vertebrates. FEBS Lett. 1999, 457, 499–504. [Google Scholar] [CrossRef]
- Entman, M.L.; Levey, G.S.; Epstein, S.E. Mechanism of action of epinephrine and glucagon on the canine heart. Evidence for increase in sarcotubular calcium stores mediated by cyclic 3′,5′-AMP. Circ. Res. 1969, 25, 429–438. [Google Scholar] [CrossRef]
- Barritt, G.J.; Spiel, P.F. Effects of glucagon on 45Ca outflow exchange in the isolated perfused rat heart. Biochem. Pharmacol. 1981, 30, 1407–1414. [Google Scholar] [CrossRef]
- Blinks, J.R.; Lee, N.M.K.; Morgan, J.P. Ca++ transients in the mammalian heart muscle: Effects of inotropic agents on aequorin signals. In Federation Proceedings; Federation of American Societies for Experimental Biology: Rockville MD, USA, 1980; Volume 39, p. 854. [Google Scholar]
- Brand, M.D.; De Selincourt, C. Effects of glucagon and Na+ on the control of extramitochondrial free Ca2+ concentration by mitochondrial from liver and heart. Biochem. Biophys. Res. Commun. 1980, 92, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Denton, R.M. The activation of pyruvate dehydrogenase in the perfused rat heart by adrenaline and other inotropic agents. Biochem. J. 1981, 194, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Keely, S.L.; Corbin, J.D.; Park, C.R. Regulation of adenosine 3:5-monophosphate-dependent protein kinase. J. Biol. Chem. 1975, 250, 4832–4840. [Google Scholar] [CrossRef]
- Anton, S.E.; Kayser, C.; Maiellaro, I.; Nemec, K.; Möller, J.; Koschinski, A.; Zaccolo, M.; Annibale, P.; Falcke, M.; Lohse, M.J.; et al. Receptor-associated independent cAMP nanodomains mediate spatiotemporal specificity of GPCR signaling. Cell 2022, 185, 1130–1142.e11. [Google Scholar] [CrossRef]
- Dolce, B.; Christ, T.; Grammatika Pavlidou, N.; Yildirim, Y.; Reichenspurner, H.; Eschenhagen, T.; Nikolaev, V.O.; Kaumann, A.J.; Molina, C.E. Impact of phosphodiesterases PDE3 and PDE4 on 5-hydroxytryptamine receptor4-mediated increase of cAMP in human atrial fibrillation. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 291–298. [Google Scholar] [CrossRef]
- Cajulao, J.M.B.; Hernandez, E.; von Zastrow, M.E.; Sanchez, E.L. Glucagon receptor-mediated regulation of gluconeogenic gene transcription is endocytosis-dependent in primary hepatocytes. Mol. Biol. Cell. 2022, 33, ar90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yao, W.; Xia, J.; Wang, T.; Huang, F. Glucagon-Induced Acetylation of Energy-Sensing Factors in Control of Hepatic Metabolism. Int. J. Mol. Sci. 2019, 20, 1885. [Google Scholar] [CrossRef] [PubMed]
- Azari, J.; Huxtable, R.J. The mechanism of the adrenergic stimulation of taurine influx in the heart. Eur. J. Pharmacol. 1980, 61, 217–223. [Google Scholar] [CrossRef]
- Wakelam, M.J.; Murphy, G.J.; Hruby, V.J.; Houslay, M.D. Activation of two signal-transduction systems in hepatocytes by glucagon. Nature 1986, 323, 68–71. [Google Scholar] [CrossRef]
- Li, X.C.; Carretero, O.A.; Shao, Y.; Zhuo, J.L. Glucagon receptor-mediated extracellular signal-regulated kinase 1/2 phosphorylation in rat mesangial cells: Role of protein kinase A and phospholipase C. Hypertension 2006, 47, 580–585. [Google Scholar] [CrossRef]
- Xu, Y.; Xie, X. Glucagon receptor mediates calcium signaling by coupling to G alpha q/11 and G alpha i/o in HEK293 cells. J. Recept. Signal Transduct. Res. 2009, 29, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Spolitu, S.; Okamoto, H.; Dai, W.; Zadroga, J.A.; Wittchen, E.S.; Gromada, J.; Ozcan, L. Hepatic Glucagon Signaling Regulates PCSK9 and Low-Density Lipoprotein Cholesterol. Circ. Res. 2019, 124, 38–51. [Google Scholar] [CrossRef]
- Lindsay, R.T.; Ambery, P.; Jermutus, L.; Murray, A.J. Glucagon and exenatide improve contractile recovery following ischaemia/reperfusion in the isolated perfused rat heart. Physiol. Rep. 2023, 11, e15597. [Google Scholar] [CrossRef]
- Janssen, P.M. Myocardial contraction-relaxation coupling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1741–H1749. [Google Scholar] [CrossRef]
- Stemmer, P.; Akera, T. Concealed positive force-frequency relationships in rat and mouse cardiac muscle revealed by ryanodine. Am. J. Physiol. 1986, 251 Pt 2, H1106–H1110. [Google Scholar] [CrossRef]
- Endoh, M. Force-frequency relationship in intact mammalian ventricular myocardium: Physiological and pathophysiological relevance. Eur. J. Pharmacol. 2004, 500, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, I.; Zlokovic, B. Protective effects of glucagon during the anaphylactic response in guinea-pig isolated heart. Br. J. Pharmacol. 1982, 76, 483–489. [Google Scholar] [CrossRef]
- Hartmann, M.; Stumpe, T.; Schrader, J. alpha 1-Adrenoceptor stimulation inhibits the isoproterenol-induced effects on myocardial contractility and protein phosphorylation. Eur. J. Pharmacol. 1995, 287, 57–64. [Google Scholar] [CrossRef]
- Gaide, M.S.; Gelband, H.; Bassett, A.L. Relaxation by glucagon of potassium contracture in cat ventricle. Experientia 1981, 37, 281–282. [Google Scholar] [CrossRef]
- Neumann, J.; Kirchhefer, U.; Dhein, S.; Hofmann, B.; Gergs, U. The Roles of Cardiovascular H2-Histamine Receptors under Normal and Pathophysiological Conditions. Front. Pharmacol. 2021, 12, 732842. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Hofmann, B.; Dhein, S.; Gergs, U. Cardiac Roles of Serotonin (5-HT) and 5-HT-Receptors in Health and Disease. Int. J. Mol. Sci. 2023, 24, 4765. [Google Scholar] [CrossRef] [PubMed]
- He, L.P.; Mears, D.; Atwater, I.; Kitasato, H. Glucagon induces suppression of ATP-sensitive K+ channel activity through a Ca2+/calmodulin-dependent pathway in mouse pancreatic beta-cells. J. Membr. Biol. 1998, 166, 237–244. [Google Scholar] [CrossRef]
- Britsch, S.; Krippeit-Drews, P.; Lang, F.; Gregor, M.; Drews, G. Glucagon-like peptide-1 modulates Ca2+ current but not K+ATP current in intact mouse pancreatic B-cells. Biochem. Biophys. Res. Commun. 1995, 207, 33–39. [Google Scholar] [CrossRef]
- Lüderitz, B.; Bolte, H.D. Zur kardialen Wirkung Wirkung von Glukagon. Elektrophysiologsiche Messungen am Papillarmuskel des Herzens [Cardiac effect of glucagon. Electrophysiologic measurements in the papillary muscle of the heart]. Z. Kreislaufforsch. 1971, 60, 130–135. (In German) [Google Scholar]
- Daniell, H.B.; Holl, J.E.; Pruett, J.K.; Bagwell, E.E.; Woods, E.F. Cardiovascular effects of glucagon in dogs with non-nodal pacemakers. J. Electrocardiol. 1970, 3, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Spodick, D.H. Annotations. Effect of glucagon on intraventricular conduction. J. Electrocardiol. 1972, 5, 207–208. [Google Scholar] [CrossRef]
- Xiao, Y.F.; Nikolskaya, A.; Jaye, D.A.; Sigg, D.C. Glucagon-like peptide-1 enhances cardiac L-type Ca2+ currents via activation of the cAMP-dependent protein kinase A pathway. Cardiovasc. Diabetol. 2011, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Widomski, D.L.; Liu, X.; Limberis, J.T.; Green, J.; Diaz, G.; Martin, R.L.; Cox, B.F.; Gintant, G.A. A novel secretagogue increases cardiac contractility by enhancement of L-type Ca2+ current. Biochem. Pharmacol. 2010, 80, 1000–1006. [Google Scholar] [CrossRef]
- Aromataris, E.C.; Roberts, M.L.; Barritt, G.J.; Rychkov, G.Y. Glucagon activates Ca2+ and Cl− channels in rat hepatocytes. J. Physiol. 2006, 573 Pt 3, 611–625. [Google Scholar] [CrossRef]
- Spinelli, V.; Sartiani, L.; Mugelli, A.; Romanelli, M.N.; Cerbai, E. Hyperpolarization-activated cyclic-nucleotide-gated channels: Pathophysiological, developmental, and pharmacological insights into their function in cellular excitability. Can. J. Physiol. Pharmacol. 2018, 96, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Robles-Flores, M.; Allende, G.; Piña, E.; García-Sáinz, J.A. Cross-talk between glucagon- and adenosine-mediated signalling systems in rat hepatocytes: Effects on cyclic AMP-phosphodiesterase activity. Biochem. J. 1995, 312 Pt 3, 763–767. [Google Scholar] [CrossRef]
- MacDougall, L.K.; Campbell, D.G.; Hubbard, M.J.; Cohen, P. Partial structure and hormonal regulation of rabbit liver inhibitor-1; distribution of inhibitor-1 and inhibitor-2 in rabbit and rat tissues. Biochim. Biophys. Acta 1989, 1010, 218–226. [Google Scholar] [CrossRef]
- Neumann, J.; Gupta, R.C.; Schmitz, W.; Scholz, H.; Nairn, A.C.; Watanabe, A.M. Evidence for isoproterenol-induced phosphorylation of phosphatase inhibitor-1 in the intact heart. Circ. Res. 1991, 69, 1450–1457. [Google Scholar] [CrossRef]
- Takemiya, M.; Miyayama, H.; Takeya, M.; Takeuchi, T.; Konno, T.; Kitoh, M.; Fukushima, H. A postmortem study of malignant glucagonoma with heart muscle hypertrophy, including chemical, histochemical, immunohistological and ultrastructural observations. Hum. Pathol. 1981, 12, 988–999. [Google Scholar] [CrossRef]
- Chang-Chretien, K.; Chew, J.T.; Judge, D.P. Reversible dilated cardiomyopathy associated with glucagonoma. Heart 2004, 90, e44. [Google Scholar] [CrossRef] [PubMed]
- Barabas, M.; Huang-Doran, I.; Pitfield, D.; Philips, H.; Goonewardene, M.; Casey, R.T.; Challis, B.G. Glucagonoma-associated dilated cardiomyopathy refractory to somatostatin analogue therapy. Endocrinol. Diabetes Metab. Case Rep. 2019, 2019, 18-0157. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.G.; Chejfec, G.; Prinz, R.A. Islet cell carcinoma of the pancreas. Am. Surg. 1989, 55, 325–332. [Google Scholar] [PubMed]
- Zhang, K.; Lehner, L.J.; Praeger, D.; Baumann, G.; Knebel, F.; Quinkler, M.; Roepke, T.K. Glucagonoma-induced acute heart failure. Endocrinol. Diabetes Metab. Case Rep. 2014, 2014, 140061. [Google Scholar] [CrossRef]
- Lvoff, R.; Wilcken, D.E. Glucagon in heart failure and in cardiogenic shock. Experience in 50 patients. Circulation 1972, 45, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Nord, H.J.; Fontanes, A.L.; Williams, J.F., Jr. Treatment of congestive heart failure with glucagon. Ann. Intern. Med. 1970, 72, 649–653. [Google Scholar] [CrossRef]
- Zhang, C.; Kaye, J.A.; Cai, Z.; Wang, Y.; Prescott, S.L.; Liberles, S.D. Area Postrema Cell Types that Mediate Nausea-Associated Behaviors. Neuron 2021, 109, 461–472.e5. [Google Scholar] [CrossRef]
- Ranganath, L.; Schaper, F.; Gama, R.; Morgan, L. Mechanism of glucagon-induced nausea. Clin. Endocrinol. 1999, 51, 260–261. [Google Scholar] [CrossRef]
- Sélley, E.; Kun, S.; Szijártó, I.A.; Kertész, M.; Wittmann, I.; Molnár, G.A. Vasodilator Effect of Glucagon: Receptorial Crosstalk Among Glucagon, GLP-1, and Receptor for Glucagon and GLP-1. Horm. Metab. Res. 2016, 48, 476–483. [Google Scholar] [CrossRef]
- Fouad, F.M.; Shimamatsu, K.; Said, S.I.; Tarazi, R.C. Inotropic responsiveness in hypertensive left ventricular hypertrophy: Impaired inotropic response to glucagon and vasoactive intestinal peptide in renal hypertensive rats. J. Cardiovasc. Pharmacol. 1986, 8, 398–405. [Google Scholar] [CrossRef]
- Vander Ark, C.R.; Reynolds, E.W., Jr. Clinical evaluation of glucagon by continuous infusion in the treatment of low cardiac output states. Am. Heart J. 1970, 79, 481–487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Farr, O.M.; Sofopoulos, M.; Tsoukas, M.A.; Dincer, F.; Thakkar, B.; Sahin-Efe, A.; Filippaios, A.; Bowers, J.; Srnka, A.; Gavrieli, A.; et al. GLP-1 receptors exist in the parietal cortex, hypothalamus and medulla of human brains and the GLP-1 analogue liraglutide alters brain activity related to highly desirable food cues in individuals with diabetes: A crossover, randomised, placebo-controlled trial. Diabetologia 2016, 59, 954–965. [Google Scholar] [CrossRef]
- Busuttil, R.W.; Paddock, R.J.; Fisher, J.W.; George, W.J. Changes in cyclic nucleotide levels and contractile force in the isolated hypoxic rat heart during perfusion with glucagon. Circ. Res. 1976, 38, 162–167. [Google Scholar] [CrossRef]
- Gao, C.; Ren, S.V.; Yu, J.; Baal, U.; Thai, D.; Lu, J.; Zeng, C.; Yan, H.; Wang, Y. Glucagon Receptor Antagonism Ameliorates Progression of Heart Failure. JACC Basic Transl. Sci. 2019, 4, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Gelling, R.W.; Du, X.Q.; Dichmann, D.S.; Romer, J.; Huang, H.; Cui, L.; Obici, S.; Tang, B.; Holst, J.J.; Fledelius, C.; et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Urthaler, F.; Isobe, J.H.; James, T.N. Comparative effects of glucagon on automaticity of the sinus node and atrioventricular junction. Am. J. Physiol. 1974, 227, 1415–1421. [Google Scholar] [CrossRef]
- Tuncok, Y.; Apaydin, S.; Kalkan, S.; Ates, M.; Guven, H. The effects of amrinone and glucagon on verapamil-induced cardiovascular toxicity in anaesthetized rats. Int. J. Exp. Pathol. 1996, 77, 207–212. [Google Scholar] [CrossRef]
- Jolly, S.R.; Kipnis, J.N.; Lucchesi, B.R. Cardiovascular depression by verapamil: Reversal by glucagon and interactions with propranolol. Pharmacology 1987, 35, 249–255. [Google Scholar] [CrossRef]
- Sensky, P.R.; Olczak, S.A. High-dose intravenous glucagon in severe tricyclic poisoning. Postgrad. Med. J. 1999, 75, 611–612. [Google Scholar] [CrossRef]
- DeWitt, C.R.; Waksman, J.C. Pharmacology, pathophysiology and management of calcium channel blocker and beta-blocker toxicity. Toxicol. Rev. 2004, 23, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Senart, A.M.; LeClair, L.A. Cardiovascular and Adverse Effects of Glucagon for the Management of Suspected Beta Blocker Toxicity: A Case Series. J. Med. Toxicol. 2023, 19, 9–15. [Google Scholar] [CrossRef]
- Avenhaus, H.; Lüderitz, B.; Nordeck, E. Einfluss von Glucagon auf die Hämodynamik des menschlichen Herzens nach Beta-Rezeptoren-Blockade [Influence of glucagon on hemodynamics of the human heart following beta-receptor blockade]. Verh. Dtsch. Ges. Kreislaufforsch. 1971, 37, 427–432. [Google Scholar]
- Ceriello, A.; Genovese, S.; Mannucci, E.; Gronda, E. Glucagon and heart in type 2 diabetes: New perspectives. Cardiovasc. Diabetol. 2016, 15, 123. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.X.; Quittner-Strom, E.B.; Lee, Y.; Johnson, J.A.; Martin, S.A.; Yu, X.; Li, J.; Lu, J.; Cai, Z.; Chen, S.; et al. Glucagon Receptor Antagonism Improves Glucose Metabolism and Cardiac Function by Promoting AMP-Mediated Protein Kinase in Diabetic Mice. Cell Rep. 2018, 22, 1760–1773. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.; Wit, A.L.; Damato, A.N. Effects of glucagon on atrioventricular conduction and ventricular automaticity in dogs. Circ. Res. 1969, 24, 167–177. [Google Scholar] [CrossRef]
- Wilkerson, R.D.; Pruett, J.K.; Woods, E.F. Glucagon-enhanced ventricular automaticity in dogs. Its concealment by positive chronotropism. Circ. Res. 1971, 29, 616–625. [Google Scholar] [CrossRef]
- Pruett, J.K.; Woods, E.F.; Daniell, H.B. Glucagon enhanced automaticity in spontaneously beating Purkinje fibres of canine false tendons. Cardiovasc. Res. 1971, 5, 436–439. [Google Scholar] [CrossRef]
- Madan, B.R. Effect of glucagon on ventricular arrhythmias after coronary artery occlusion and on ventricular automaticity in the dog. Br. J. Pharmacol. 1971, 43, 279–286. [Google Scholar]
- Singh, J.; Bala, S.; Kaur, A.H.; Garg, K.N. Effect of glucagon on arrhythmias induced by coronary artery occlusion and ouabain in dogs. Indian J. Physiol. Pharmacol. 1980, 24, 329–334. [Google Scholar]
- Einzig, S.; Todd, E.P.; Nicoloff, D.M.; Lucas, R.V., Jr. Glucagon in prevention and abolition of ouabain-induced ventricular tachycardia in normokalemic and hypokalemic dogs. Circ. Res. 1971, 29, 88–95. [Google Scholar] [CrossRef]
- Kusumoto, F.M.; Schoenfeld, M.H.; Barrett, C.; Edgerton, J.R.; Ellenbogen, K.A.; Gold, M.R.; Goldschlager, N.F.; Hamilton, R.M.; Joglar, J.A.; Kim, R.J.; et al. 2018 ACC/AHA/HRS Guideline on the Evaluation and Management of Patients With Bradycardia and Cardiac Conduction Delay: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines, and the Heart Rhythm Society. Circulation 2019, 140, e333–e381, Erratum in Circulation 2019, 140, e504–e505. [Google Scholar] [CrossRef] [PubMed]
- Niemann, J.T.; Haynes, K.S.; Garner, D.; Jagels, G.; Rennie, C.J., 3rd. Postcountershock pulseless rhythms: Hemodynamic effects of glucagon in a canine model. Crit Care Med. 1987, 15, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, K.; Cholewa, M.; Górski, L. Cardiac arrhythmias after intravenous administration of glucagon. Eur. J. Cardiol. 1978, 6, 449–458. [Google Scholar] [PubMed]
- Jaca, I.J.; Desai, D.; Barkin, J.S. Paroxysmal supraventricular tachycardia after administration of glucagon during upper endoscopy. Gastrointest Endosc. 2002, 56, 304. [Google Scholar] [CrossRef] [PubMed]
- Podzuweit, T.; Els, D.J.; McCarthy, J. Cyclic AMP mediated arrhythmias induced in the ischaemic pig heart. Basic Res. Cardiol. 1981, 76, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Sanna, S.; Leo, P.; Netter, R. Azione terapeutica del “glucagone” su stati patologici del miocardio particolarmente nei disturbi del tessuto specifico di conduzione [Therapeutic action of glucagon on pathological states of the myocardium, especially on specific conduction tissue disorders]. Boll. Soc. Ital. Cardiol. 1975, 20, 1233–1249. (In Italian) [Google Scholar]
- Hurwitz, R.A. Effect of glucagon on infants and children with atrioventricular heart block. Br. Heart J. 1973, 35, 1260–1264. [Google Scholar] [CrossRef]
- Wu, S.; Lu, W.; Chen, Z.; Dai, Y.; Chen, K.; Zhang, S. Association of glucagon-like peptide-1 receptor agonists with cardiac arrhythmias in patients with type 2 diabetes or obesity: A systematic review and meta-analysis of randomized controlled trials. Diabetol. Metab. Syndr. 2022, 14, 195. [Google Scholar] [CrossRef]
- Raubenheimer, P.J.; Cushman, W.C.; Avezum, A.; Basile, J.; Conget, I.; Dagenais, G.; Hoover, A.; Jansky, P.; Lanas, F.; Leiter, L.A.; et al. Dulaglutide and incident atrial fibrillation or flutter in patients with type 2 diabetes: A post hoc analysis from the REWIND randomized trial. Diabetes Obes. Metab. 2022, 24, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Kai, Z.; Yongbo, W.; Lin, Z.; Jie, G.; Daoqun, J.; Zhiqiang, C. Exendin-4 attenuates ischemia-induced ventricular arrhythmias in rats. Cardiol. J. 2013, 20, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Yu, R. Mahvash Disease: 10 Years After Discovery. Pancreas 2018, 47, s511–s515. [Google Scholar] [CrossRef]
- Hager, J.; Hansen, L.; Vaisse, C.; Vionnet, N.; Philippi, A.; Poller, W.; Velho, G.; Carcassi, C.; Contu, L.; Julier, C.; et al. A missense mutation in the glucagon receptor gene is associated with non-insulin-dependent diabetes mellitus. Nat. Genet. 1995, 9, 299–304. [Google Scholar] [CrossRef]
- Chambers, S.M.; Morris, B.J. Glucagon receptor gene mutation in essential hypertension. Nat. Genet. 1996, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, W.J.C.; Lindquist, P.; Madsen, J.S.; Stassen, R.H.M.J.; Wewer Albrechtsen, N.J.; Holst, J.J.; Hauser, A.S.; Rosenkilde, M.M. Molecular and in vivo phenotyping of missense variants of the human glucagon receptor. J. Biol. Chem. 2022, 298, 101413. [Google Scholar] [CrossRef]
- Cefalu, W.T.; Stenlöf, K.; Leiter, L.A.; Wilding, J.P.H.; Blonde, L.; Polidori, D.; Xie, J.; Sullivan, D.; Usiskin, K.; Canovatchel, W.; et al. Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia 2015, 58, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Al-Jobori, H.; Ali, A.M.; Adams, J.; Abdul-Ghani, M.; Triplitt, C.; DeFronzo, R.A.; Cersosimo, E. Endogenous glucose production and hormonal changes in response to canagliflozin and liraglutide combination therapy. Diabetes 2018, 67, 1182–1189. [Google Scholar] [CrossRef]
- Saponaro, C.; Pattou, F.; Bonner, C. SGLT2 inhibition and glucagon secretion in humans. Diabetes Metab. 2018, 44, 383–385. [Google Scholar] [CrossRef]
- Wang, M.Y.; Yu, X.; Lee, Y.; McCorkle, S.K.; Chen, S.; Li, J.; Wang, Z.V.; Davidson, J.A.; Scherer, P.E.; Holland, W.L.; et al. Dapagliflozin suppresses glucagon signaling in rodent models of diabetes. Proc. Natl. Acad. Sci. USA 2017, 114, 6611–6616. [Google Scholar] [CrossRef]
- Zinman, B.; Bhosekar, V.; Busch, R.; Holst, I.; Ludvik, B.; Thielke, D.; Thrasher, J.; Woo, V.; Philis-Tsimikas, A. Semaglutide once weekly as add-on to SGLT-2 inhibitor therapy in type 2 diabetes (SUSTAIN 9): A randomised, placebocontrolled trial. Lancet. Diabetes Endocrinol. 2019, 7, 356–367. [Google Scholar] [CrossRef]
- Pearson, M.J.; Unger, R.H.; Holland, W.L. Clinical trials, triumphs, and tribulations of glucagon receptor antagonists. Diabetes Care 2016, 39, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Nahra, R.; Wang, T.; Gadde, K.M.; Oscarsson, J.; Stumvoll, M.; Jermutus, L.; Hirshberg, B.; Ambery, P. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults with Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care 2021, 44, 1433–1442, Erratum in Diabetes Care 2022, 45, 3112. [Google Scholar] [CrossRef] [PubMed]
- Parker, V.E.R.; Robertson, D.; Wang, T.; Hornigold, D.C.; Petrone, M.; Cooper, A.T.; Posch, M.G.; Heise, T.; Plum-Moerschel, L.; Schlichthaar, H.; et al. Efficacy, Safety, and Mechanistic Insights of Cotadutide, a Dual Receptor Glucagon-Like Peptide-1 and Glucagon Agonist. J. Clin. Endocrinol. Metab. 2020, 105, dgz047. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Left Atrium | Right Atrium | Ventricle | Remarks | |
|---|---|---|---|---|
| Cat | [3,5,25] PCE [25] PIE | [5,25,26] In vivo: PIE [5,27] PIE: papillary muscle [27] PIE: perfused heart [28] No PIE in heart failure [25,29] PIE in heart failure | ||
| Dog | [3,4] PIE | [3,4,30,31,32,33,34,35,36,37,38,39,40] PCE [31,36] PIE | [3,4,34,35,39,40] PIE [4] failing dog ventricle | [40] Coronary perfusion enhanced |
| Embryon-ic chick heart | [41] PCE | |||
| Frog | [42] PIE | |||
| Guinea pig | [3,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57] PIE | [43,44,45,47,58,59] PCE | [44,45] No inotropic effect | |
| Human cardiac tissue (isolated) | [50] No inotropic effect | [23,50] No inotropic effect | [50,60,61] No inotropic effect, [60] APD shortened, in vivo: PIE [49,62] PIE in left ventricular papillary muscle strips from failing hearts | |
| Human patient or healthy volunteer In vivo | [38,51,52,53,63,64] PCE | [38,51,52,53,63,64] PIE (1–5 mg i.v.) | [38,52,63,64] Vascular peripheral resistance decreased, [38,51,52,63] nausea [51,63] vomiting, [64] flushing, [64] palpitations, [64] diarrhoea, and [64] hyperglycemia [52] Coronary flow increased, [52] oxygen consumption increased, [52] Blood glucose increased | |
| Monkey | [42] PIE | |||
| Mouse adult | [18] PCE, [54] No PCE | [50] No PIE | ||
| Mouse fetal and neo-natal | [65] PCE late-term fetal mouse heart | Neonatal mouse cardiomyocyte [54,65] PIE, [55,56,57] PCE | ||
| Rabbit | [66] No PCE | [3,66] No effect | ||
| Rat, adult | [3] No inotropic effect [67,68] PIE | [23,44,67,68,69,70] PCE [3] No inotropic effect | [3,44,45,68,70,71] PIE | [44,68] Relaxation shortened |
| Rat, fetal | [54,72] No PCE |
| Species | Right Atrium | Left Atrium | Ventricle | |
|---|---|---|---|---|
| Cat | [28] AC stimulation in normal heart [28] No stimulation of AC in failing hearts | [74] AC stimulation [74] PDE not inhibited | ||
| Chicken embryonic ventricular cardio-myocytes | [75] Calcium transients increased | |||
| Dog | [31] AC not stimulated | [31] AC not stimulated | [76] cAMP increase [31] AC stimulated | |
| Embryonic chick heart | [41] AC stimulated | [41] Glucagon binding increased with age | ||
| Frog heart | [77] PDE inhibition | [77] LTCC stimulation | ||
| Guinea pig ventricle | [45,77] cAMP not increased | [59,76] AC not stimulated | [42,77] PDE inhibition | |
| Human atrium | [78] AC stimulation and inhibition | |||
| Human | [20] AC stimulation in normal adult heart, [74] No stimulation of AC in adult failing hearts, [74] AC stimulation human fetal heart | [79] AC stimulation, [79] PDE not inhibited | ||
| Rabbit | [76] AC not stimulated in membranes | [76] AC not stimulated in membranes, [80] AC stimulated in nuclei | ||
| Monkey | [81] PDE inhibition [42] AC not stimulated | [42] AC not stimulated | [42] AC not stimulated | |
| Mouse | [77] PDE inhibition | |||
| Neonatal rat heart | [72] No AC stimulation | |||
| Rat heart | [42,45,82,83] AC stimulation | [45,84] LTCC stimulation [84] cAMP increase [85] AC not stimulated [82] fetal rat AC not stimulated | [86] TnI phosphorylation increased [85] AC stimulated | Augmentation by PDE III [71] or IV [72] [87,88,89,90] cAMP |
| Compound: | Affinity at GR | Organism Cells | Half Life |
|---|---|---|---|
| Antagonist HM15136 | [95] Human GR: EC50 = 92 pM | CHO, Mice | In mice: 136 h |
| Antagonist: nonpeptide:2-(4-pyridyl)-5-(4-chlorophenyl)-3-(5-bromo-2-propyloxy-phenyl)pyrrole (L-168,049) | Human GR [96] IC50 = 3.7 nM | Mice | |
| Antagonist: nonpeptide: N-[3-cyano-6-(1,1-dimethylpropyl)-4,5,6,7-tetrahydro-1-benzothien-2-yl]-2-ethylbutanamide (SC203972) | [96] Human GR IC50 = 181 nM | Mice | |
| Antagonist, peptide: desHis1-Pro4-glucagon | [97] Human GR IC50 = 1 nM | Mice | persistent biological effects |
| Antagonist, peptide, des-His1-[Glu9]glucagon, GR specific, | [98] Rat GR | HEK | |
| LY2490921 GR unspecific | [98] Rat GR: 1.3 µM, [98] Human GLP-1-R: 1.2 µM | HEK | |
| Antagonist, peptide desHis1Glu9(Lys30PAL)-glucagon | [99] GR: 170 pM | Mice, HEK293 | persistent biological effects |
| Antibody REMD2.29 | [100] 30 pM | ||
| Glucagon: physiological agonist | [95] Human GR: 800 pM [98] Rat GR: 400 pM [98] Rat GLP-1R: 4.9 nM | [95] 5 min | |
| Sacubitril: inhibitor of glucagon degradation | [101] Inhibition of glucagon metabolism: about 1 nM sacubitrilat | ||
| Anti-sense for GR | [102] Mice, [103] patients T2DM | ||
| Antagonist: Exenadin 9–39 | [98] IC50: Human GLP-1-R: 17 nM | ||
| Exenadin4: GLP-1-receptor agonist | [98] EC50: Human GLP-1-R: 30 pM | ||
| GLP-1 | [98] EC50: Human GLP-1-R: 65 pM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, J.; Hofmann, B.; Dhein, S.; Gergs, U. Glucagon and Its Receptors in the Mammalian Heart. Int. J. Mol. Sci. 2023, 24, 12829. https://doi.org/10.3390/ijms241612829
Neumann J, Hofmann B, Dhein S, Gergs U. Glucagon and Its Receptors in the Mammalian Heart. International Journal of Molecular Sciences. 2023; 24(16):12829. https://doi.org/10.3390/ijms241612829
Chicago/Turabian StyleNeumann, Joachim, Britt Hofmann, Stefan Dhein, and Ulrich Gergs. 2023. "Glucagon and Its Receptors in the Mammalian Heart" International Journal of Molecular Sciences 24, no. 16: 12829. https://doi.org/10.3390/ijms241612829
APA StyleNeumann, J., Hofmann, B., Dhein, S., & Gergs, U. (2023). Glucagon and Its Receptors in the Mammalian Heart. International Journal of Molecular Sciences, 24(16), 12829. https://doi.org/10.3390/ijms241612829