
Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms



Abstract
1. Introduction
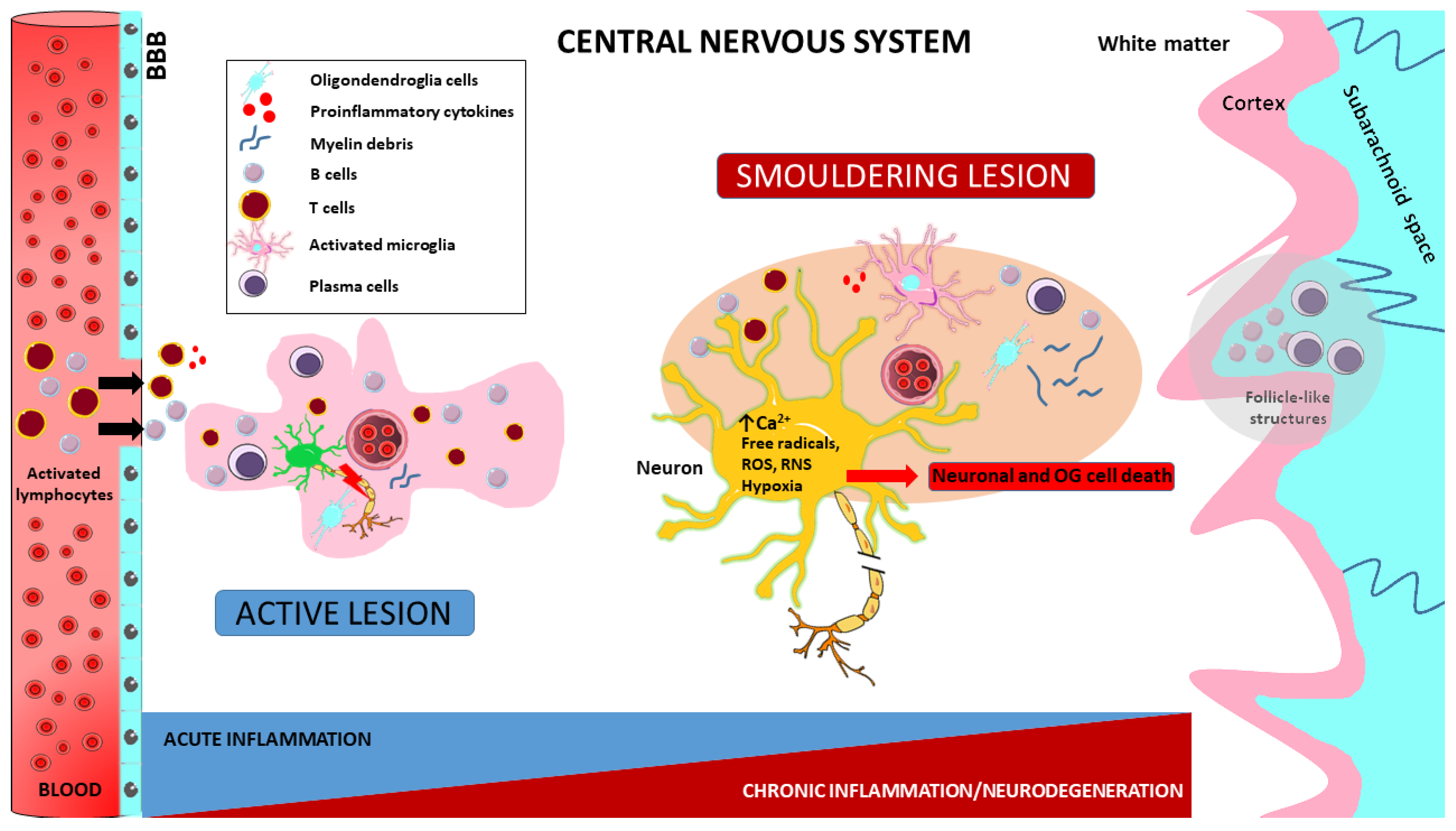
2. MS Lesion Pathology
3. Immunopathology of MS
3.1. T-Cells in the Immunopathology of MS
3.2. B-Cells in the Immunopathology of MS
3.3. Microglia in the Immunopathology of MS
3.3.1. Neurotoxic Microglia
3.3.2. Available Biomarkers of Microglia
3.3.3. Neuroprotective Microglia
4. Pathogenetic Implications in MS
4.1. Neuroinflammation in MS
4.2. Oxidative Stress
4.3. Chronic Toxic Environment in MS Lesions
4.3.1. Glutamate Excitotoxicity
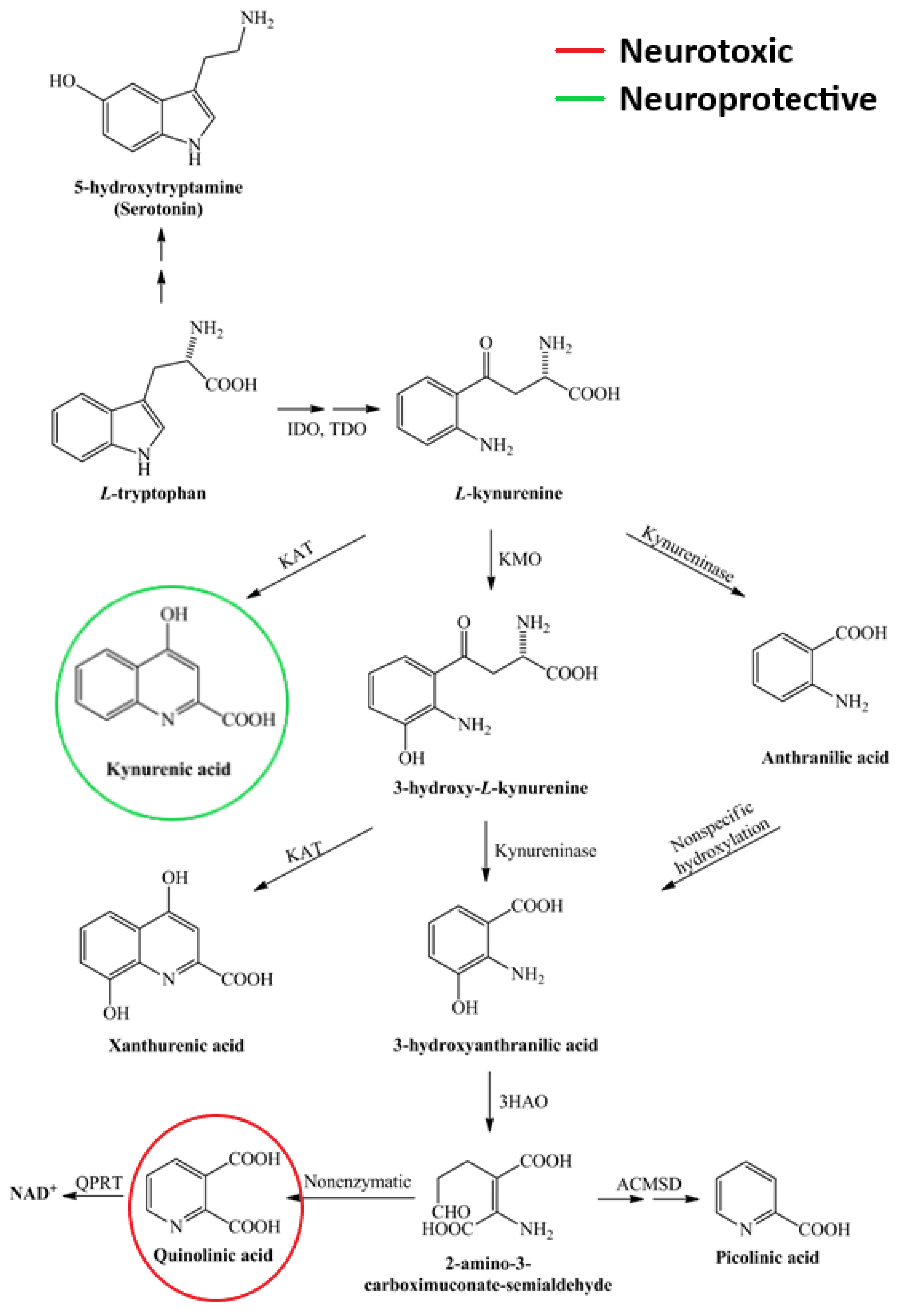
4.3.2. Kynurenines
4.3.3. Mitochondrial Dysfunction
4.3.4. Essential Metal Homeostasis Disruption in MS
5. PIRA and Future Perspectives of MS Research
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lassmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Biström, M.; Jons, D.; Engdahl, E.; Gustafsson, R.; Huang, J.; Brenner, N.; Butt, J.; Alonso-Magdalena, L.; Gunnarsson, M.; Vrethem, M.; et al. Epstein–Barr virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur. J. Neurol. 2020, 28, 579–586. [Google Scholar] [CrossRef]
- Lublin, F.D. New Multiple Sclerosis Phenotypic Classification. Eur. Neurol. 2014, 72, 1–5. [Google Scholar] [CrossRef]
- Cree, B.A.; Hollenbach, J.A.; Bove, R.; Kirkish, G.; Sacco, S.; Caverzasi, E.; Bischof, A.; Gundel, T.; Zhu, A.H.; Papinutto, N.; et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann. Neurol. 2019, 85, 653–666. [Google Scholar] [PubMed]
- Jonkman, L.E.; Soriano, A.L.; Amor, S.; Barkhof, F.; van der Valk, P.; Vrenken, H.; Geurts, J.J.G. Can MS lesion stages be distinguished with MRI? A postmortem MRI and histopathology study. J. Neurol. 2015, 262, 1074–1080. [Google Scholar] [CrossRef]
- Kipp, M.; van der Valk, P.; Amor, S. Pathology of multiple sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 506–517. [Google Scholar] [CrossRef]
- Haase, S.; Linker, R.A. Inflammation in multiple sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007687. [Google Scholar] [CrossRef] [PubMed]
- Machado-Santos, J.; Saji, E.; Tröscher, A.R.; Paunovic, M.; Liblau, R.; Gabriely, G.; Bien, C.G.; Bauer, J.; Lassmann, H. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018, 141, 2066–2082. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain J. Neurol. 2009, 132 Pt 5, 1175–1189. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lassmann, H. Pathology of multiple sclerosis and related inflammatory demyelinating diseases. Handb. Clin. Neurol. 2014, 122, 15–58. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.M.; Reich, D.S. Association of Chronic Active Multiple Sclerosis Lesions with Disability In Vivo. JAMA Neurol. 2019, 76, 1474–1483. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2016, 133, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G.; Popescu, V.; Wuerfel, J.; Hellwig, K.; Iacobaeus, E.; Jensen, M.B.; García-Domínguez, J.M.; Sousa, L.; De Rossi, N.; Hupperts, R.; et al. Smouldering multiple sclerosis: The ‘real MS’. Ther. Adv. Neurol. Disord. 2022, 15, 17562864211066751. [Google Scholar]
- Absinta, M.; Sati, P.; Fechner, A.; Schindler, M.; Nair, G.; Reich, D. Identification of Chronic Active Multiple Sclerosis Lesions on 3T MRI. Am. J. Neuroradiol. 2018, 39, 1233–1238. [Google Scholar] [CrossRef]
- Absinta, M.; Sati, P.; Schindler, M.; Leibovitch, E.C.; Ohayon, J.; Wu, T.; Meani, A.; Filippi, M.; Jacobson, S.; Cortese, I.C.; et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J. Clin. Investig. 2016, 126, 2597–2609. [Google Scholar] [CrossRef]
- Hemond, C.C.; Reich, D.S.; Dundamadappa, S.K. Paramagnetic Rim Lesions in Multiple Sclerosis: Comparison of Visualization at 1.5-T and 3-T MRI. Am. J. Roentgenol. 2022, 219, 120–131. [Google Scholar] [CrossRef]
- Calvi, A.; Haider, L.; Prados, F.; Tur, C.; Chard, D.; Barkhof, F. In vivo imaging of chronic active lesions in multiple sclerosis. Mult. Scler. J. 2020, 28, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Wei, W.; Belachew, S.; Arnold, D.L. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult. Scler. J. 2018, 25, 1915–1925. [Google Scholar] [CrossRef]
- Bagnato, F.; Hametner, S.; Yao, B.; Van Gelderen, P.; Merkle, H.; Cantor, F.K.; Lassmann, H.; Duyn, J.H. Tracking iron in multiple sclerosis: A combined imaging and histopathological study at 7 Tesla. Brain 2011, 134, 3602–3615. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Höftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow expansion of multiple sclerosis iron rim lesions: Pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 2016, 133, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.L.; Belachew, S.; Gafson, A.R.; Gaetano, L.; Bernasconi, C.; Elliott, C. Slowly expanding lesions are a marker of progressive MS—No. Mult. Scler. J. 2021, 27, 1681–1683. [Google Scholar] [CrossRef]
- Altokhis, A.I.; Hibbert, A.M.; Allen, C.M.; Mougin, O.; Alotaibi, A.; Lim, S.-Y.; Constantinescu, C.S.; Abdel-Fahim, R.; Evangelou, N. Longitudinal clinical study of patients with iron rim lesions in multiple sclerosis. Mult. Scler. J. 2022, 28, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Preziosa, P.; Pagani, E.; Meani, A.; Moiola, L.; Rodegher, M.; Filippi, M.; Rocca, M.A. Slowly Expanding Lesions Predict 9-Year Multiple Sclerosis Disease Progression. Neurol.-Neuroimmunol. Neuroinflamm. 2022, 9, e1139. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.W.; Metz, L.M.; Agrawal, S.M.; Yong, V.W. Environmental factors and their regulation of immunity in multiple sclerosis. J. Neurol. Sci. 2013, 324, 10–16. [Google Scholar] [CrossRef]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2016, 133, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Popescu, B.F.; Bunyan, R.F.; Moll, N.M.; Roemer, S.F.; Lassmann, H.; Brück, W.; Parisi, J.E.; Scheithauer, B.W.; Giannini, C.; et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 2011, 365, 2188–2197. [Google Scholar] [CrossRef]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Berge, I.J.M.T.; van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef]
- Van Nierop, G.P.; van Luijn, M.M.; Michels, S.S.; Melief, M.-J.; Janssen, M.; Langerak, A.W.; Ouwendijk, W.J.D.; Hintzen, R.Q.; Verjans, G.M.G.M. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. 2017, 134, 383–401. [Google Scholar] [CrossRef]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schröder, R.; Deckert, M.; Schmidt, S.; et al. Clonal Expansions of Cd8+ T Cells Dominate the T Cell Infiltrate in Active Multiple Sclerosis Lesions as Shown by Micromanipulation and Single Cell Polymerase Chain Reaction. J. Exp. Med. 2000, 192, 393–404. [Google Scholar] [CrossRef]
- Denic, A.; Wootla, B.; Rodriguez, M. CD8(+) T cells in multiple sclerosis. Expert Opin. Ther. Targets 2013, 17, 1053–1066. [Google Scholar] [PubMed]
- Vukmanovic-Stejic, M.; Thomas, M.J.; Noble, A.; Kemeny, D.M. Specificity, restriction and effector mechanisms of immunoregulatory CD8 T cells. Immunology 2001, 102, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 Cell, the New Player of Neuroinflammatory Process in Multiple Sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef]
- Ramgolam, V.S.; Sha, Y.; Jin, J.; Zhang, X.; Markovic-Plese, S. IFN-beta inhibits human Th17 cell differentiation. J. Immunol. 2009, 183, 5418–5427. [Google Scholar] [PubMed]
- Zhang, X.; Markovic-Plese, S. Interferon beta inhibits the Th17 cell-mediated autoimmune response in patients with relapsing–remitting multiple sclerosis. Clin. Neurol. Neurosurg. 2010, 112, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, A.L.; Wiendl, H. The role of regulatory T cells in multiple sclerosis. Nat. Clin. Pr. Neurol. 2008, 4, 384–398. [Google Scholar] [CrossRef]
- Chen, M.; Chen, G.; Deng, S.; Liu, X.; Hutton, G.J.; Hong, J. IFN-β induces the proliferation of CD4+CD25+Foxp3+ regulatory T cells through upregulation of GITRL on dendritic cells in the treatment of multiple sclerosis. J. Neuroimmunol. 2012, 242, 39–46. [Google Scholar] [CrossRef]
- Haas, J.; Korporal, M.; Balint, B.; Fritzsching, B.; Schwarz, A.; Wildemann, B. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4+CD25+FOXP3+CD31+ T-cells in patients with multiple sclerosis. J. Neuroimmunol. 2009, 216, 113–117. [Google Scholar] [CrossRef]
- Haas, J.; Würthwein, C.; Korporal-Kuhnke, M.; Viehoever, A.; Jarius, S.; Ruck, T.; Pfeuffer, S.; Meuth, S.G.; Wildemann, B. Alemtuzumab in Multiple Sclerosis: Short- and Long-Term Effects of Immunodepletion on the Peripheral Treg Compartment. Front. Immunol. 2019, 10, 1204. [Google Scholar] [CrossRef]
- Trinschek, B.; Luessi, F.; Gross, C.C.; Wiendl, H.; Jonuleit, H. Interferon-Beta Therapy of Multiple Sclerosis Patients Improves the Responsiveness of T Cells for Immune Suppression by Regulatory T Cells. Int. J. Mol. Sci. 2015, 16, 16330–16346. [Google Scholar] [CrossRef]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [PubMed]
- Miyazaki, Y.; Niino, M. B-cell depletion therapy for multiple sclerosis. Immunol. Med. 2021, 45, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Marta, M.; Pryce, G.; Giovannoni, G.; Schmierer, K. Memory B Cells are Major Targets for Effective Immunotherapy in Relapsing Multiple Sclerosis. Ebiomedicine 2017, 16, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Ramagopalan, S.; Davis, A.; Giovannoni, G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: A meta-analysis of prevalence, prognosis and effect of latitude. J. Neurol. Neurosurg. Psychiatry 2013, 84, 909–914. [Google Scholar] [CrossRef]
- Mulero, P.; Midaglia, L.; Montalban, X. Ocrelizumab: A new milestone in multiple sclerosis therapy. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418773025. [Google Scholar] [CrossRef]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010, 68, 477–493. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of Ectopic B-cell Follicles with Germinal Centers in the Meninges of Patients with Secondary Progressive Multiple Sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef]
- Lisak, R.P.; Benjamins, J.A.; Nedelkoska, L.; Barger, J.L.; Ragheb, S.; Fan, B.; Ouamara, N.; Johnson, T.A.; Rajasekharan, S.; Bar-Or, A. Secretory products of multiple sclerosis B cells are cytotoxic to oligodendroglia in vitro. J. Neuroimmunol. 2012, 246, 85–95. [Google Scholar] [CrossRef]
- Owens, G.P.; Bennett, J.L.; Lassmann, H.; O’Connor, K.C.; Ritchie, A.M.; Shearer, A.; Lam, C.; Yu, X.; Birlea, M.; DuPree, C.; et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann. Neurol. 2009, 65, 639–649. [Google Scholar] [CrossRef]
- Kumar, G.; Axtell, R.C. Dual Role of B Cells in Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 2336. [Google Scholar] [PubMed]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Ding, A.H.; Nathan, C.F.; Stuehr, D.J. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J. Immunol. 1988, 141, 2407–2412. [Google Scholar] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. J. Virtual Libr. 2008, 13, 453–461. [Google Scholar]
- Böttcher, C.; van der Poel, M.; Fernández-Zapata, C.; Schlickeiser, S.; Leman, J.K.H.; Hsiao, C.-C.; Mizee, M.R.; Adelia; Vincenten, M.C.J.; Kunkel, D.; et al. Single-cell mass cytometry reveals complex myeloid cell composition in active lesions of progressive multiple sclerosis. Acta Neuropathol. Commun. 2020, 8, 136. [Google Scholar] [CrossRef]
- O’loughlin, E.; Madore, C.; Lassmann, H.; Butovsky, O. Microglial Phenotypes and Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028993. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; De Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of neuropathology-associated reactive astrocytes: A systematic review. Acta Neuropathol. Commun. 2023, 11, 42. [Google Scholar] [CrossRef]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef]
- Fischer, M.-T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; Van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef]
- Lisak, R.P.; Benjamins, J.A.; Bealmear, B.; Nedelkoska, L.; Studzinski, D.; Retland, E.; Yao, B.; Land, S. Differential effects of Th1, monocyte/macrophage and Th2 cytokine mixtures on early gene expression for molecules associated with metabolism, signaling and regulation in central nervous system mixed glial cell cultures. J. Neuroinflamm. 2009, 6, 4. [Google Scholar] [CrossRef]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Brück, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef]
- Gao, Z.; Tsirka, S.E. Animal Models of MS Reveal Multiple Roles of Microglia in Disease Pathogenesis. Neurol. Res. Int. 2011, 2011, 383087. [Google Scholar] [CrossRef]
- McMahon, E.J.; Bailey, S.L.; Castenada, C.V.; Waldner, H.; Miller, S.D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005, 11, 335–339. [Google Scholar] [PubMed]
- Krumbholz, M.; Theil, D.; Derfuss, T.; Rosenwald, A.; Schrader, F.; Monoranu, C.-M.; Kalled, S.L.; Hess, D.M.; Serafini, B.; Aloisi, F.; et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J. Exp. Med. 2005, 201, 195–200. [Google Scholar] [CrossRef]
- Breij, E.C.W.; Brink, B.P.; Veerhuis, R.; Bsc, C.v.D.B.; Vloet, R.; Yan, R.; Dijkstra, C.D.; van der Valk, P.; Bö, L. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann. Neurol. 2008, 63, 16–25. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain J. Neurol. 2005, 28 Pt 11, 2705–2712. [Google Scholar] [CrossRef]
- Airas, L.; Nylund, M.; Rissanen, E. Evaluation of Microglial Activation in Multiple Sclerosis Patients Using Positron Emission Tomography. Front. Neurol. 2018, 9, 181. [Google Scholar] [CrossRef]
- Karamita, M.; Barnum, C.; Möbius, W.; Tansey, M.G.; Szymkowski, D.E.; Lassmann, H.; Probert, L. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. J. Clin. Investig. 2017, 2, e87455. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef]
- Franklin, R.J.; Goldman, S.A. Glia Disease and Repair-Remyelination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020594. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Mado, H.; Adamczyk-Sowa, M.; Sowa, P. Role of Microglial Cells in the Pathophysiology of MS: Synergistic or Antagonistic? Int. J. Mol. Sci. 2023, 24, 1861. [Google Scholar]
- Veremeyko, T.; Siddiqui, S.; Sotnikov, I.; Yung, A.; Ponomarev, E.D. IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS ONE 2013, 8, e81774. [Google Scholar]
- Zhang, L.; Zhang, J.; You, Z. Switching of the Microglial Activation Phenotype Is a Possible Treatment for Depression Disorder. Front. Cell. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Davies, C.L.; Holloway, R.K.; Labrak, Y.; Ireland, G.; Carradori, D.; Dillenburg, A.; Borger, E.; Soong, D.; Richardson, J.C.; et al. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 2019, 22, 1046–1052. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Howell, O.W.; Rundle, J.L.; Garg, A.; Komada, M.; Brophy, P.J.; Reynolds, R. Activated Microglia Mediate Axoglial Disruption That Contributes to Axonal Injury in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Korczyn, A.D.; Manouchehri, N.; Stüve, O. The temporal and causal relationship between inflammation and neurodegeneration in multiple sclerosis. Mult. Scler. J. 2019, 26, 876–886. [Google Scholar] [CrossRef]
- Calahorra, L.; Camacho-Toledano, C.; Serrano-Regal, M.P.; Ortega, M.C.; Clemente, D. Regulatory Cells in Multiple Sclerosis: From Blood to Brain. Biomedicines 2022, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Kutzelnigg, A.; Lassmann, H. Cortical demyelination in multiple sclerosis: A substrate for cognitive deficits? J. Neurol. Sci. 2006, 245, 123–126. [Google Scholar]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [PubMed]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.; Gay, D. Immunological and neuropathological significance of the Virchow-Robin space. J. Neurol. Sci. 1990, 100, 3–8. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain J. Neurol. 2007, 130 Pt 4, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Makshakov, G.; Magonov, E.; Totolyan, N.; Nazarov, V.; Lapin, S.; Mazing, A.; Verbitskaya, E.; Trofimova, T.; Krasnov, V.; Shumilina, M.; et al. Leptomeningeal Contrast Enhancement Is Associated with Disability Progression and Grey Matter Atrophy in Multiple Sclerosis. Neurol. Res. Int. 2017, 2017, 8652463. [Google Scholar] [CrossRef] [PubMed]
- Zivadinov, R.; Ramasamy, D.P.; Vaneckova, M.; Gandhi, S.; Chandra, A.; Hagemeier, J.; Bergsland, N.; Polak, P.; Benedict, R.H.; Hojnacki, D.; et al. Leptomeningeal contrast enhancement is associated with progression of cortical atrophy in MS: A retrospective, pilot, observational longitudinal study. Mult. Scler. J. 2016, 23, 1336–1345. [Google Scholar] [CrossRef]
- Fransen, N.L.; de Jong, B.A.; Heß, K.; Kuhlmann, T.; Vincenten, M.C.; Hamann, J.; Huitinga, I.; Smolders, J. Absence of B Cells in Brainstem and White Matter Lesions Associates with Less Severe Disease and Absence of Oligoclonal Bands in MS. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e955. [Google Scholar] [CrossRef]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar]
- Lassmann, H. Multiple sclerosis: Lessons from molecular neuropathology. Exp. Neurol. 2014, 262, 2–7. [Google Scholar] [CrossRef]
- De Groot, C.J.A.; Bergers, E.; Kamphorst, W.; Ravid, R.; Polman, C.H.; Barkhof, F.; van der Valk, P. Post-mortem MRI-guided sampling of multiple sclerosis brain lesions: Increased yield of active demyelinating and (p)reactive lesions. Brain 2001, 124, 1635–1645. [Google Scholar] [CrossRef]
- Singh, S.; Metz, I.; Amor, S.; van der Valk, P.; Stadelmann, C.; Brück, W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 2013, 125, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- van Horssen, J.; Witte, M.E.; Schreibelt, G.; de Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002, 1, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Gilbert, D.L. Microglia, an in vivo source of reactive oxygen species in the brain. Adv. Neurol. 1993, 59, 321–326. [Google Scholar] [PubMed]
- Smith, K.J. Newly lesioned tissue in multiple sclerosis—A role for oxidative damage? Brain J. Neurol. 2011, 134 Pt 7, 1877–1881. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R. Kynurenine pathway metabolism and neuroinflammatory disease. Neural Regen. Res. 2017, 12, 39–42. [Google Scholar] [CrossRef]
- Murphy, M.P. Nitric oxide and cell death. Biochim. Biophys. Acta 1999, 1411, 401–414. [Google Scholar] [CrossRef]
- Martinelli, R.; Gegg, M.; Longbottom, R.; Adamson, P.; Turowski, P.; Greenwood, J. ICAM-1–mediated Endothelial Nitric Oxide Synthase Activation via Calcium and AMP-activated Protein Kinase Is Required for Transendothelial Lymphocyte Migration. Mol. Biol. Cell 2009, 20, 995–1005. [Google Scholar] [CrossRef]
- Karg, E.; Klivényi, P.; Németh, I.; Bencsik, K.; Pintér, S.; Vécsei, L. Nonenzymatic antioxidants of blood in multiple sclerosis. J. Neurol. 1999, 246, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Pukoli, D.; Polyák, H.; Rajda, C.; Vécsei, L. Kynurenines and Neurofilament Light Chain in Multiple Sclerosis. Front. Neurosci. 2021, 15, 658202. [Google Scholar] [CrossRef] [PubMed]
- Smerjac, S.M.; Bizzozero, O.A. Cytoskeletal protein carbonylation and degradation in experimental autoimmune encephalomyelitis. J. Neurochem. 2007, 105, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Seven, A.; Aslan, M.; Incir, S.; Altıntaș, A. Original article Evaluation of oxidative and nitrosative stress in relapsing remitting multiple sclerosis: Effect of corticosteroid therapy. Folia Neuropathol. 2013, 1, 58–64. [Google Scholar] [CrossRef]
- Haider, L.; Zrzavy, T.; Hametner, S.; Höftberger, R.; Bagnato, F.; Grabner, G.; Trattnig, S.; Pfeifenbring, S.; Brück, W.; Lassmann, H. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016, 139, 807–815. [Google Scholar] [CrossRef]
- Azevedo, C.J.; Kornak, J.; Chu, P.; Sampat, M.; Okuda, D.T.; Cree, B.A.; Nelson, S.J.; Hauser, S.L.; Pelletier, D. In vivo evidence of glutamate toxicity in multiple sclerosis. Ann. Neurol. 2014, 76, 269–278. [Google Scholar] [CrossRef]
- Rajda, C.; Pukoli, D.; Bende, Z.; Majláth, Z.; Vécsei, L. Excitotoxins, Mitochondrial and Redox Disturbances in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 353. [Google Scholar] [CrossRef] [PubMed]
- Cawley, N.; Solanky, B.S.; Muhlert, N.; Tur, C.; Edden, R.A.E.; Wheeler-Kingshott, C.A.M.; Miller, D.H.; Thompson, A.J.; Ciccarelli, O. Reduced gamma-aminobutyric acid concentration is associated with physical disability in progressive multiple sclerosis. Brain 2015, 138, 2584–2595. [Google Scholar] [CrossRef]
- Stojanovic, I.R.; Kostic, M.; Ljubisavljevic, S. The role of glutamate and its receptors in multiple sclerosis. J. Neural Transm. 2014, 121, 945–955. [Google Scholar] [CrossRef]
- Sriram, S.; Rodriguez, M. Indictment of the microglia as the villain in multiple sclerosis. Neurology 1997, 48, 464–470. [Google Scholar] [CrossRef]
- Piani, D.; Frei, K.; Do, K.Q.; Cuénod, M.; Fontana, A. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci. Lett. 1991, 133, 159–162. [Google Scholar] [CrossRef]
- Fuchs, S.A.; Peeters-Scholte, C.M.P.C.D.; de Barse, M.M.J.; Roeleveld, M.W.; Klomp, L.W.J.; Berger, R.; de Koning, T.J. Increased concentrations of both NMDA receptor co-agonists d-serine and glycine in global ischemia: A potential novel treatment target for perinatal asphyxia. Amino Acids 2011, 43, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; Le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.; Perkins, M. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.G.; Tasca, C.I.; Santos, C.E.; Alves, L.B.; Porciúncula, L.O.; Emanuelli, T.; Souza, D.O. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem. Int. 2002, 40, 621–627. [Google Scholar] [CrossRef]
- Tavares, R.G.; Tasca, C.I.; Santos, C.E.S.; Wajner, M.; Souza, D.; Dutra-Filho, C. Quinolinic acid inhibits glutamate uptake into synaptic vesicles from rat brain. Neuroreport 2000, 11, 249–254. [Google Scholar] [CrossRef]
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in Alzheimer’s disease. J. Neuroinflamm. 2009, 6, 36. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Yang, H.C.; Horrocks, L. Involvement of phospholipase A2 in neurodegeneration. Neurochem. Int. 1997, 30, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 2009, 37, 1147–1160. [Google Scholar] [CrossRef]
- Sekine, A.; Okamoto, M.; Kanatani, Y.; Sano, M.; Shibata, K.; Fukuwatari, T. Amino acids inhibit kynurenic acid formation via suppression of kynurenine uptake or kynurenic acid synthesis in rat brain in vitro. Springerplus 2015, 4, 48. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for Quinolinic Acid Cytotoxicity in Human Astrocytes and Neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Cammer, W. Oligodendrocyte killing by quinolinic acid in vitro. Brain Res. 2001, 896, 157–160. [Google Scholar] [CrossRef]
- Cammer, W. Protection of cultured oligodendrocytes against tumor necrosis factor-α by the antioxidants coenzyme Q10 and N-acetyl cysteine. Brain Res. Bull. 2002, 58, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brew, B.J.; Guillemin, G.J. Characterization of the kynurenine pathway in NSC-34 cell line: Implications for amyotrophic lateral sclerosis. J. Neurochem. 2010, 118, 816–825. [Google Scholar] [CrossRef]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2012, 12, 64–82. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef] [PubMed]
- Rajda, C.; Galla, Z.; Polyák, H.; Maróti, Z.; Babarczy, K.; Pukoli, D.; Vécsei, L. Cerebrospinal Fluid Neurofilament Light Chain Is Associated with Kynurenine Pathway Metabolite Changes in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 2665. [Google Scholar] [CrossRef]
- Saraste, M.; Matilainen, M.; Rajda, C.; Galla, Z.; Sucksdorff, M.; Vécsei, L.; Airas, L. Association between microglial activation and serum kynurenine pathway metabolites in multiple sclerosis patients. Mult. Scler. Relat. Disord. 2022, 59, 103667. [Google Scholar] [CrossRef]
- Szabo, M.; Lajkó, N.; Dulka, K.; Barczánfalvi, G.; Lőrinczi, B.; Szatmári, I.; Mihály, A.; Vécsei, L.; Gulya, K. The kynurenic acid analog SZR104 induces cytomorphological changes associated with the anti-inflammatory phenotype in cultured microglia. Sci. Rep. 2023, 13, 11328. [Google Scholar] [CrossRef]
- Szabo, M.; Lajkó, N.; Dulka, K.; Szatmári, I.; Fülöp, F.; Mihály, A.; Vécsei, L.; Gulya, K. Kynurenic Acid and Its Analog SZR104 Exhibit Strong Antiinflammatory Effects and Alter the Intracellular Distribution and Methylation Patterns of H3 Histones in Immunochallenged Microglia-Enriched Cultures of Newborn Rat Brains. Int. J. Mol. Sci. 2022, 23, 1079. [Google Scholar] [CrossRef]
- Tai, Y.-H.; Engels, D.; Locatelli, G.; Emmanouilidis, I.; Fecher, C.; Theodorou, D.; Müller, S.A.; Licht-Mayer, S.; Kreutzfeldt, M.; Wagner, I.; et al. Targeting the TCA cycle can ameliorate widespread axonal energy deficiency in neuroinflammatory lesions. Nat. Metab. 2023, 1–18. [Google Scholar] [CrossRef]
- Simkins, T.J.; Duncan, G.J.; Bourdette, D. Chronic Demyelination and Axonal Degeneration in Multiple Sclerosis: Pathogenesis and Therapeutic Implications. Curr. Neurol. Neurosci. Rep. 2021, 21, 26. [Google Scholar] [CrossRef]
- Waxman, S.G. Axonal conduction and injury in multiple sclerosis: The role of sodium channels. Nat. Rev. Neurosci. 2006, 7, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Nikić, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Ziabreva, I.; Campbell, G.; Rist, J.; Zambonin, J.; Rorbach, J.; Wydro, M.M.; Lassmann, H.; Franklin, R.J.M.; Mahad, D. Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia 2010, 58, 1827–1837. [Google Scholar] [CrossRef]
- Giorgio, A.; De Stefano, N. Advanced Structural and Functional Brain MRI in Multiple Sclerosis. Semin. Neurol. 2016, 36, 163–176. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef]
- Kwakye, G.F.; Paoliello, M.M.; Mukhopadhyay, S.; Bowman, A.B.; Aschner, M. Manganese-Induced Parkinsonism and Parkinson’s Disease: Shared and Distinguishable Features. Int. J. Environ. Res. Public Health 2015, 12, 7519–7540. [Google Scholar] [CrossRef]
- Chtourou, Y.; Trabelsi, K.; Fetoui, H.; Mkannez, G.; Kallel, H.; Zeghal, N. Manganese Induces Oxidative Stress, Redox State Unbalance and Disrupts Membrane Bound ATPases on Murine Neuroblastoma Cells In Vitro: Protective Role of Silymarin. Neurochem. Res. 2011, 36, 1546–1557. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Hider, R.C.; Gaeta, A.; Williams, R.; Francis, P. Metals ions and neurodegeneration. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2007, 20, 639–654. [Google Scholar] [CrossRef]
- Sayre, L.M.; Smith, M.A.; Perry, G. Chemistry and Biochemistry of Oxidative Stress in Neurodegenerative Disease. Curr. Med. Chem. 2001, 8, 721–738. [Google Scholar] [CrossRef]
- Bénardais, K.; Kotsiari, A.; Škuljec, J.; Koutsoudaki, P.N.; Gudi, V.; Singh, V.; Vulinović, F.; Skripuletz, T.; Stangel, M. Cuprizone [Bis(Cyclohexylidenehydrazide)] is Selectively Toxic for Mature Oligodendrocytes. Neurotox. Res. 2013, 24, 244–250. [Google Scholar] [CrossRef]
- Rossi-George, A.; Guo, C.-J.; Oakes, B.L.; Gow, A.J. Copper modulates the phenotypic response of activated BV2 microglia through the release of nitric oxide. Nitric Oxide 2012, 27, 201–209. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Teunissen, C.; Langkammer, C. Iron and Neurodegeneration in Multiple Sclerosis. Mult. Scler. Int. 2011, 2011, 606807. [Google Scholar] [CrossRef]
- Lehmann, I.; Sack, U.; Lehmann, J. Metal ions affecting the immune system. Met. Ions Life Sci. 2011, 8, 157–185. [Google Scholar] [PubMed]
- Pihl-Jensen, G.; Tsakiri, A.; Frederiksen, J.L. Statin Treatment in Multiple Sclerosis: A Systematic Review and Meta-Analysis. CNS Drugs 2015, 29, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Schoeps, V.A.; Graves, J.S.; Stern, W.A.; Zhang, L.; Nourbakhsh, B.; Mowry, E.M.; Henry, R.G.; Waubant, E. N-Acetyl Cysteine as a Neuroprotective Agent in Progressive Multiple Sclerosis (NACPMS) trial: Study protocol for a randomized, double-blind, placebo-controlled add-on phase 2 trial. Contemp. Clin. Trials 2022, 122, 106941. [Google Scholar] [CrossRef]
- Fitzgerald, K.C.; Morris, B.; Soroosh, A.; Balshi, A.; Maher, D.; Kaplin, A.; Nourbakhsh, B. Pilot randomized active-placebo-controlled trial of low-dose ketamine for the treatment of multiple sclerosis-related fatigue. Mult. Scler. 2021, 27, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Metz, L.M.; Li, D.; Traboulsee, A.; Myles, M.L.; Duquette, P.; Godin, J.; Constantin, M.; Yong, V.W. Glatiramer acetate in combination with minocycline in patients with relapsing--remitting multiple sclerosis: Results of a Canadian, multicenter, double-blind, placebo-controlled trial. Mult. Scler. 2009, 15, 1183–1194. [Google Scholar]
- Metz, L.M.; Li, D.K.B.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Sicotte, N.L.; Giesser, B.S.; Tandon, V.; Klutch, R.; Steiner, B.; Drain, A.E.; Shattuck, D.W.; Hull, L.; Wang, H.J.; Elashoff, R.M.; et al. Testosterone treatment in multiple sclerosis: A pilot study. Arch. Neurol. 2007, 64, 683–688. [Google Scholar] [CrossRef]
- Rajendran, R.; Böttiger, G.; Dentzien, N.; Rajendran, V.; Sharifi, B.; Ergün, S.; Stadelmann, C.; Karnati, S.; Berghoff, M. Effects of FGFR Tyrosine Kinase Inhibition in OLN-93 Oligodendrocytes. Cells 2021, 10, 1318. [Google Scholar] [CrossRef]
- Lagrèze, W.A.; Küchlin, S.; Ihorst, G.; Grotejohann, B.; Beisse, F.; Volkmann, M.; Albrecht, P.; Ungewiss, J.; Wörner, M.; Wolf, S.; et al. Safety and efficacy of erythropoietin for the treatment of patients with optic neuritis (TONE): A randomised, double-blind, multicentre, placebo-controlled study. Lancet Neurol. 2021, 20, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin regulates the in vitro and in vivo production of neuronal pro-genitors by mammalian forebrain neural stem cells. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 9733–9743. [Google Scholar] [CrossRef]
- Sanoobar, M.; Eghtesadi, S.; Azimi, A.; Khalili, M.; Khodadadi, B.; Jazayeri, S.; Gohari, M.R.; Aryaeian, N. Coenzyme Q10 supplementation ameliorates inflammatory markers in patients with multiple sclerosis: A double blind, placebo, controlled randomized clinical trial. Nutr. Neurosci. 2014, 18, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Fiebiger, S.M.; Bros, H.; Grobosch, T.; Janssen, A.; Chanvillard, C.; Paul, F.; Dörr, J.; Millward, J.M.; Infante-Duarte, C. The antioxidant idebenone fails to prevent or attenuate chronic experimental autoimmune encephalomyelitis in the mouse. J. Neuroimmunol. 2013, 262, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Manczak, M.; Shirendeb, U.P.; Reddy, P.H. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 2322–2331. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Galea, E.; Gavrilyuk, V.; Brosnan, C.F.; Whitacre, C.C.; Dumitrescu-Ozimek, L.; Landreth, G.E.; Pershadsingh, H.A.; Weinberg, G.; Heneka, M.T. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann. Neurol. 2002, 51, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, O.H.; Lebrun, C.; Siva, A.; Keegan, M.B.; Azevedo, C.J.; Inglese, M.; Tintoré, M.; Newton, B.D.; Durand-Dubief, F.; Amato, M.P.; et al. Primary Progressive Multiple Sclerosis Evolving from Radiologically Isolated Syndrome. Ann. Neurol. 2015, 79, 288–294. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Pharmacological Actions and Mechanisms | References |
|---|---|---|
| Simvastatin | Pleiotropic effects, including modulation of excitotoxicity | [159] |
| N-acetyl cysteine | Glutathione (GSH) precursor with antioxidant properties | [160] |
| Ketamine | Glutamate antagonists | [161] |
| Clemastine fumarate | Antihistamine | [162] |
| Minocycline | Second-generation tetracycline antibiotic with immunomodulating properties | [163,164] |
| Ibudilast | Anti-apoptotic agent (non-selective phosphodiesterase inhibitor) | [165] |
| Bruton’s tyrosine kinase inhibitors (BTKi) | Agent acting on microglia | [166] |
| Testosterone | Immunomodulatory effects | [167] |
| Basic fibroblast growth factor (bFGF) | Promotes proliferation and migration of OPCs | [168] |
| Erythropoietin (EPO) | Blocking of ROS production and related apoptosis, neuroprotective effects, and stimulation of neurogenesis | [169,170] |
| Coenzyme Q10 | Antioxidant agent | [171] |
| Idebenone | Synthetic analogue of coenzyme Q10 | [172] |
| Mitoquinone (MitoQ) | Mitochondria-targeted antioxidant | [173] |
| Pioglitazone | Agonists of the peroxisome proliferator-activated receptors (PPARγ) | [174] |
| SZR104 | Anti-inflammatory phenotype in cultured microglia | [140,141] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pukoli, D.; Vécsei, L. Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms. Int. J. Mol. Sci. 2023, 24, 12631. https://doi.org/10.3390/ijms241612631
Pukoli D, Vécsei L. Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms. International Journal of Molecular Sciences. 2023; 24(16):12631. https://doi.org/10.3390/ijms241612631
Chicago/Turabian StylePukoli, Dániel, and László Vécsei. 2023. "Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms" International Journal of Molecular Sciences 24, no. 16: 12631. https://doi.org/10.3390/ijms241612631
APA StylePukoli, D., & Vécsei, L. (2023). Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms. International Journal of Molecular Sciences, 24(16), 12631. https://doi.org/10.3390/ijms241612631