


Beyond Motor Deficits: Environmental Enrichment Mitigates Huntington’s Disease Effects in YAC128 Mice

 ,
,  , ,
, ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Body Weight
2.2. Effects of Genotype and Environment on Motor Performance
2.3. Effects of Genotype and Environment on Locomotion and Anxious-like Behavior
2.4. Effects of Genotype and Environment on Depressive-like Behavior
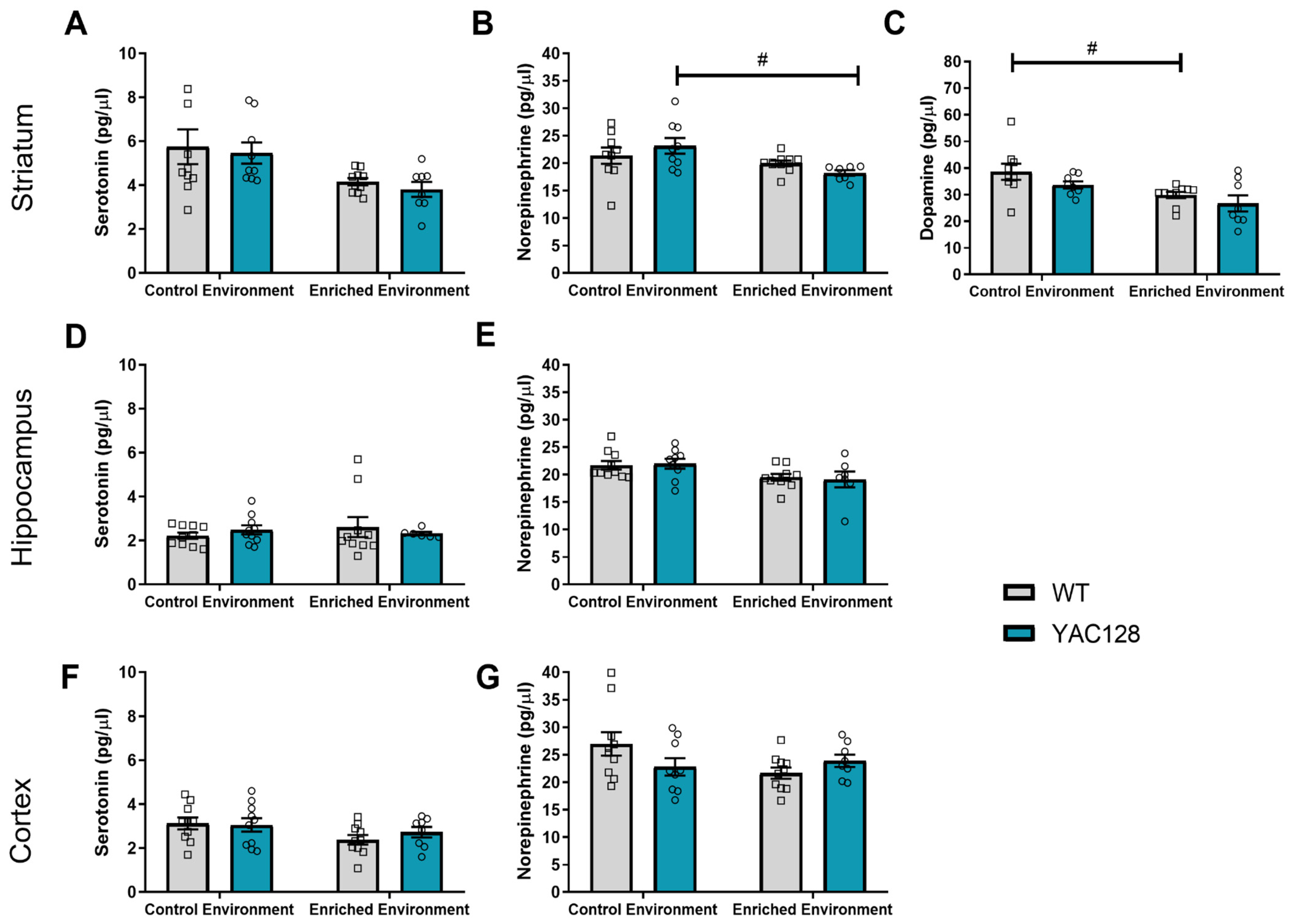
2.5. Effects of Genotype and Environment on Monoamine Levels
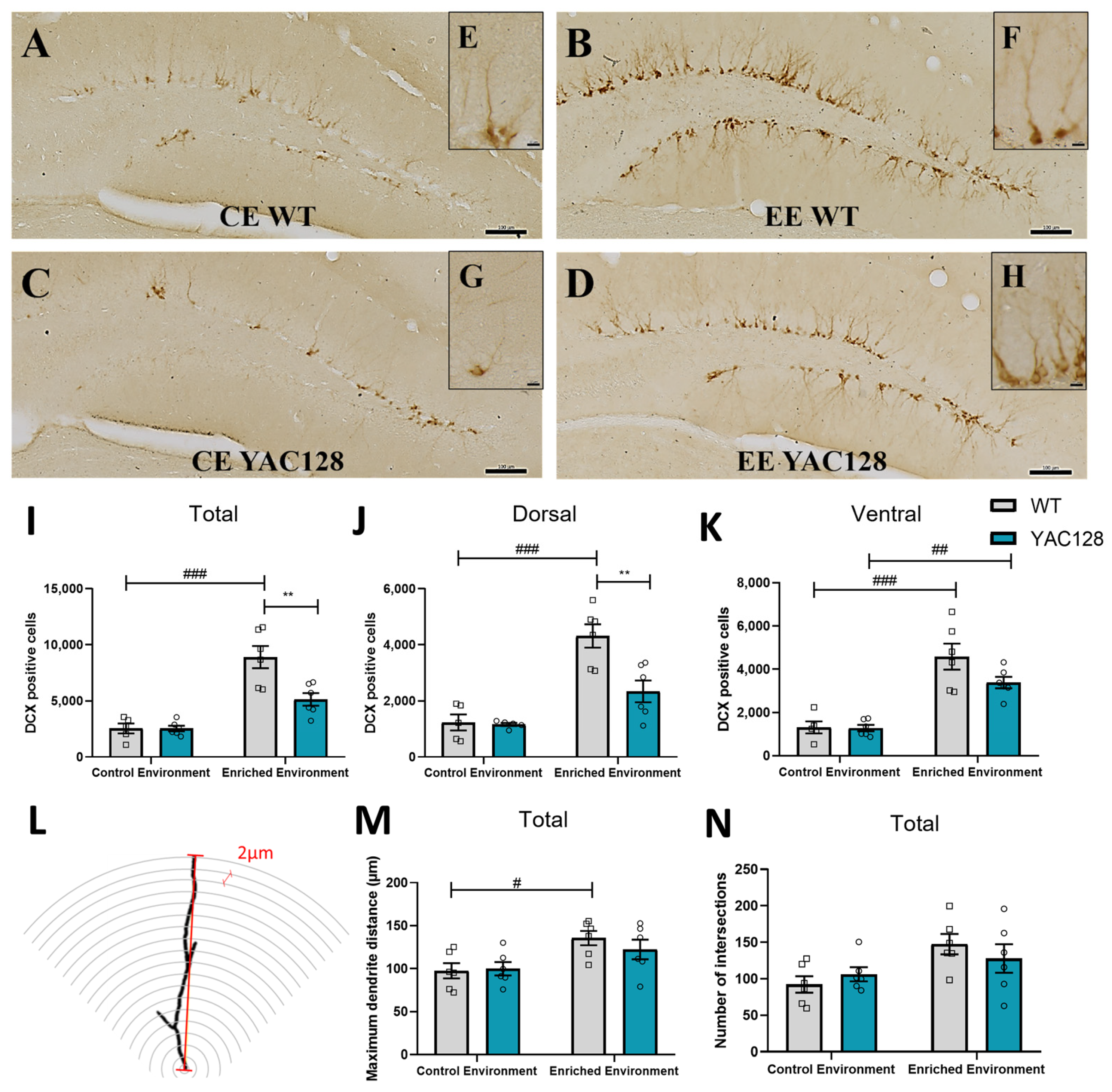
2.6. Effects of Genotype and Environment on Hippocampal Cell Proliferation
2.7. Effects of Genotype and Environment on Hippocampal Neuronal Differentiation and Dendritic Arborization
3. Discussion
3.1. Effects of EE Exposure on Non-Motor Symptoms in YAC128 Mice
3.2. Effects of EE Exposure on Motor Symptoms in YAC128 Mice
3.3. Effects of EE Exposure on Monoamine Neurotransmission in YAC128 Mice
3.4. Effects of EE Exposure on Neuronal Plasticity in YAC128 Mice
4. Material and Methods
4.1. Animals
4.2. Genotyping
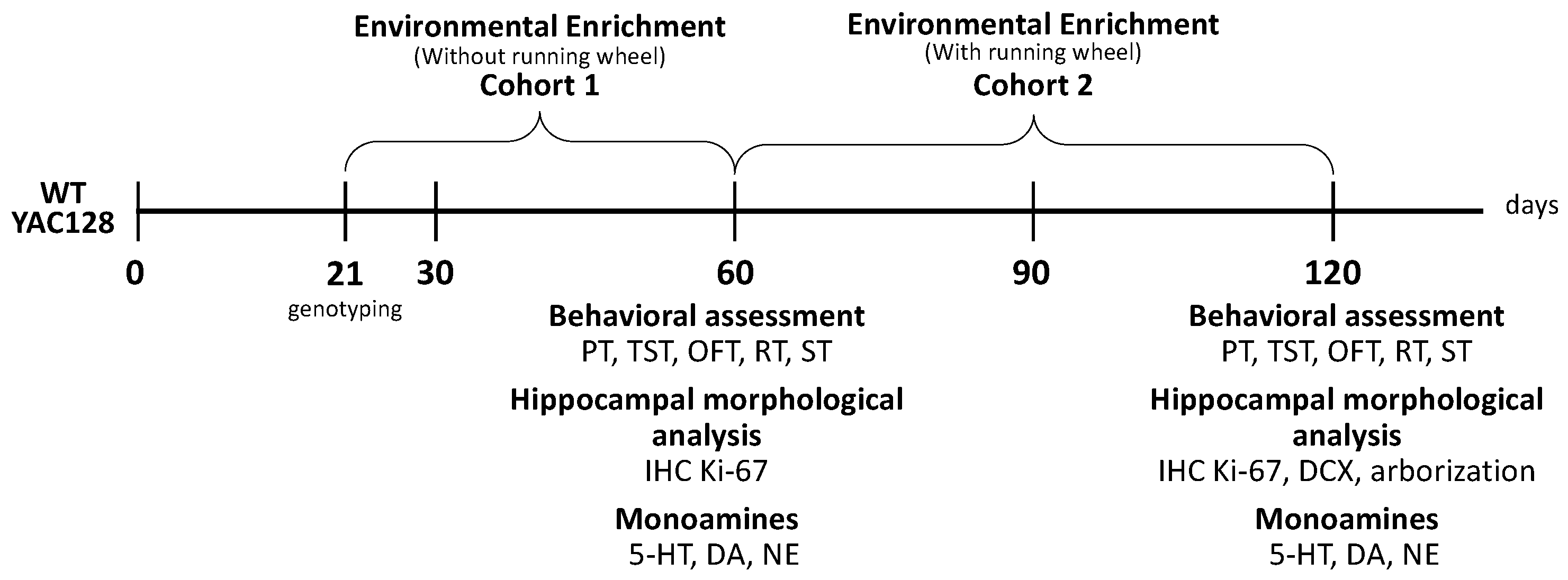
4.3. Experimental Procedure
4.4. Behavioral Tests
4.4.1. Locomotor Activity and Motor Function
- (a)
- Open field test (OFT): Locomotor activity was assessed in the open field apparatus, which consisted of a wooden box measuring 40 × 60 × 52 cm. The floor was divided into central and periphery regions, where activity in the center measures vs. the periphery of the apparatus is considered a proxy for anxiety-like behavior [92]. The distance traveled and the time spent in the center of the arena (13.5 × 30 cm) were measured for 6 min. Data were analyzed using the ANY-maze video-tracking system (Stoelting Co., Wood Dale, IL, USA).
- (b)
- Pole test (PT): The motor function was assessed by the PT as previously described [93] with some modifications. The testing apparatus consisted of a vertical rough-surfaced pole (54 cm height, 1 cm diameter), and the mouse was placed head-upward on top. The time between orienting downward and descending to the floor (time to descend) was measured. Each animal completed five trials, and the best performance was used. If the mouse could not turn entirely downward, fell, or slipped down, the default time of 120 s was recorded and taken as the maximal severity of impairment.
- (c)
- Accelerating rotarod test: The motor performance on the rotarod (Insight®, São Paulo, Brazil) was assessed as previously described [94,95] with some modifications. For the accelerating task, mice were once acclimatized to the apparatus for 120 s at a constant speed of 5 rpm. After a 120 min resting interval, each animal received 4 trials, with a progressively increasing speed, from 5 to 37 rpm over 5 min, with a 60 min interval between each trial. The time that each animal stayed on the rod before falling (the latency time for the first fall) and the number of falls were measured. Results were expressed as the average of the four sessions.
4.4.2. Depressive-like Behavior Analyses
- (a)
- Tail suspension test (TST): The TST was initially proposed in [96], and it is based on the evaluation of immobility in a situation of “behavioral despair”. Briefly, mice were suspended by their tail about 50 cm above the floor using adhesive tape. Mice were considered immobile only when they hung passively and were completely motionless. Immobility time was assessed during a 6 min period for each mouse.
- (b)
- Splash test (ST): The ST was performed using a previously described protocol [97] with some modifications. Animals were individually placed in a clear cylinder for 5 min for acclimatization to the apparatus. After habituation, a 10% sucrose solution was squirted onto the dorsal coat of each mouse. This procedure usually induces grooming behaviors because of its viscosity and palatable flavor. After applying sucrose solution, the latency to initiate grooming and the time spent grooming were recorded for 5 min as an index of self-care and motivational behavior.
4.5. Measurement of Brain Monoamine Levels
4.6. Tissue Processing for Immunohistochemistry
4.6.1. Hippocampal Cell Proliferation and Neuronal Differentiation
4.6.2. Morphological Quantification
4.6.3. Arborization Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huntington, G. On Chorea. Med. Surg. Rep. 1872, 15, 317–321. [Google Scholar]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington Disease: Natural History, Biomarkers and Prospects for Therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef]
- Macdonald, M.E.; Haines, J.; Zimmer, M.; Cheng, S.V.; Youngman, S.; Whaley, W.; Wexler, N.; Bucan, M.; Allitto, B.A.; Smith, B.; et al. Recombination Events Suggest Potential Sites for the Huntington’s Disease Gene. Neuron 1989, 3, 183–190. [Google Scholar] [CrossRef]
- Strong, T.V.; Tagle, D.A.; Valdes, J.M.; Elmer, L.W.; Boehm, K.; Swaroop, M.; Kaatz, K.W.; Collins, F.S.; Albin, R.L. Widespread Expression of the Human and Rat Huntington’s Disease Gene in Brain and Nonneural Tissues. Nat. Genet. 1993, 5, 259–265. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, V.; Singh, K.; Kumar, S.; Kim, Y.; Lee, Y.-M.; Kim, J.-J. Therapeutic Advances for Huntington’s Disease. Brain Sci. 2020, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef]
- Vonsattel, J.P.G.; DiFiglia, M. Huntington Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Ransome, M.I.; Renoir, T.; Hannan, A.J. Hippocampal Neurogenesis, Cognitive Deficits and Affective Disorder in Huntington’s Disease. Neural Plast. 2012, 2012, 874387. [Google Scholar] [CrossRef] [PubMed]
- Rosas, H.D.; Liu, A.K.; Hersch, S.; Glessner, M.; Ferrante, R.J.; Salat, D.H.; van der Kouwe, A.; Jenkins, B.G.; Dale, A.M.; Fischl, B. Regional and Progressive Thinning of the Cortical Ribbon in Huntington’s Disease. Neurology 2002, 58, 695–701. [Google Scholar] [CrossRef]
- Paoli, R.A.; Botturi, A.; Ciammola, A.; Silani, V.; Prunas, C.; Lucchiari, C.; Zugno, E.; Caletti, E. Neuropsychiatric Burden in Huntington’s Disease. Brain Sci. 2017, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Arenas, J.; Campos, Y.; Ribacoba, R.; Martín, M.A.; Rubio, J.C.; Ablanedo, P.; Cabello, A. Complex I Defect in Muscle from Patients with Huntington’s Disease. Ann. Neurol. 1998, 43, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.J.; Aziz, N.A.; van der Burg, J.M.M.; Landwehrmeyer, G.B.; Brundin, P.; Stijnen, T.; Roos, R.A.C. Weight Loss in Huntington Disease Increases with Higher CAG Repeat Number. Neurology 2009, 73, 572. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, J.M.M.; Gardiner, S.L.; Ludolph, A.C.; Landwehrmeyer, G.B.; Roos, R.A.C.; Aziz, N.A. Body Weight Is a Robust Predictor of Clinical Progression in Huntington Disease. Ann. Neurol. 2017, 82, 479–483. [Google Scholar] [CrossRef]
- Spokes, E.G.S. Neurochemical Alterations in Huntington’s Chorea: A Study of Post-Mortem Brain Tissue. Brain 1980, 103, 179–210. [Google Scholar] [CrossRef] [PubMed]
- Spokes, E.G.S. The Neurochemistry of Huntington’s Chorea. Trends Neurosci. 1981, 4, 115–118. [Google Scholar] [CrossRef]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain Dopamine and the Syndromes of Parkinson and Huntington Clinical, Morphological and Neurochemical Correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Elevated Serotonin and Reduced Dopamine in Subregionally Divided Huntington’s Disease Striatum. Ann. Neurol. 1987, 22, 386–389. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wang, E.A.; Cepeda, C.; Levine, M.S. Dopamine Imbalance in Huntington’s Disease: A Mechanism for the Lack of Behavioral Flexibility. Front. Neurosci. 2013, 7, 114. [Google Scholar] [CrossRef]
- Cepeda, C.; Murphy, K.P.S.; Parent, M.; Levine, M.S. The Role of Dopamine in Huntington’s Disease. In Progress in Brain Research; Elsevier B.V.: Amsterdam, The Netherlands, 2014; Volume 211, pp. 235–254. ISBN 9780444634252. [Google Scholar]
- Hayden, M.R.; Leavitt, B.R.; Yasothan, U.; Kirkpatrick, P. Tetrabenazine. Nat. Rev. Drug Discov. 2009, 8, 17–18. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Austedo (Deutetrabenazine); U.S. Food and Drug Administration: Silver Spring, MD, USA, 2017; pp. 1–17. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/208082s011lbl.pdf (accessed on 1 June 2023).
- U.S. Food and Drug Administration. FDA Xenazine (Tetrabenazine); U.S. Food and Drug Administration: Silver Spring, MD, USA, 2008; pp. 1–63. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021894lbl.pdf (accessed on 1 June 2023).
- Langbehn, D.R.; Stout, J.C.; Gregory, S.; Mills, J.A.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Long, J.D.; Owen, G.; Johnson, H.J.; et al. Association of CAG Repeats with Long-Term Progression in Huntington Disease. JAMA Neurol. 2019, 76, 1375–1385. [Google Scholar] [CrossRef]
- Djoussé, L.; Knowlton, B.; Hayden, M.; Almqvist, E.W.; Brinkman, R.; Ross, C.; Margolis, R.; Rosenblatt, A.; Durr, A.; Dode, C.; et al. Interaction of Normal and Expanded CAG Repeat Sizes Influences Age at Onset of Huntington Disease. Am. J. Med. Genet. Part A 2003, 119, 279–282. [Google Scholar] [CrossRef]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayan, J.; et al. Venezuelan Kindreds Reveal That Genetic and Environmental Factors Modulate Huntington’s Disease Age of Onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, N.; Bradshaw, J.L.; Chiu, E.; Tudor, A.; O’Gorman, L.; Phillips, J.G. Differential Clinical and Motor Control Function in a Pair of Monozygotic Twins with Huntington’s Disease. Mov. Disord. 1999, 14, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Trembath, M.K.; Horton, Z.A.; Tippett, L.; Hogg, V.; Collins, V.R.; Churchyard, A.; Velakoulis, D.; Roxburgh, R.; Delatycki, M.B. A Retrospective Study of the Impact of Lifestyle on Age at Onset of Huntington Disease. Mov. Disord. 2010, 25, 1444–1450. [Google Scholar] [CrossRef]
- Mo, C.; Renoir, T.; Hannan, A.J. What’s Wrong with My Mouse Cage? Methodological Considerations for Modeling Lifestyle Factors and Gene-Environment Interactions in Mice. J. Neurosci. Methods 2016, 265, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. More Hippocampal Neurons in Adult Mice Living in an Enriched Environment. Nature 1997, 386, 493–495. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-Derived Neurotrophic Factor in Neurodegenerative Diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Spargo, E.; Everall, I.P.; Lantos, P.L. Neuronal Loss in the Hippocampus in Huntington’s Disease: A Comparison with HIV Infection. J. Neurol. Neurosurg. Psychiatry 1993, 56, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Kee, N.; Sivalingam, S.; Boonstra, R.; Wojtowicz, J.M. The Utility of Ki-67 and BrdU as Proliferative Markers of Adult Neurogenesis. J. Neurosci. Methods 2002, 115, 97–105. [Google Scholar] [CrossRef]
- Brown, J.P.; Couillard-Després, S.; Cooper-Kuhn, C.M.; Winkler, J.; Aigner, L.; Kuhn, H.G. Transient Expression of Doublecortin during Adult Neurogenesis. J. Comp. Neurol. 2003, 467, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A.C. Huntington’s Disease: A Clinical Review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Glidden, A.M.; Luebbe, E.A.; Elson, M.J.; Goldenthal, S.B.; Snyder, C.W.; Zizzi, C.E.; Dorsey, E.R.; Heatwole, C.R. Patient-Reported Impact of Symptoms in Huntington Disease: PRISM-HD. Neurology 2020, 94, e2045–e2053. [Google Scholar] [CrossRef]
- Last, J.M. A Dictionary of Epidemiology. J. Epidemiol. Community Health 2001, 47, 1–196. [Google Scholar] [CrossRef][Green Version]
- Wang, H.; Chen, X.; Li, Y.; Tang, T.S.; Bezprozvanny, I. Tetrabenazine Is Neuroprotective in Huntington’s Disease Mice. Mol. Neurodegener. 2010, 5, 18. [Google Scholar] [CrossRef]
- Yang, X.; Gray, M. Mouse Models for Validating Preclinical Candidates for Huntington’s Disease; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2010; pp. 165–195. ISBN 978084939000. [Google Scholar]
- Gil-Mohapel, J.M. Screening of Therapeutic Strategies for Huntington’s Disease in YAC128 Transgenic Mice. CNS Neurosci. Ther. 2012, 18, 77–86. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Slow, E.J.; Hossain, S.M.; Leavitt, B.R.; Hayden, M.R. Cognitive Dysfunction Precedes Neuropathology and Motor Abnormalities in the YAC128 Mouse Model of Huntington’s Disease. J. Neurosci. 2005, 25, 4169–4180. [Google Scholar] [CrossRef]
- Djoussé, L.; Knowlton, B.; Cupples, L.A.; Marder, K.; Shoulson, I.; Myers, R.H. Weight Loss in Early Stage of Huntington’s Disease. Neurology 2002, 59, 1325–1330. [Google Scholar] [CrossRef]
- Sanberg, P.R.; Fibiger, H.C.; Mark, R.F. Body Weight and Dietary Factors in Huntington’s Disease Patients Compared with Matched Controls. Med. J. Aust. 1981, 1, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Pouladi, M.A.; Xie, Y.; Skotte, N.H.; Ehrnhoefer, D.E.; Graham, R.K.; Kim, J.E.; Bissada, N.; Yang, X.W.; Paganetti, P.; Friedlander, R.M.; et al. Full-Length Huntingtin Levels Modulate Body Weight by Influencing Insulin-like Growth Factor 1 Expression. Hum. Mol. Genet. 2010, 19, 1528–1538. [Google Scholar] [CrossRef]
- Sjögren, M.; Soylu-Kucharz, R.; Dandunna, U.; Stan, T.L.; Cavalera, M.; Sandelius, Å.; Zetterberg, H.; Björkqvist, M. Leptin Deficiency Reverses High Metabolic State and Weight Loss without Affecting Central Pathology in the R6/2 Mouse Model of Huntington’s Disease. Neurobiol. Dis. 2019, 132, 104560. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Warby, S.C.; Hayden, M.R. Selective Degeneration in YAC Mouse Models of Huntington Disease. Brain Res. Bull. 2007, 72, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Zocher, S.; Schilling, S.; Grzyb, A.N.; Adusumilli, V.S.; Lopes, J.B.; Günther, S.; Overall, R.W.; Kempermann, G. Early-Life Environmental Enrichment Generates Persistent Individualized Behavior in Mice. bioRxiv 2019. [Google Scholar] [CrossRef] [PubMed]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.Z.; et al. Selective Striatal Neuronal Loss in a YAC128 Mouse Model of Huntington Disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.L.; Thompson, J.C.; Wild, S.; Yardumian, P.; Snowden, J.S.; Turner, G.; Craufurd, D. Psychiatric Disorders in Preclinical Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 939–943. [Google Scholar] [CrossRef]
- Paulsen, J.S.; Hoth, K.F.; Nehl, C.; Stierman, L. Critical Periods of Suicide Risk in Huntington’s Disease. Am. J. Psychiatry 2005, 162, 725–731. [Google Scholar] [CrossRef]
- Slaughter, J.R.; Martens, M.P.; Slaughter, K.A. Depression and Huntington’s Disease: Prevalence, Clinical Manifestations, Etiology, and Treatment. CNS Spectr. 2001, 6, 306–326. [Google Scholar] [CrossRef]
- Van Duijn, E.; Kingma, E.M.; Van Der Mast, R.C. Psychopathology in Verified Huntington’s Disease Gene Carriers. J. Neuropsychiatry Clin. Neurosci. 2007, 19, 441–448. [Google Scholar] [CrossRef]
- Plácido, E.; de Paula Nascimento-Castro, C.; Welter, P.G.; Gil-Mohapel, J.; Brocardo, P.S. Linking Huntington Disease, Brain-Derived Neurotrophic Factor, and Depressive-like Behaviors. In The Neuroscience of Depression; Elsevier: Amsterdam, The Netherlands, 2021; pp. 161–177. [Google Scholar]
- Duff, K.; Paulsen, J.S.; Beglinger, L.J.; Langbehn, D.R.; Stout, J.C. Psychiatric Symptoms in Huntington’s Disease before Diagnosis: The Predict-HD Study. Biol. Psychiatry 2007, 62, 1341–1346. [Google Scholar] [CrossRef]
- De Paula Nascimento-Castro, C.; Wink, A.C.; Da Fônseca, V.S.; Bianco, C.D.; Winkelmann-Duarte, E.C.; Farina, M.; Rodrigues, A.L.S.; Gil-Mohapel, J.; De Bem, A.F.; Brocardo, P.S. Antidepressant Effects of Probucol on Early-Symptomatic YAC128 Transgenic Mice for Huntington’s Disease. Neural Plast. 2018, 2018, 4056383. [Google Scholar] [CrossRef]
- Dale, M.; van Duijn, E. Anxiety in Huntington’s Disease. J. Neuropsychiatry Clin. Neurosci. 2015, 27, 262–271. [Google Scholar] [CrossRef]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A Rotarod Test for Evaluation of Motor Skill Learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef]
- Pinto, W.B.V.d.R.; Ko, G.M. Teste de Rotarod: Contribuições No Estudo Das Doenças Neuromusculares, Das Síndromes Extrapiramidais Das Ataxias Cerebelares. RESBCAL 2012, 1, 201–210. [Google Scholar]
- Brooks, S.P.; Dunnett, S.B. Tests to Assess Motor Phenotype in Mice: A User’s Guide. Nat. Rev. Neurosci. 2009, 10, 519–529. [Google Scholar] [CrossRef]
- McFadyen, M.P.; Kusek, G.; Bolivar, V.J.; Flaherty, L. Differences among Eight Inbred Strains of Mice in Motor Ability and Motor Learning on a Rotorod. Genes Brain Behav. 2003, 2, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Vuong, K.; Canning, C.G.; Menant, J.C.; Loy, C.T. Gait, Balance, and Falls in Huntington Disease, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 159, ISBN 9780444639165. [Google Scholar]
- Heindel, W.C.; Butters, N.; Salmon, D.P. Impaired Learning of a Motor Skill in Patients with Huntington’s Disease. Behav. Neurosci. 1988, 102, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Pearson, J.; Rogers, D.A.; Bissada, N.; Vogl, A.W.; Hayden, M.R.; Leavitt, B.R. Loss of Wild-Type Huntingtin Influences Motor Dysfunction and Survival in the YAC128 Mouse Model of Huntington Disease. Hum. Mol. Genet. 2005, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pouladi, M.A.; Stanek, L.M.; Xie, Y.; Franciosi, S.; Southwell, A.L.; Deng, Y.; Butland, S.; Zhang, W.; Cheng, S.H.; Shihabuddin, L.S.; et al. Marked Differences in Neurochemistry and Aggregates despite Similar Behavioural and Neuropathological Features of Huntington Disease in the Full-Length BACHD and YAC128 Mice. Hum. Mol. Genet. 2012, 21, 2219–2232. [Google Scholar] [CrossRef]
- Pouladi, M.A.; Graham, R.K.; Karasinska, J.M.; Xie, Y.; Santos, R.D.; Petersn, Â.; Hayden, M.R.; Petersen, A.; Hayden, M.R. Prevention of Depressive Behaviour in the YAC128 Mouse Model of Huntington Disease by Mutation at Residue 586 of Huntingtin. Brain 2008, 132, 919–932. [Google Scholar] [CrossRef]
- Vonsattel, J.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P. Neuropathological Classification of Huntington’s Disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.W.; Abercrombie, E.D. In Vivo Dopamine Efflux Is Decreased in Striatum of Both Fragment (R6/2) and Full-Length (YAC128) Transgenic Mouse Models of Huntington’s Disease. Front. Syst. Neurosci. 2011, 5, 61. [Google Scholar] [CrossRef]
- Yi Teo, R.T.; Hong, X.; Yu-Taeger, L.; Huang, Y.; Tan, L.J.; Xie, Y.; To, X.V.; Guo, L.; Rajendran, R.; Novati, A.; et al. Structural and Molecular Myelination Deficits Occur Prior to Neuronal Loss in the YAC128 and BACHD Models of Huntington Disease. Hum. Mol. Genet. 2016, 25, 2621–2632. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Ooi, J.; Ferrari Bardile, C.; Tan, L.J.; George, M.; Drum, C.L.; Lin, R.Y.; Hayden, M.R.; Pouladi, M.A. Treatment with the MAO-A Inhibitor Clorgyline Elevates Monoamine Neurotransmitter Levels and Improves Affective Phenotypes in a Mouse Model of Huntington Disease. Exp. Neurol. 2016, 278, 4–10. [Google Scholar] [CrossRef]
- Garrett, M.C.; Soares-da-Silva, P. Increased Cerebrospinal Fluid Dopamine and 3,4-Dihydroxyphenylacetic Acid Levels in Huntington’s Disease: Evidence for an Overactive Dopaminergic Brain Transmission. J. Neurochem. 1992, 58, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Rosas, H.D.; Doros, G.; Bhasin, S.; Thomas, B.; Gevorkian, S.; Malarick, K.; Matson, W.; Hersch, S.M. A Systems-Level “Misunderstanding”: The Plasma Metabolome in Huntington’s Disease. Ann. Clin. Transl. Neurol. 2015, 2, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Ghilan, M.; Bostrom, C.A.; Hryciw, B.N.; Simpson, J.M.; Christie, B.R.; Gil-Mohapel, J. YAC128 Huntington’s Disease Transgenic Mice Show Enhanced Short-Term Hippocampal Synaptic Plasticity Early in the Course of the Disease. Brain Res. 2014, 1581, 117–128. [Google Scholar] [CrossRef]
- Ranjbar-Slamloo, Y.; Fazlali, Z. Dopamine and Noradrenaline in the Brain; Overlapping or Dissociate Functions? Front. Mol. Neurosci. 2020, 12, 334. [Google Scholar] [CrossRef]
- Bureau, G.; Carrier, M.; Lebel, M.; Cyr, M. Intrastriatal Inhibition of Extracellular Signal-Regulated Kinases Impaired the Consolidation Phase of Motor Skill Learning. Neurobiol. Learn. Mem. 2010, 94, 107–115. [Google Scholar] [CrossRef]
- Gould, T.J.; Rowe, W.B.; Heman, K.L.; Mesches, M.H.; Young, D.A.; Rose, G.M.; Bickford, P.C. Effects of Hippocampal Lesions on Patterned Motor Learning in the Rat. Brain Res. Bull. 2002, 58, 581–586. [Google Scholar] [CrossRef]
- Scholz, J.; Niibori, Y.; Frankland, P.W.; Lerch, J.P. Rotarod Training in Mice Is Associated with Changes in Brain Structure Observable with Multimodal MRI. NeuroImage 2015, 107, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Bruel-Jungerman, E.; Rampon, C.; Laroche, S. Adult Hippocampal Neurogenesis, Synaptic Plasticity and Memory: Facts and Hypotheses. Rev. Neurosci. 2007, 18, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Jurkowski, M.P.; Bettio, L.; Woo, E.K.; Patten, A.; Yau, S.-Y.; Gil-Mohapel, J. Beyond the Hippocampus and the SVZ: Adult Neurogenesis Throughout the Brain. Front. Cell. Neurosci. 2020, 14, 576444. [Google Scholar] [CrossRef] [PubMed]
- Gil-mohapel, J.M.; Rego, A.C. Doença de Huntington: Uma Revisão Dos Aspectos Fisiopatológicos. Rev. Neurocienc. 2011, 19, 724–734. [Google Scholar] [CrossRef][Green Version]
- Kempermann, G.; Brandon, E.P.; Gage, F.H. Environmental Stimulation of 129/SvJ Mice Causes Increased Cell Proliferation and Neurogenesis in the Adult Dentate Gyrus. Curr. Biol. 1998, 8, 939–944. [Google Scholar] [CrossRef]
- van Praag, H.; Kempermann, G.; Gage, F.H. Running Increases Cell Proliferation and Neurogenesis in the Adult Mouse Dentate Gyrus. Nat. Neurosci. 1999, 2, 266–270. [Google Scholar] [CrossRef]
- Bjørnebekk, A.; Mathé, A.A.; Brené, S. The Antidepressant Effect of Running Is Associated with Increased Hippocampal Cell Proliferation. Int. J. Neuropsychopharmacol. 2005, 8, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Leggio, M.G.; Mandolesi, L.; Federico, F.; Spirito, F.; Ricci, B.; Gelfo, F.; Petrosini, L. Environmental Enrichment Promotes Improved Spatial Abilities and Enhanced Dendritic Growth in the Rat. Behav. Brain Res. 2005, 163, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Bindu, B.; Alladi, P.A.; Mansooralikhan, B.M.; Srikumar, B.N.; Raju, T.R.; Kutty, B.M. Short-Term Exposure to an Enriched Environment Enhances Dendritic Branching but Not Brain-Derived Neurotrophic Factor Expression in the Hippocampus of Rats with Ventral Subicular Lesions. Neuroscience 2007, 144, 412–423. [Google Scholar] [CrossRef]
- Spires, T.L.; Grote, H.E.; Garry, S.; Cordery, P.M.; Van Dellen, A.; Blakemore, C.; Hannan, A.J. Dendritic Spine Pathology and Deficits in Experience-Dependent Dendritic Plasticity in R6/1 Huntington’s Disease Transgenic Mice. Eur. J. Neurosci. 2004, 19, 2799–2807. [Google Scholar] [CrossRef]
- Rao, M.S.; Shetty, A.K. Efficacy of Doublecortin as a Marker to Analyse the Absolute Number Anddendritic Growth of Newly Generated Neurons in the Adult Dentate Gyrus. Eur. J. Neurosci. 2004, 19, 234–246. [Google Scholar] [CrossRef]
- Sholl, D.A. Dendritic Organization in the Neurons of the Visual and Motor Cortices of the Cat. J. Anat. 1953, 87, 387–406. [Google Scholar] [PubMed]
- Lazic, S.E.; Grote, H.E.; Blakemore, C.; Hannan, A.J.; Van Dellen, A.; Phillips, W.; Barker, R.A. Neurogenesis in the R6/1 Transgenic Mouse Model of Huntington’s Disease: Effects of Environmental Enrichment. Eur. J. Neurosci. 2006, 23, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Steventon, J.J.; Harrison, D.J.; Trueman, R.C.; Rosser, A.E.; Jones, D.K.; Brooks, S.P. In Vivo MRI Evidence That Neuropathology Is Attenuated by Cognitive Enrichment in the Yac128 Huntington’s Disease Mouse Model. J. Huntingtons. Dis. 2015, 4, 149–160. [Google Scholar] [CrossRef]
- da Fonsêca, V.S.; da Silva Colla, A.R.; de Paula Nascimento-Castro, C.; Plácido, E.; Rosa, J.M.; Farina, M.; Gil-Mohapel, J.; Rodrigues, A.L.S.; Brocardo, P.S. Brain-Derived Neurotrophic Factor Prevents Depressive-Like Behaviors in Early-Symptomatic YAC128 Huntington’s Disease Mice. Mol. Neurobiol. 2018, 55, 7201–7215. [Google Scholar] [CrossRef]
- Bailey, K.R.; Crawley, J.N.; Bailey, K.R. Behaviors in Mice. In Analysis; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; pp. 77–101. ISBN 9781420052343. [Google Scholar]
- Ogawa, N.; Hirose, Y.; Ohara, S.; Ono, T.; Watanabe, Y. A Simple Quantitative Bradykinesia Test in MPTP-Treated Mice. Res. Commun. Chem. Pathol. Pharmacol. 1985, 50, 435–441. [Google Scholar]
- Dunham, N.W.; Miya, T.S. A Note on a Simple Apparatus for Detecting Neurological Deficit in Rats and Mice. J. Am. Pharm. Assoc. (Sci. Ed.) 1957, 46, 208–209. [Google Scholar] [CrossRef]
- Carter, R.J.; Morton, J.; Dunnett, S.B. Motor Coordination and Balance in Rodents. Curr. Protoc. Neurosci. 2001, 15, 8.12.1–8.12.14. [Google Scholar] [CrossRef]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The Tail Suspension Test: A New Method for Screening Antidepressants in Mice. Psychopharmacology 1985, 85, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Ducottet, C.; Belzung, C. Behaviour in the Elevated Plus-Maze Predicts Coping after Subchronic Mild Stress in Mice. Physiol. Behav. 2004, 81, 417–426. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, G.E.; Fico, D.; Pennetta, A.; Malitesta, C.; Nicolardi, G.; Lofrumento, D.D.; De Nuccio, F.; La Pesa, V. A Rapid and Simple Method for the Determination of 3,4-Dihydroxyphenylacetic Acid, Norepinephrine, Dopamine, and Serotonin in Mouse Brain Homogenate by HPLC with Fluorimetric Detection. J. Pharm. Biomed. Anal. 2014, 98, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Scholzen, T.; Gerdes, J. The Ki-67 Protein: From the Known and the Unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Franklin, K.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates, Compact, 3rd ed.; Elsevier: New York, NY, USA, 2008; pp. 1–256. ISBN 9780123742445. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.E.; Heinrich, I.A.; Moura, T.M.; Fraga, D.B.; Costa, A.P.; Azevedo, D.; Brocardo, P.S.; Kaster, M.P.; Leal, R.B.; Rodrigues, A.L.S. Agmatine Potentiates Antidepressant and Synaptic Actions of Ketamine: Effects on Dendritic Arbors and Spines Architecture and Akt/S6 Kinase Signaling. Exp. Neurol. 2020, 333, 113398. [Google Scholar] [CrossRef] [PubMed]
- Fraga, D.B.; Costa, A.P.; Olescowicz, G.; Camargo, A.; Pazini, F.L.; Freitas, A.E.; Moretti, M.; Brocardo, P.S.; Ana, A.L. Ascorbic Acid Presents Rapid Behavioral and Hippocampal Synaptic Plasticity Effects. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 96, 109757. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Environment | Genotype | Body Weight (g) | N |
|---|---|---|---|---|
| 2 months | Control | WT | 22.3 ± 0.47 | 10 (5F–5M) |
| YAC128 | 25.5 ± 1.07 * | 10 (5F–5M) | ||
| Enriched Environment | WT | 23 ± 0.67 | 10 (5F–5M) | |
| YAC128 | 23.4 ± 0.62 | 08 (3F–5M) | ||
| 4 months | Control | WT | 26.78 ± 0.74 | 09 (4F–5M) |
| YAC128 | 26.9 ± 1.00 | 10 (6F–4M) | ||
| Enriched Environment | WT | 25.29 ± 0.68 | 07 (4F–3M) | |
| YAC128 | 27.90 ± 0.74 | 10 (4F–5M) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plácido, E.; Gomes Welter, P.; Wink, A.; Karasiak, G.D.; Outeiro, T.F.; Dafre, A.L.; Gil-Mohapel, J.; Brocardo, P.S. Beyond Motor Deficits: Environmental Enrichment Mitigates Huntington’s Disease Effects in YAC128 Mice. Int. J. Mol. Sci. 2023, 24, 12607. https://doi.org/10.3390/ijms241612607
Plácido E, Gomes Welter P, Wink A, Karasiak GD, Outeiro TF, Dafre AL, Gil-Mohapel J, Brocardo PS. Beyond Motor Deficits: Environmental Enrichment Mitigates Huntington’s Disease Effects in YAC128 Mice. International Journal of Molecular Sciences. 2023; 24(16):12607. https://doi.org/10.3390/ijms241612607
Chicago/Turabian StylePlácido, Evelini, Priscilla Gomes Welter, Ana Wink, Gabriela Duarte Karasiak, Tiago Fleming Outeiro, Alcir Luiz Dafre, Joana Gil-Mohapel, and Patricia S. Brocardo. 2023. "Beyond Motor Deficits: Environmental Enrichment Mitigates Huntington’s Disease Effects in YAC128 Mice" International Journal of Molecular Sciences 24, no. 16: 12607. https://doi.org/10.3390/ijms241612607
APA StylePlácido, E., Gomes Welter, P., Wink, A., Karasiak, G. D., Outeiro, T. F., Dafre, A. L., Gil-Mohapel, J., & Brocardo, P. S. (2023). Beyond Motor Deficits: Environmental Enrichment Mitigates Huntington’s Disease Effects in YAC128 Mice. International Journal of Molecular Sciences, 24(16), 12607. https://doi.org/10.3390/ijms241612607