
Anticancer Study of a Novel Pan-HDAC Inhibitor MPT0G236 in Colorectal Cancer Cells

Abstract
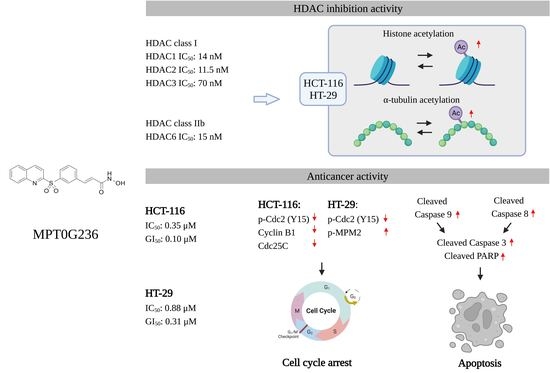
1. Introduction
2. Results
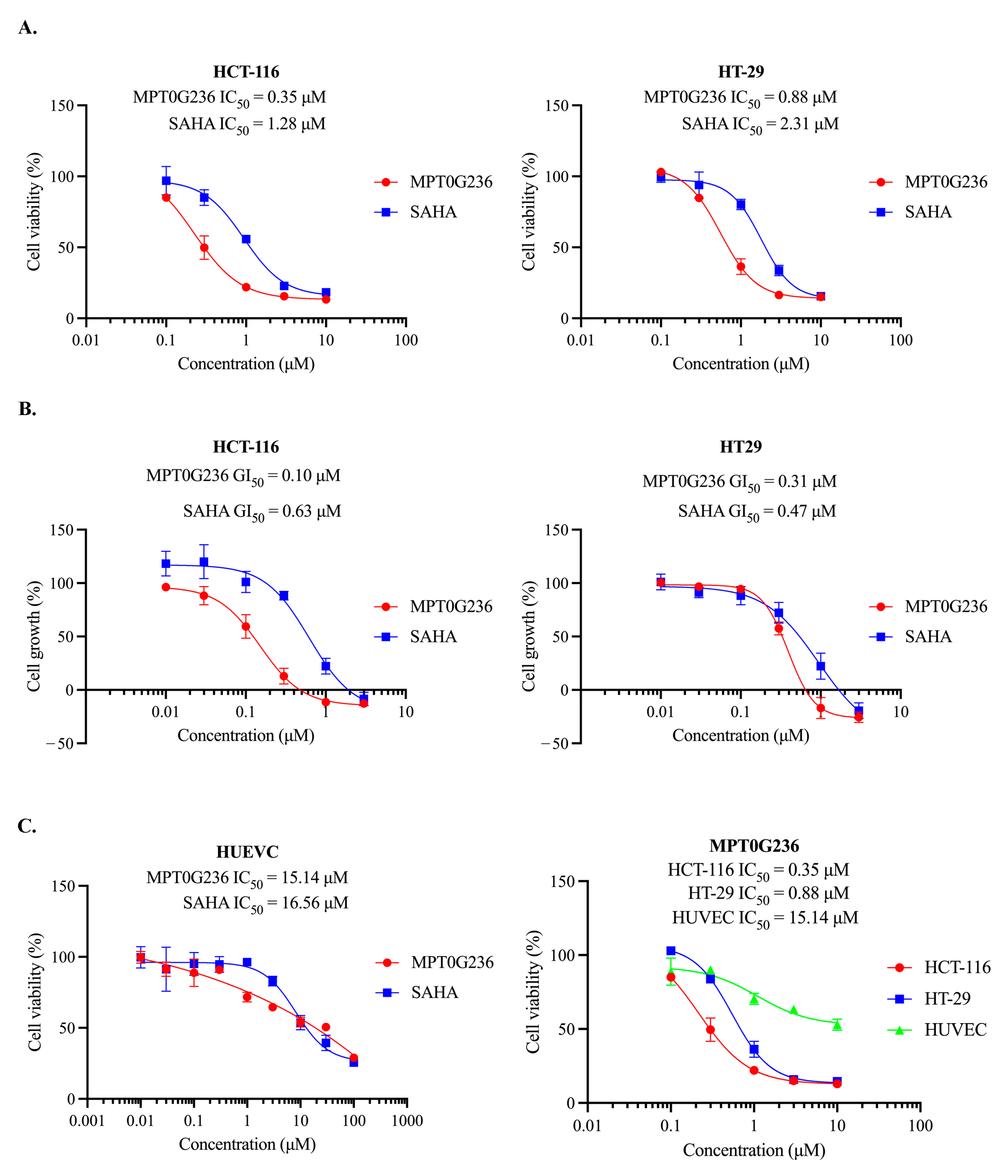
2.1. Antiproliferative Effect of MPT0G236 on Cancer Cells
2.2. HDAC Activity Inhibited by MPT0G236
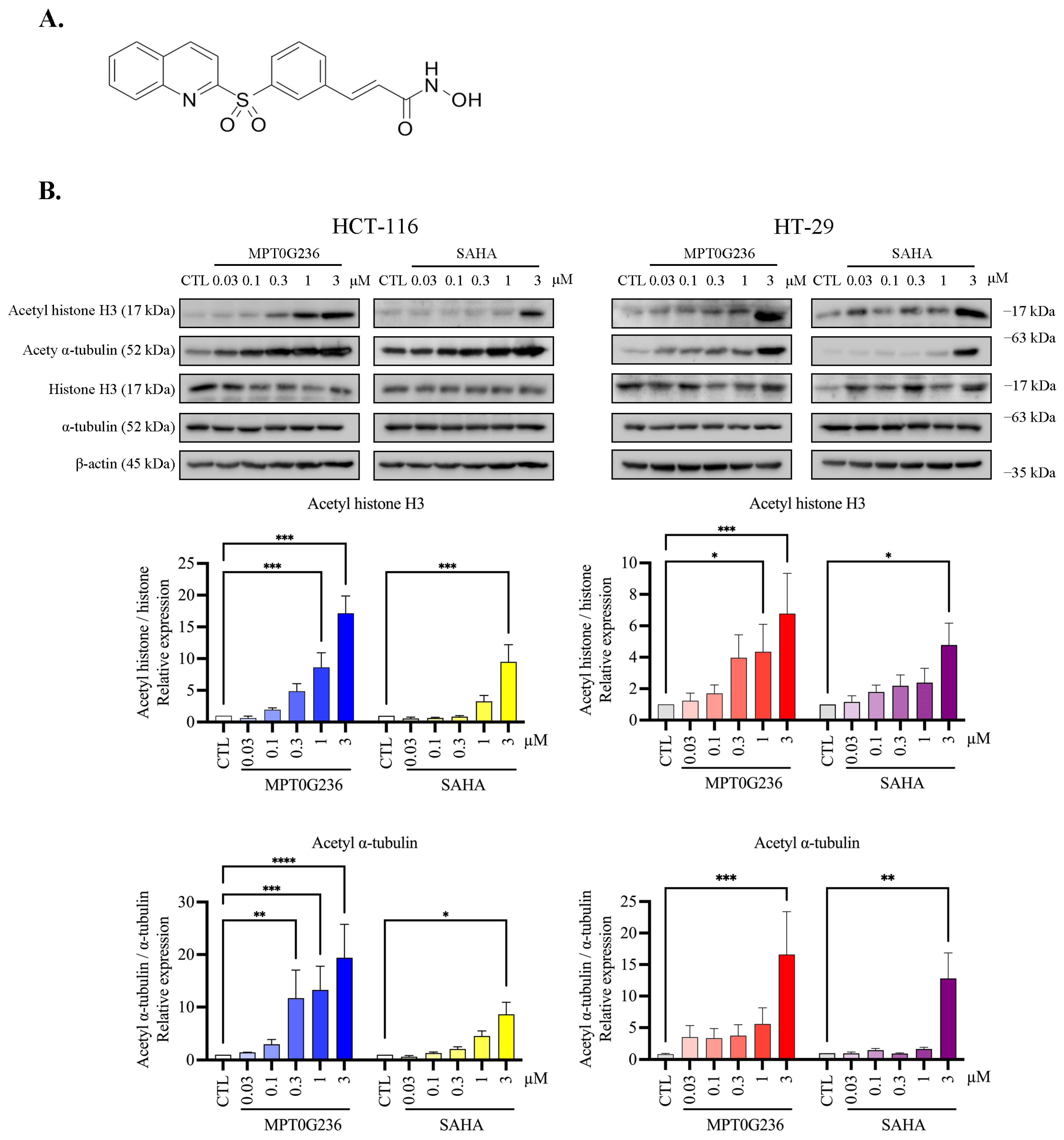
2.3. MPT0G236 Increased α-Tubulin and Histone H3 Acetylation Abundance in CRC Cells
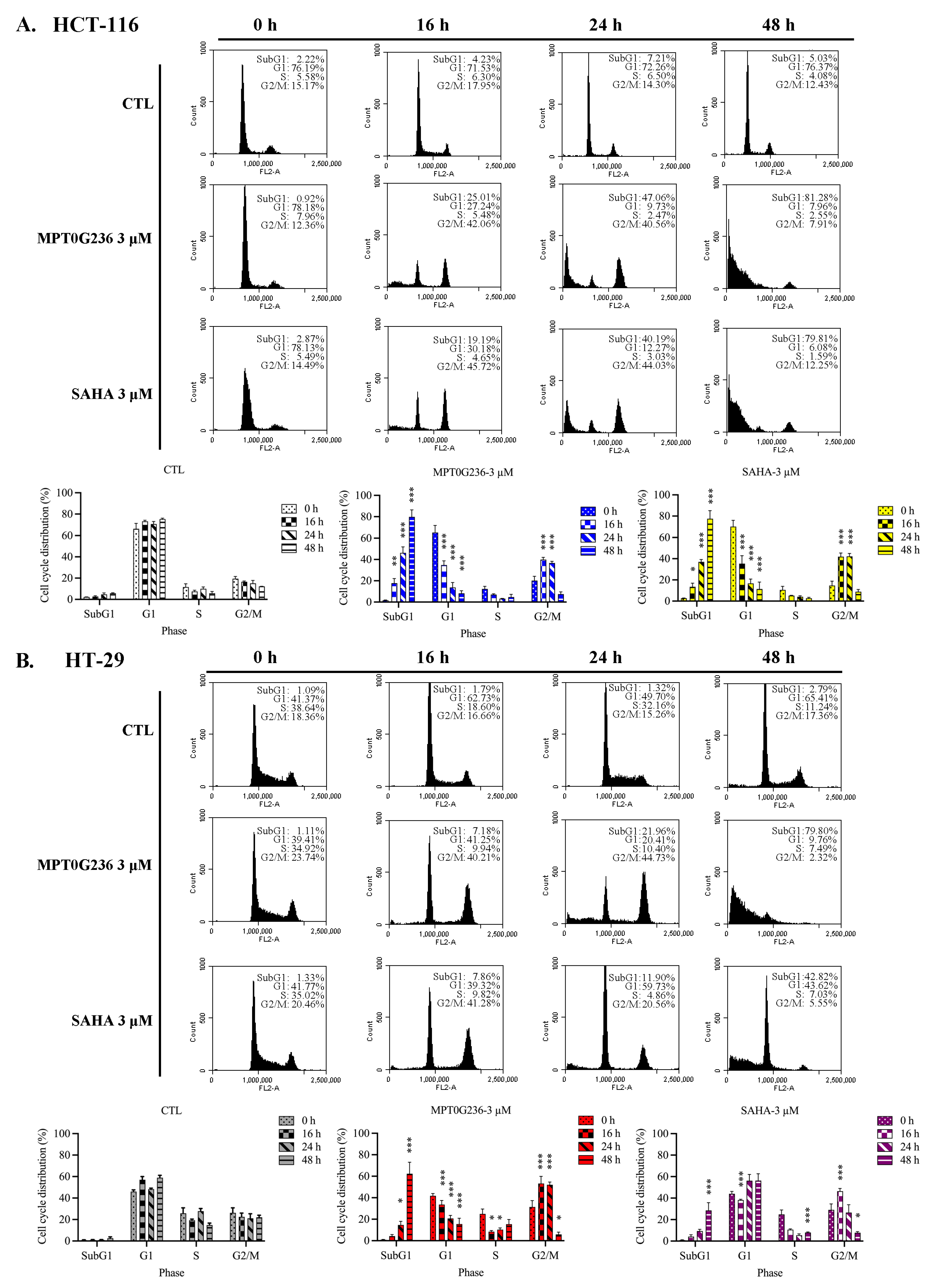
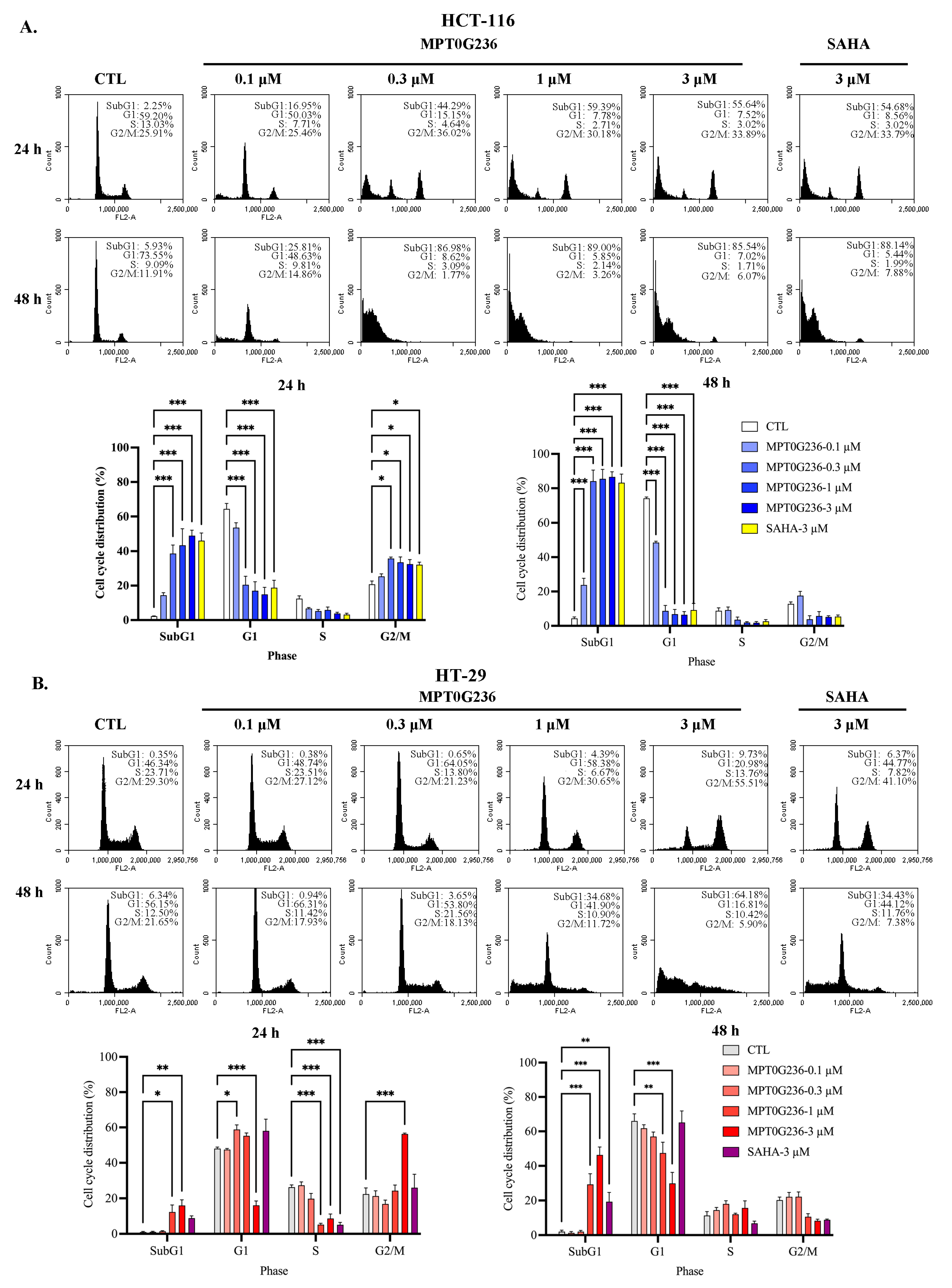
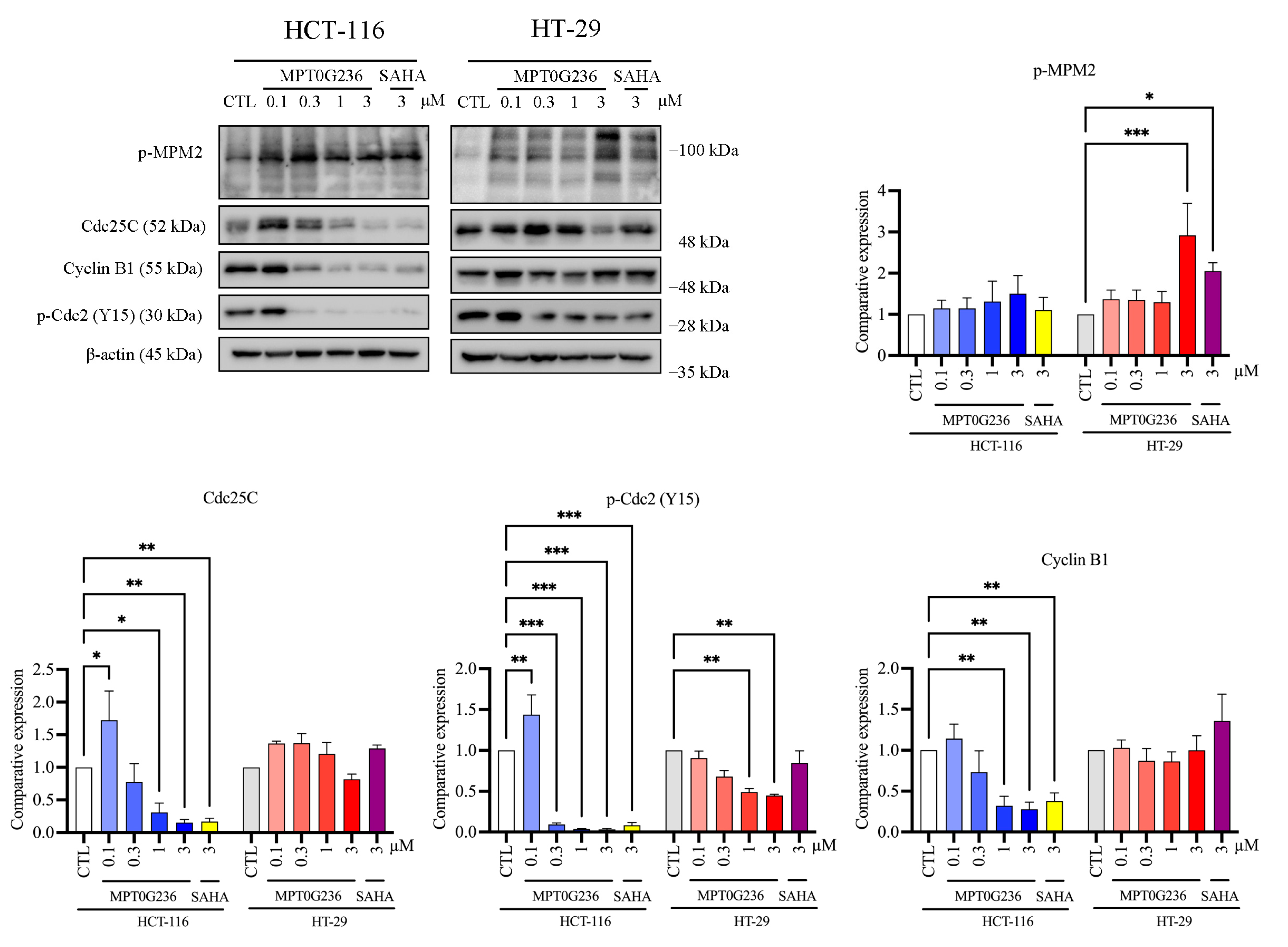
2.4. Effects of MPT0G236 on Mitotic Arrest in HCT-116 and HT-29 Cells
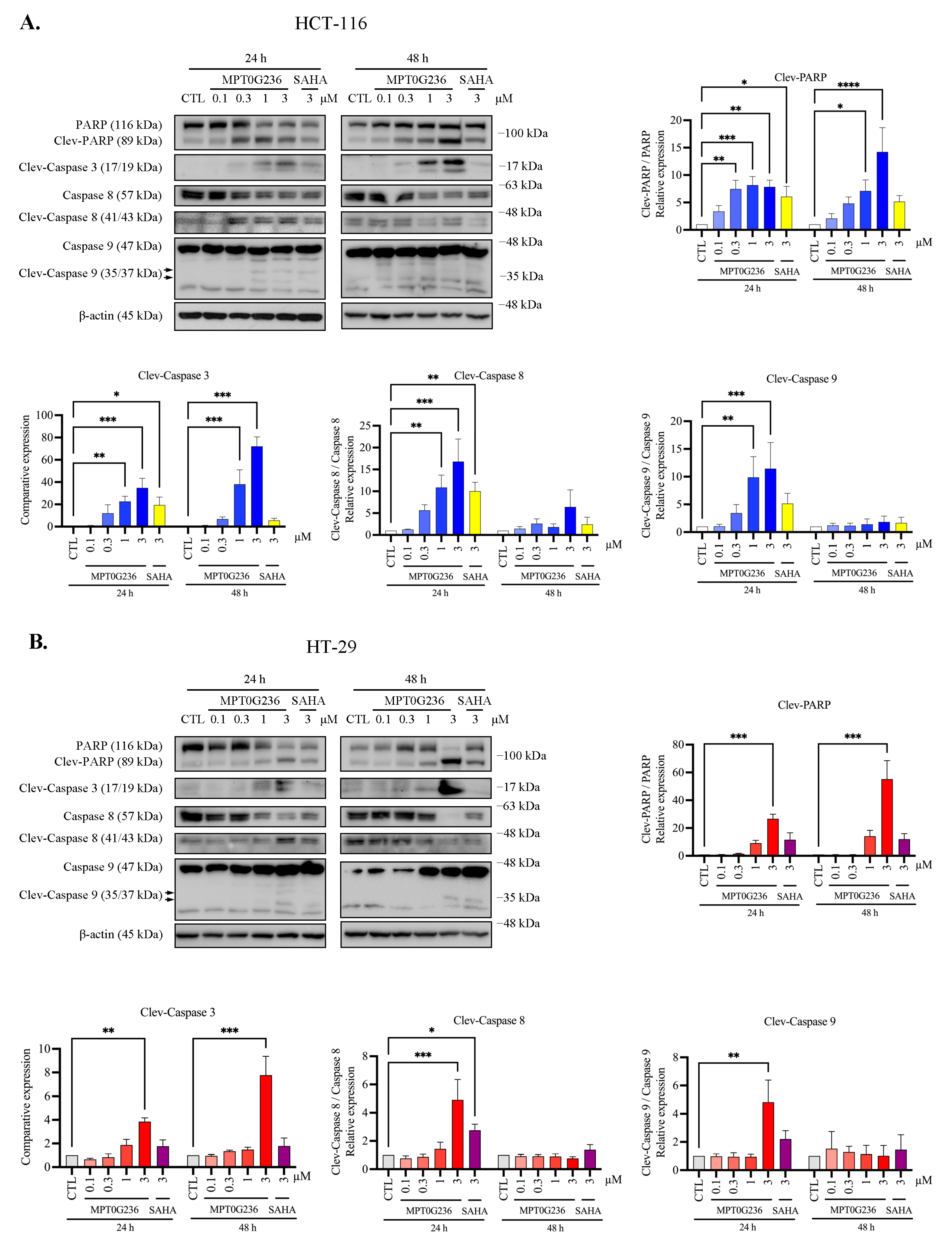
2.5. MPT0G236 Induced Apoptosis in Human Colorectal Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. HDAC Enzyme Inhibition Assay
4.4. Cell Proliferation Assay
4.5. Cell Viability Assay
4.6. Flow Cytometry
4.7. Immunoblot Analyses
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Biller, L.H.; Schrag, D. Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Gastroenterol. Rev. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; He, Q.; Yu, W. The histone deacetylase HDAC1 activates HIF1α/VEGFA signal pathway in colorectal cancer. Gene 2020, 754, 144851. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef]
- Chen, C.H.; Lee, C.H.; Liou, J.P.; Teng, C.M.; Pan, S.L. Molecular mechanisms underlying the antitumor activity of (E)-N-hydroxy-3-(1-(4-methoxyphenylsulfonyl)-1,2,3,4-tetrahydroquinolin-6-yl)acrylamide in human colorectal cancer cells in vitro and in vivo. Oncotarget 2015, 6, 35991–36002. [Google Scholar] [CrossRef]
- He, P.; Li, K.; Li, S.B.; Hu, T.T.; Guan, M.; Sun, F.Y.; Liu, W.W. Upregulation of AKAP12 with HDAC3 depletion suppresses the progression and migration of colorectal cancer. Int. J. Oncol. 2018, 52, 1305–1316. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yang, Y.; Liu, S.; Lu, J.; Huang, B.; Zhang, Y. HDAC inhibitor PAC-320 induces G2/M cell cycle arrest and apoptosis in human prostate cancer. Oncotarget 2017, 9, 512–523. [Google Scholar] [CrossRef] [PubMed]
- HuangFu, W.C.; Chao, M.W.; Cheng, C.C.; Wei, Y.C.; Wu, Y.W.; Liou, J.P.; Hsiao, G.; Lee, Y.C.; Yang, C.R. Anti-leukemia effects of the novel synthetic 1-benzylindole derivative 21-900 in vitro and in vivo. Sci. Rep. 2017, 7, 42291. [Google Scholar] [CrossRef]
- Jin, H.; Liang, L.; Liu, L.; Deng, W.; Liu, J. HDAC inhibitor DWP0016 activates p53 transcription and acetylation to inhibit cell growth in U251 glioblastoma cells. J. Cell. Biochem. 2013, 114, 1498–1509. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Lee, H.Y.; Nepali, K.; Huang, F.I.; Chang, C.Y.; Lai, M.J.; Li, Y.H.; Huang, H.L.; Yang, C.R.; Liou, J.P. (N-Hydroxycarbonylbenylamino)quinolines as selective histone deacetylase 6 inhibitors suppress growth of multiple myeloma in vitro and in vivo. J. Med. Chem. 2018, 61, 905–917. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Khan, O.; La Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Chang, C.Y.; Su, C.J.; Huang, H.L.; Mehndiratta, S.; Chao, Y.H.; Hsu, C.M.; Kumar, S.; Sung, T.Y.; Huang, Y.Z.; et al. 2-(Phenylsulfonyl)quinoline N-hydroxyacrylamides as potent anticancer agents inhibiting histone deacetylase. Eur. J. Med. Chem. 2016, 122, 92–101. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Dash, B.C.; El-Deiry, W.S. Phosphorylation of p21 in G2/M promotes cyclin B-Cdc2 kinase activity. Mol. Cell. Biol. 2005, 25, 3364–3387. [Google Scholar] [CrossRef] [PubMed]
- Escargueil, A.E.; Larsen, A.K. Mitosis-specific MPM-2 phosphorylation of DNA topoisomerase IIalpha is regulated directly by protein phosphatase 2A. Biochem. J. 2007, 403, 235–242. [Google Scholar] [CrossRef]
- Huang, Y.C.; Huang, F.I.; Mehndiratta, S.; Lai, S.C.; Liou, J.P.; Yang, C.R. Anticancer activity of MPT0G157, a derivative of indolylbenzenesulfonamide, inhibits tumor growth and angiogenesis. Oncotarget 2015, 6, 18590–18601. [Google Scholar] [CrossRef]
- Tu, H.J.; Lin, Y.J.; Chao, M.W.; Sung, T.Y.; Wu, Y.W.; Chen, Y.Y.; Lin, M.H.; Liou, J.P.; Pan, S.L.; Yang, C.R. The anticancer effects of MPT0G211, a novel HDAC6 inhibitor, combined with chemotherapeutic agents in human acute leukemia cells. Clin. Epigenet. 2018, 10, 162. [Google Scholar] [CrossRef]
- Qin, J.; Wen, B.; Liang, Y.; Yu, W.; Li, H. Histone modifications and their role in colorectal cancer (Review). Pathol. Oncol. Res. 2020, 26, 2023–2033. [Google Scholar] [CrossRef]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Klaus-Jurgen, W.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Quint, K.; Agaimy, A.; Di Fazio, P.; Montalbano, R.; Steindorf, C.; Jung, R.; Hellerbrand, C.; Hartmann, A.; Sitter, H.; Neureiter, D.; et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011, 459, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Stypula-Cyrus, Y.; Damania, D.; Kunte, D.P.; Cruz, M.D.; Subramanian, H.; Roy, H.K.; Backman, V. HDAC up-regulation in early colon field carcinogenesis is involved in cell tumorigenicity through regulation of chromatin structure. PLoS ONE 2013, 8, e64600. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C.; et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol. Cell. Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the crossroads between different types of HDAC inhibitor-mediated cancer cell death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef]
- Bao, L.; Diao, H.; Dong, N.; Su, X.; Wang, B.; Mo, Q.; Yu, H.; Wang, X.; Chen, C. Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biol. Toxicol. 2016, 32, 469–482. [Google Scholar] [CrossRef]
- Ryu, J.K.; Lee, W.J.; Lee, K.H.; Hwang, J.H.; Kim, Y.T.; Yoon, Y.B.; Kim, C.Y. SK-7041, a new histone deacetylase inhibitor, induces G2-M cell cycle arrest and apoptosis in pancreatic cancer cell lines. Cancer Lett. 2006, 237, 143–154. [Google Scholar] [CrossRef]
- Luchenko, V.L.; Litman, T.; Chakraborty, A.R.; Heffner, A.; Devor, C.; Wilkerson, J.; Stein, W.; Robey, R.W.; Bangiolo, L.; Levens, D.; et al. Histone deacetylase inhibitor-mediated cell death is distinct from its global effect on chromatin. Mol. Oncol. 2014, 8, 1379–1392. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- LaBonte, M.J.; Wilson, P.M.; Fazzone, W.; Groshen, S.; Lenz, H.J.; Ladner, R.D. DNA microarray profiling of genes differentially regulated by the histone deacetylase inhibitors vorinostat and LBH589 in colon cancer cell lines. BMC Med. Genom. 2009, 2, 67. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.; Mizzau, M.; Paroni, G.; Maestro, R.; Schneider, C.; Brancolini, C. Role of caspases, Bid, and p53 in the apoptotic response triggered by histone deacetylase inhibitors trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA). J. Biol. Chem. 2003, 278, 12579–12589. [Google Scholar] [CrossRef]
- Lee, H.Y.; Tang, D.W.; Liu, C.Y.; Cho, E.C. A novel HDAC1/2 inhibitor suppresses colorectal cancer through apoptosis induction and cell cycle regulation. Chem. Biol. Interact. 2022, 352, 109778. [Google Scholar] [CrossRef] [PubMed]
- Bär, S.I.; Pradhan, R.; Biersack, B.; Nitzsche, B.; Höpfner, M.; Schobert, R. New chimeric HDAC inhibitors for the treatment of colorectal cancer. Arch. Pharm. 2023, 356, e2200422. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Nepali, K.; Chen, C.H.; Chuang, K.H.; Wu, T.Y.; Lin, T.E.; Hsu, K.C.; Chao, M.W.; Lai, M.J.; Lin, M.H.; et al. Isoindoline scaffold-based dual inhibitors of HDAC6 and HSP90 suppressing the growth of lung cancer in vitro and in vivo. Eur. J. Med. Chem. 2020, 190, 112086. [Google Scholar] [CrossRef]
- Xu, W.; Xu, B.; Yao, Y.; Yu, X.; Shen, J. The novel HDAC inhibitor AR-42-induced anti-colon cancer cell activity is associated with ceramide production. Biochem. Biophys. Res. Commun. 2015, 463, 545–550. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDAC Isoforms (IC50 (nM)) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Class | Class I | Class IIa | Class IIb | ||||||
| HDAC Isoform | HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | HDAC5 | HDAC7 | HDAC9 | HDAC6 |
| MPT0G236 | 14 a | 11.5 a | 70 | 130 a | 19,650 | 15,500 | 12,300 | 12,600 | 15 a |
| SAHA b | 110 a | 120 a | 64.3 b | 306 b | 76,000 b | 27,200 b | 105,000 b | 141,000 b | 110 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, F.-L.; Huang, H.-L.; Lai, M.-J.; Liou, J.-P.; Pan, S.-L.; Yang, C.-R. Anticancer Study of a Novel Pan-HDAC Inhibitor MPT0G236 in Colorectal Cancer Cells. Int. J. Mol. Sci. 2023, 24, 12588. https://doi.org/10.3390/ijms241612588
Tsai F-L, Huang H-L, Lai M-J, Liou J-P, Pan S-L, Yang C-R. Anticancer Study of a Novel Pan-HDAC Inhibitor MPT0G236 in Colorectal Cancer Cells. International Journal of Molecular Sciences. 2023; 24(16):12588. https://doi.org/10.3390/ijms241612588
Chicago/Turabian StyleTsai, Feng-Lung, Han-Li Huang, Mei-Jung Lai, Jing-Ping Liou, Shiow-Lin Pan, and Chia-Ron Yang. 2023. "Anticancer Study of a Novel Pan-HDAC Inhibitor MPT0G236 in Colorectal Cancer Cells" International Journal of Molecular Sciences 24, no. 16: 12588. https://doi.org/10.3390/ijms241612588
APA StyleTsai, F.-L., Huang, H.-L., Lai, M.-J., Liou, J.-P., Pan, S.-L., & Yang, C.-R. (2023). Anticancer Study of a Novel Pan-HDAC Inhibitor MPT0G236 in Colorectal Cancer Cells. International Journal of Molecular Sciences, 24(16), 12588. https://doi.org/10.3390/ijms241612588