
Classification of Promoter Sequences from Human Genome


Abstract
1. Introduction
2. Results
2.1. Human Genome Promoter Sequence Classes Creation
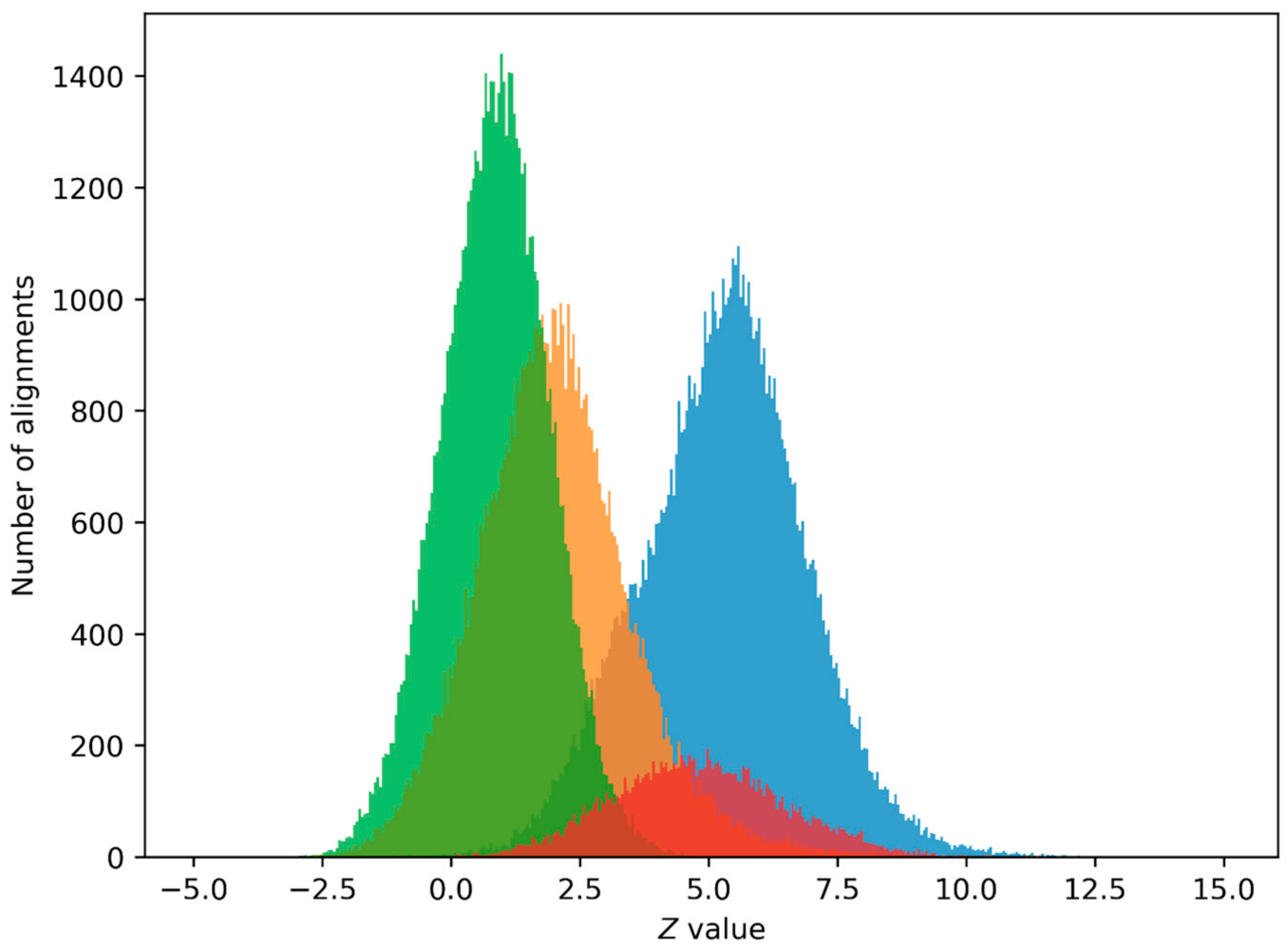
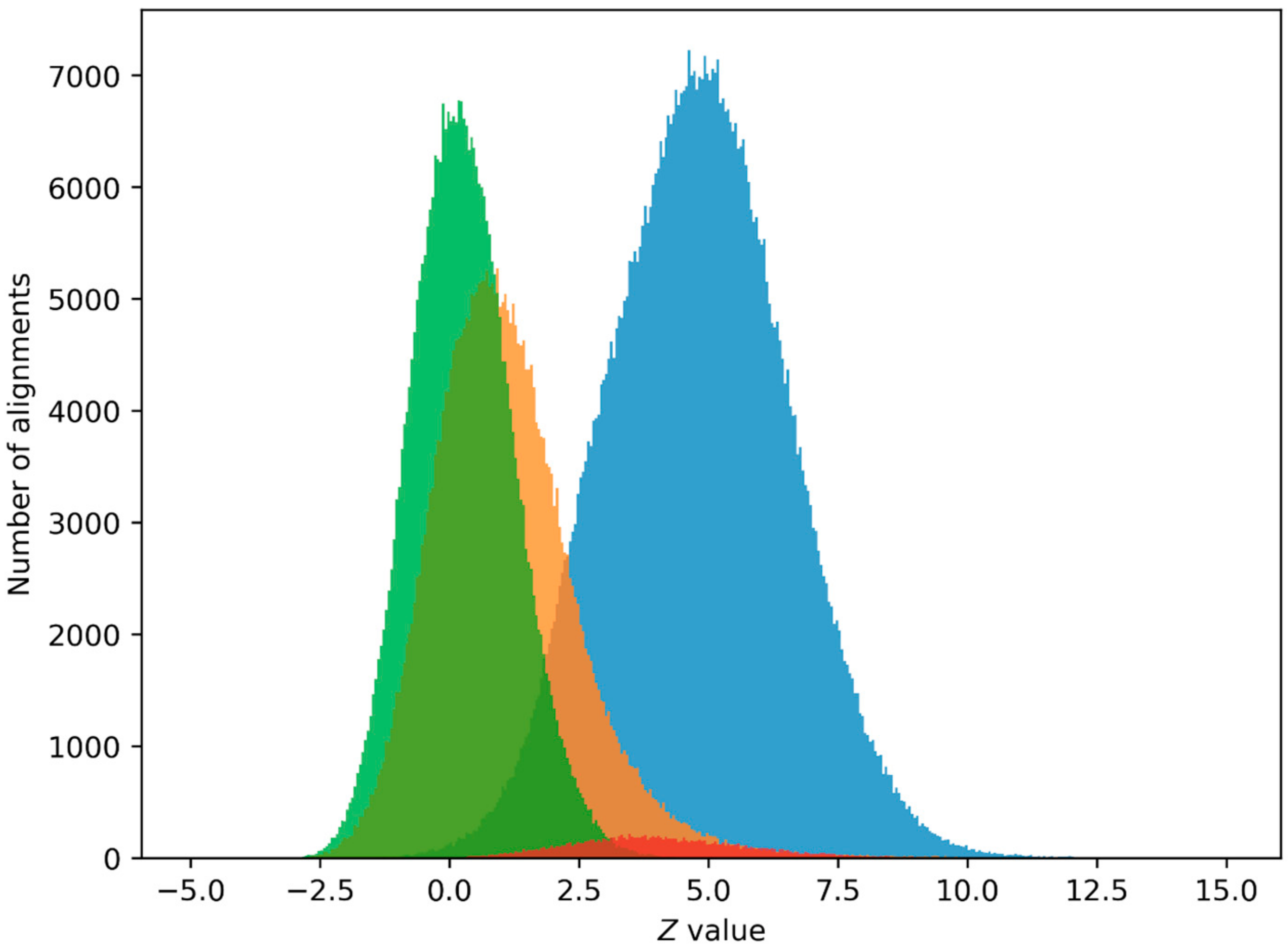
2.2. PPS Search in Human Genome
2.3. PPS Intersection with Annotated Sequences from the Human Genome
2.4. Comparison with Existing Methods
3. Discussion
4. Materials and Methods
4.1. Promoter Sequences
4.2. Classification of Promoter Sequences from the Human Genome
4.3. Alignment of Promoter Sequences with Position Weight Matrix W(i, j)
4.4. Estimation of the Statistical Significance of a Local Alignment
4.5. Potential Promoter Sequence (PPS) Search in Human Genome
4.6. Z0 Value Selection
4.7. Intersection of PPSs with Annotated Sequences from Human Genome
4.8. Comparison with Other Methods for Promoter Prediction
4.9. Computational Resources
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smale, S.T.; Kadonaga, J.T. The RNA Polymerase II Core Promoter. Annu. Rev. Biochem. 2003, 72, 449–479. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Juven-Gershon, T.; Kadonaga, J.T. Regulation of Gene Expression via the Core Promoter and the Basal Transcriptional Machinery. Dev. Biol. 2010, 339, 225–229. [Google Scholar] [CrossRef]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of Applications of High-Throughput Sequencing in Personalized Medicine: Barriers and Facilitators of Future Progress in Research and Clinical Application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, G.K.; Perdikopanis, N.; Hatzigeorgiou, A. Solving the Transcription Start Site Identification Problem with ADAPT-CAGE: A Machine Learning Algorithm for the Analysis of CAGE Data. Sci. Rep. 2020, 10, 877. [Google Scholar] [CrossRef]
- Valen, E.; Pascarella, G.; Chalk, A.; Maeda, N.; Kojima, M.; Kawazu, C.; Murata, M.; Nishiyori, H.; Lazarevic, D.; Motti, D.; et al. Genome-Wide Detection and Analysis of Hippocampus Core Promoters Using DeepCAGE. Genome Res. 2009, 19, 255. [Google Scholar] [CrossRef]
- Cassiano, M.H.A.; Silva-Rocha, R. Benchmarking Bacterial Promoter Prediction Tools: Potentialities and Limitations. mSystems 2020, 5, e00439-20. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Bhandary, P.; Woodhouse, M.; Sen, T.Z.; Wise, R.P.; Andorf, C.M. FINDER: An Automated Software Package to Annotate Eukaryotic Genes from RNA-Seq Data and Associated Protein Sequences. BMC Bioinform. 2021, 22, 205. [Google Scholar] [CrossRef]
- Martin, R.G.; Gillette, W.K.; Rosner, J.L. Promoter Discrimination by the Related Transcriptional Activators MarA and SoxS: Differential Regulation by Differential Binding. Mol. Microbiol. 2000, 35, 623–634. [Google Scholar] [CrossRef]
- Shir-Shapira, H.; Sloutskin, A.; Adato, O.; Ovadia-Shochat, A.; Ideses, D.; Zehavi, Y.; Kassavetis, G.; Kadonaga, J.T.; Unger, R.; Juven-Gershon, T. Identification of Evolutionarily Conserved Downstream Core Promoter Elements Required for the Transcriptional Regulation of Fushi Tarazu Target Genes. PLoS ONE 2019, 14, e0215695. [Google Scholar] [CrossRef]
- Oubounyt, M.; Louadi, Z.; Tayara, H.; To Chong, K. Deepromoter: Robust Promoter Predictor Using Deep Learning. Front. Genet. 2019, 10, 286. [Google Scholar] [CrossRef]
- Périer, R.C.; Junier, T.; Bucher, P. The Eukaryotic Promoter Database EPD. Nucleic Acids Res. 1998, 26, 353–357. [Google Scholar] [CrossRef]
- Dreos, R.R.; Ambrosini, G.; Groux, R.; Cavin Périer, R.; Bucher, P.; Perier, R.C.; Bucher, P. The Eukaryotic Promoter Database in Its 30th Year: Focus on Non-Vertebrate Organisms. Nucleic Acids Res. 2017, 45, D51–D55. [Google Scholar] [CrossRef]
- Datta, S.; Mukhopadhyay, S. A Composite Method Based on Formal Grammar and DNA Structural Features in Detecting Human Polymerase II Promoter Region. PLoS ONE 2013, 8, e54843. [Google Scholar] [CrossRef][Green Version]
- Amin, R.; Rahman, C.R.; Ahmed, S.; Sifat, M.H.R.; Liton, M.N.K.; Rahman, M.M.; Khan, M.Z.H.; Shatabda, S.; Ahmed, S. IPromoter-BnCNN: A Novel Branched CNN-Based Predictor for Identifying and Classifying Sigma Promoters. Bioinformatics 2020, 36, 4869–4875. [Google Scholar] [CrossRef] [PubMed]
- Shujaat, M.; Wahab, A.; Tayara, H.; Chong, K.T. PcPromoter-CNN: A CNN-Based Prediction and Classification of Promoters. Genes 2020, 11, 1529. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.V.; Shahmuradov, I.A.; Salamov, A.A. Identification of Promoter Regions and Regulatory Sites. Methods Mol. Biol. 2010, 674, 57–83. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A.; Pietersma, H.; Cordes, M.; Kuipers, O.P.; Kok, J. PePPER: A Webserver for Prediction of Prokaryote Promoter Elements and Regulons. BMC Genom. 2012, 13, 299. [Google Scholar] [CrossRef]
- Di Salvo, M.; Pinatel, E.; Talà, A.; Fondi, M.; Peano, C.; Alifano, P. G4PromFinder: An Algorithm for Predicting Transcription Promoters in GC-Rich Bacterial Genomes Based on AT-Rich Elements and G-Quadruplex Motifs. BMC Bioinform. 2018, 19, 36. [Google Scholar] [CrossRef]
- Umarov, R.; Kuwahara, H.; Li, Y.; Gao, X.; Solovyev, V.; Hancock, J. Promoter Analysis and Prediction in the Human Genome Using Sequence-Based Deep Learning Models. Bioinformatics 2019, 35, 2730–2737. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, X.; Li, Y.; Wu, M.; Zhao, Y. Image-Based Promoter Prediction: A Promoter Prediction Method Based on Evolutionarily Generated Patterns. Sci. Rep. 2018, 8, 17695. [Google Scholar] [CrossRef]
- De Medeiros Oliveira, M.; Bonadio, I.; Lie De Melo, A.; Mendes Souza, G.; Durham, A.M. TSSFinder-Fast and Accurate Ab Initio Prediction of the Core Promoter in Eukaryotic Genomes. Brief. Bioinform. 2021, 22, bbab198. [Google Scholar] [CrossRef] [PubMed]
- Bondar, E.I.; Troukhan, M.E.; Krutovsky, K.V.; Tatarinova, T.V. Genome-Wide Prediction of Transcription Start Sites in Conifers. Int. J. Mol. Sci. 2022, 23, 1735. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, E.V.; Suvorova, Y.M.; Kostenko, D.O.; Korotkova, M.A. Multiple Alignment of Promoter Sequences from the Arabidopsis thalianal. Genome. Genes 2021, 12, 135. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2014, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Frith, M.C. Adding Unaligned Sequences into an Existing Alignment Using MAFFT and LAST. Bioinformatics 2012, 28, 3144–3146. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kostenko, D.O.; Korotkov, E.V. Application of the MAHDS Method for Multiple Alignment of Highly Diverged Amino Acid Sequences. Int. J. Mol. Sci. 2022, 23, 3764. [Google Scholar] [CrossRef]
- Korotkov, E.V.; Suvorova, Y.M.; Nezhdanova, A.V.; Gaidukova, S.E.; Yakovleva, I.V.; Kamionskaya, A.M.; Korotkova, M.A. Mathematical Algorithm for Identification of Eukaryotic Promoter Sequences. Symmetry 2021, 13, 917. [Google Scholar] [CrossRef]
- Frenkel, F.E.; Korotkov, E.V. Using Triplet Periodicity of Nucleotide Sequences for Finding Potential Reading Frame Shifts in Genes. DNA Res. 2009, 16, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Eukaryotic Promoter Database. Available online: https://epd.expasy.org/epd/ (accessed on 1 September 2021).
- Ensembl Genome Browser. Available online: http://ftp.ensembl.org/pub/release-103/fasta/homo_sapiens/dna/ (accessed on 3 March 2021).
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- The Dfam Community Resource of Transposable Element Families, Sequence Models, and Genome Annotations. Available online: https://www.dfam.org/releases/Dfam_3.3/annotations/ (accessed on 21 April 2021).
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam Community Resource of Transposable Element Families, Sequence Models, and Genome Annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- A Reference Data Set for Human and Mouse Transcription Start Sites. Available online: http://reftss.clst.riken.jp/datafiles/current/human/ (accessed on 24 May 2022).
- Abugessaisa, I.; Noguchi, S.; Hasegawa, A.; Kondo, A.; Kawaji, H.; Carninci, P.; Kasukawa, T. RefTSS: A Reference Data Set for Human and Mouse Transcription Start Sites. J. Mol. Biol. 2019, 431, 2407–2422. [Google Scholar] [CrossRef]
- Koenigsberger, C.; Chicca, J.J., 2nd; Amoureux, M.C.; Edelman, G.M.; Jones, F.S. Differential Regulation by Multiple Promoters of the Gene Encoding the Neuron-Restrictive Silencer Factor. Proc. Natl. Acad. Sci. USA 2000, 97, 2291–2296. [Google Scholar] [CrossRef]
- Vanderperre, B.; Lucier, J.-F.; Bissonnette, C.; Motard, J.; Tremblay, G.; Vanderperre, S.; Wisztorski, M.; Salzet, M.; Boisvert, F.-M.; Roucou, X. Direct Detection of Alternative Open Reading Frames Translation Products in Human Significantly Expands the Proteome. PLoS ONE 2013, 8, e70698. [Google Scholar] [CrossRef]
- Deininger, P. Alu Elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG Islands and the Regulation of Transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Polak, P.; Domany, E. Alu Elements Contain Many Binding Sites for Transcription Factors and May Play a Role in Regulation of Developmental Processes. BMC Genom. 2006, 7, 133. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Häsler, J.; Strub, K. Alu Elements as Regulators of Gene Expression. Nucleic Acids Res. 2006, 34, 5491–5497. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Macfarlan, T.S.; Lorincz, M.C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 2016, 62, 766–776. [Google Scholar] [CrossRef]
- Soloviev, V.; Salamov, A. The Gene-Finder Computer Tools for Analysis of Human and Model Organisms Genome Sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997, 5, 294–302. [Google Scholar]
- Solovyev, V.V.; Shahmuradov, I.A. PromH: Promoters Identification Using Orthologous Genomic Sequences. Nucleic Acids Res. 2003, 31, 3540–3545. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; MG, R.; Reese, M.G. Application of a Time-Delay Neural Network to Promoter Annotation in the Drosophila Melanogaster Genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Umarov, R.K.; Solovyev, V.V. Recognition of Prokaryotic and Eukaryotic Promoters Using Convolutional Deep Learning Neural Networks. PLoS ONE 2017, 12, e0171410. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA Genes Are Transcribed by RNA Polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of Post-Transcriptional Regulation by MicroRNAs: Are the Answers in Sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, V.; Korotkov, A.; Korotkov, E. Search of Latent Periodicity in Amino Acid Sequences by Means of Genetic Algorithm and Dynamic Programming. Stat. Appl. Genet. Mol. Biol. 2016, 15, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1998; ISBN 0521629713. [Google Scholar]



{kind=link}
{kind=link}
| Class | N | R |
|---|---|---|
| 1 | 11,780 | 1.077 |
| 2 | 3139 | 1.063 |
| 3 | 1624 | 0.748 |
| 4 | 767 | 0.648 |
| 5 | 464 | 0.447 |
| 6 | 161 | 0.267 |
| Chr.№ | Number of PPSs | Inverted Chromosome | Random | |||
|---|---|---|---|---|---|---|
| + | − | + | − | + | − | |
| 1 | 133,073 | 133,437 | 3374 | 3252 | 8 | 8 |
| 2 | 126,853 | 128,025 | 3113 | 3122 | 8 | 9 |
| 3 | 94,729 | 94,889 | 2218 | 2182 | 3 | 7 |
| 4 | 81,690 | 82,344 | 2125 | 2049 | 4 | 6 |
| 5 | 84,473 | 84,996 | 1886 | 1814 | 5 | 7 |
| 6 | 84,193 | 84,668 | 2308 | 2310 | 6 | 2 |
| 7 | 82,033 | 82,165 | 2242 | 2152 | 3 | 4 |
| 8 | 72,840 | 72,414 | 1763 | 1794 | 7 | 5 |
| 9 | 63,751 | 63,647 | 1511 | 1573 | 2 | 2 |
| 10 | 73,439 | 73,275 | 1739 | 1724 | 2 | 4 |
| 11 | 72,425 | 72,705 | 1623 | 1567 | 2 | 4 |
| 12 | 72,578 | 72,840 | 1952 | 2008 | 9 | 9 |
| 13 | 45,296 | 45,786 | 1133 | 1131 | 7 | 3 |
| 14 | 47,289 | 47,762 | 1099 | 1126 | 2 | 3 |
| 15 | 47,703 | 47,644 | 1024 | 987 | 0 | 3 |
| 16 | 47,886 | 47,962 | 1133 | 1177 | 4 | 1 |
| 17 | 52,660 | 54,031 | 1194 | 1289 | 3 | 1 |
| 18 | 39,048 | 39,345 | 1031 | 986 | 3 | 5 |
| 19 | 40,174 | 40,246 | 1603 | 1592 | 1 | 2 |
| 20 | 39,638 | 39,855 | 930 | 939 | 2 | 2 |
| 21 | 20,795 | 20,958 | 724 | 711 | 0 | 1 |
| 22 | 25,307 | 25,335 | 515 | 507 | 0 | 3 |
| X | 69,891 | 71,404 | 1987 | 1837 | 3 | 8 |
| Y | 10,767 | 10,767 | 295 | 305 | 2 | 1 |
| Total | 3,065,317 | 76,656 | 186 | |||
| j | Annotated Sequences | Zr(j) | Number of PPSs |
|---|---|---|---|
| 1 | Promoters ++/−− | 57 | 10,755 |
| 2 | Promoters +−/−+ | −12 | 4458 |
| 3 | NC promoters | 8 | 673 |
| 4 | TSS | 58 | 50,895 |
| 5 | Exons | 39 | 120,285 |
| 6 | Introns | 60 | 856,456 |
| 7 | MIR | −93 | 66,542 |
| 8 | AluS | 286 | 196,167 |
| 9 | AluY | 135 | 38,734 |
| 10 | AluJ | 151 | 66,919 |
| 11 | MER | −31 | 53,258 |
| 12 | LTR | 58 | 41,789 |
| 13 | MLT | 3 | 42,846 |
| 14 | SVA | 94 | 31,567 |
| 15 | Charlie | −34 | 6403 |
| 16 | Tigger | −61 | 6047 |
| 17 | MamRep | −16 | 2885 |
| 18 | HERV | 43 | 10,963 |
| 19 | THE | 20 | 12,186 |
| 20 | L1 | −176 | 111,531 |
| 21 | L2 | −36 | 70,368 |
| 22 | L3 | −13 | 4313 |
| 23 | L4 | −22 | 1313 |
| 24 | FLAM | 21 | 5604 |
| 25 | FRAM | 12 | 2526 |
| 26 | UCON | −12 | 1502 |
| 27 | MST | 9 | 7126 |
| Method | Ns | Set1 | Set2 | Set3 |
|---|---|---|---|---|
| FPROM | 1000 | 327 | 11 | 24 |
| TSSW | 400 | 219 | 45 | 75 |
| NNPP | 100 | 47 | 42 | 31 |
| Our method | 1000 | 584 | 0 | 1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaytsev, K.; Fedorov, A.; Korotkov, E. Classification of Promoter Sequences from Human Genome. Int. J. Mol. Sci. 2023, 24, 12561. https://doi.org/10.3390/ijms241612561
Zaytsev K, Fedorov A, Korotkov E. Classification of Promoter Sequences from Human Genome. International Journal of Molecular Sciences. 2023; 24(16):12561. https://doi.org/10.3390/ijms241612561
Chicago/Turabian StyleZaytsev, Konstantin, Alexey Fedorov, and Eugene Korotkov. 2023. "Classification of Promoter Sequences from Human Genome" International Journal of Molecular Sciences 24, no. 16: 12561. https://doi.org/10.3390/ijms241612561
APA StyleZaytsev, K., Fedorov, A., & Korotkov, E. (2023). Classification of Promoter Sequences from Human Genome. International Journal of Molecular Sciences, 24(16), 12561. https://doi.org/10.3390/ijms241612561