
Six Functions of Respiration: Isn’t It Time to Take Control over ROS Production in Mitochondria, and Aging Along with It?

 ,
,  ,
,  ,
,  ,
,  , and
, and 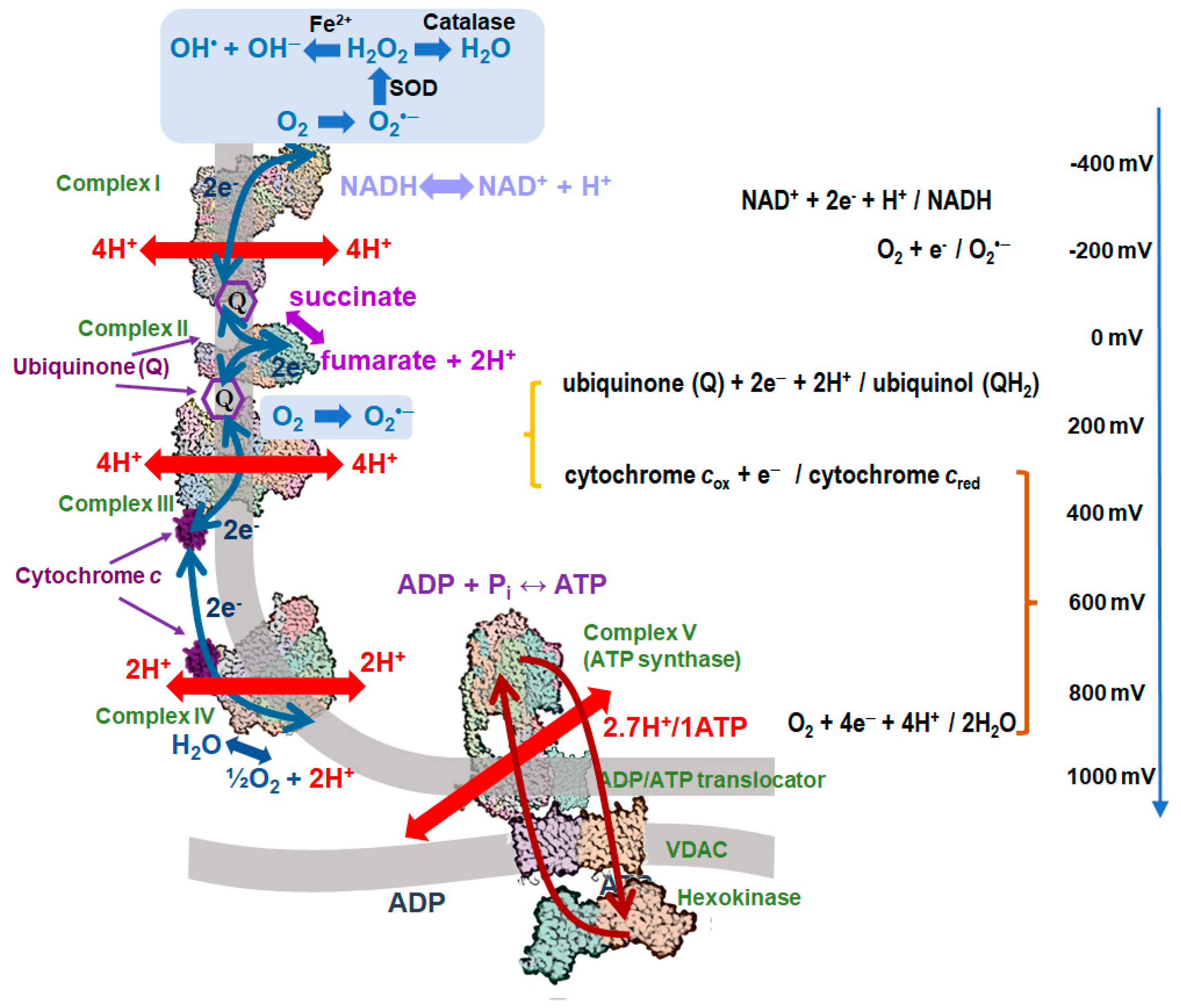{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Generation of mtROS in the Respiratory Chain
2.1. Mechanisms of Protection against the mtROS Formation in Mitochondria
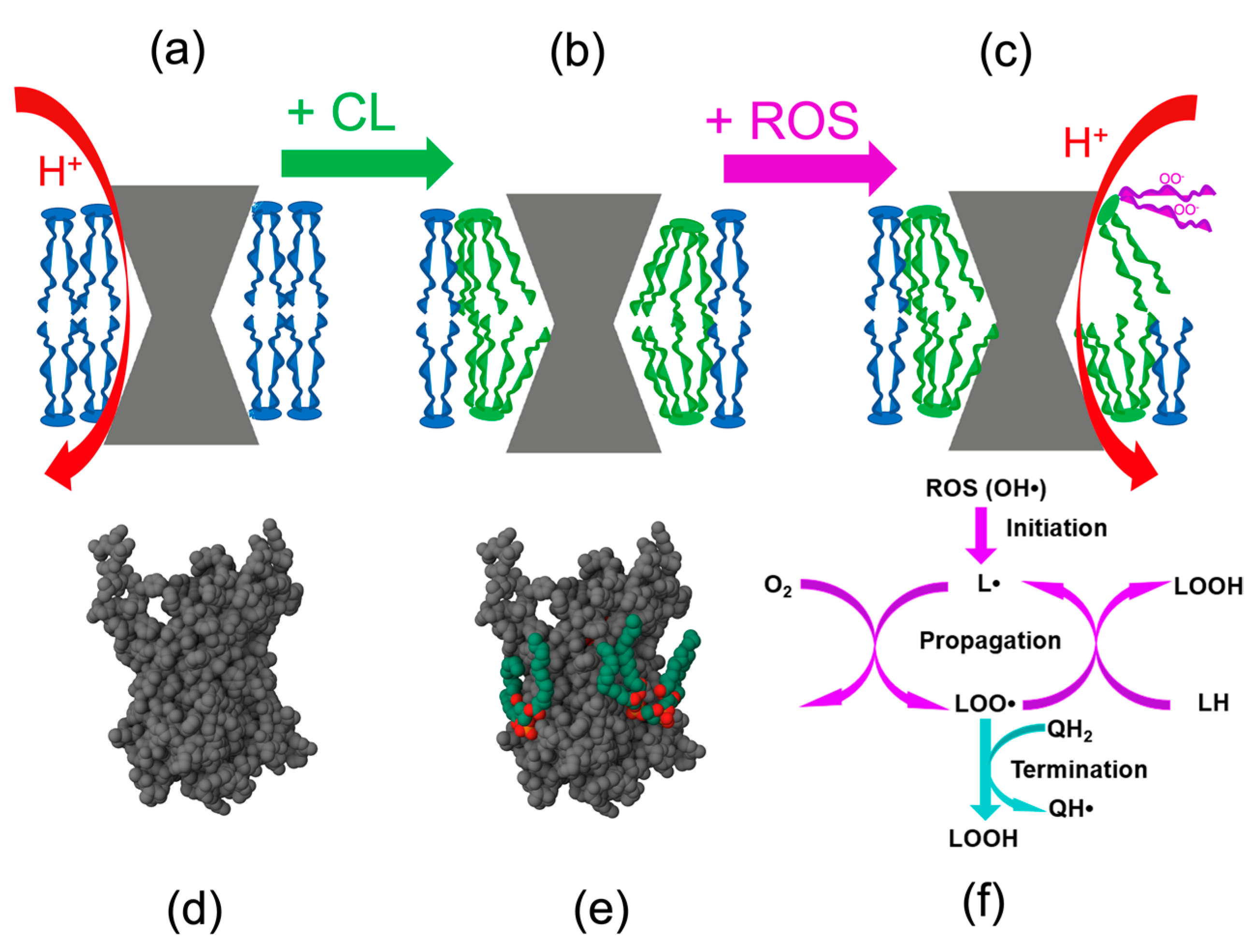
2.2. Cardiolipin and the mtROS-Signaling
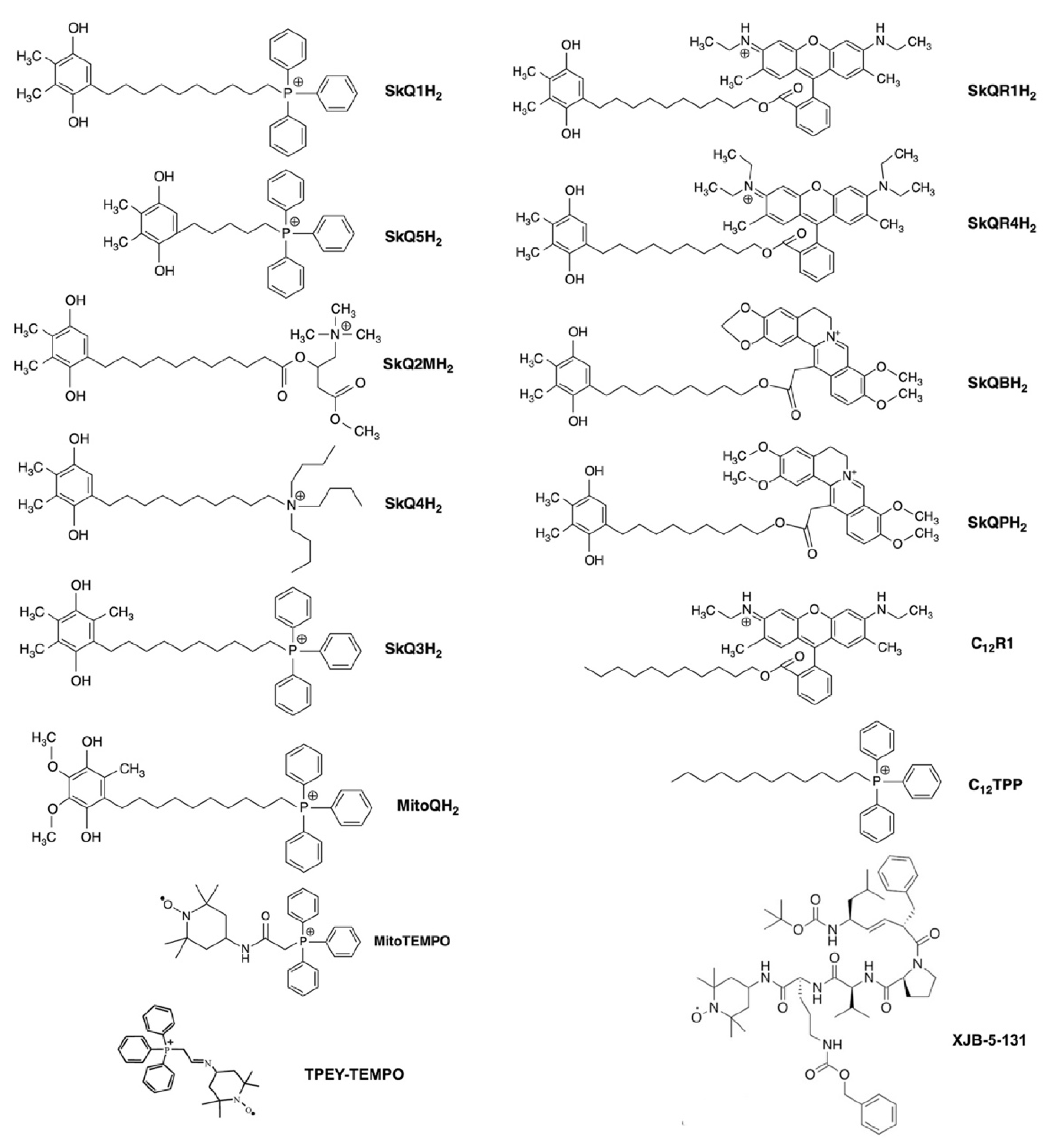
3. Modulation of mtROS Signaling by Mitochondria-Targeted Antioxidants
3.1. Prevention of Cardiolipin Oxidation
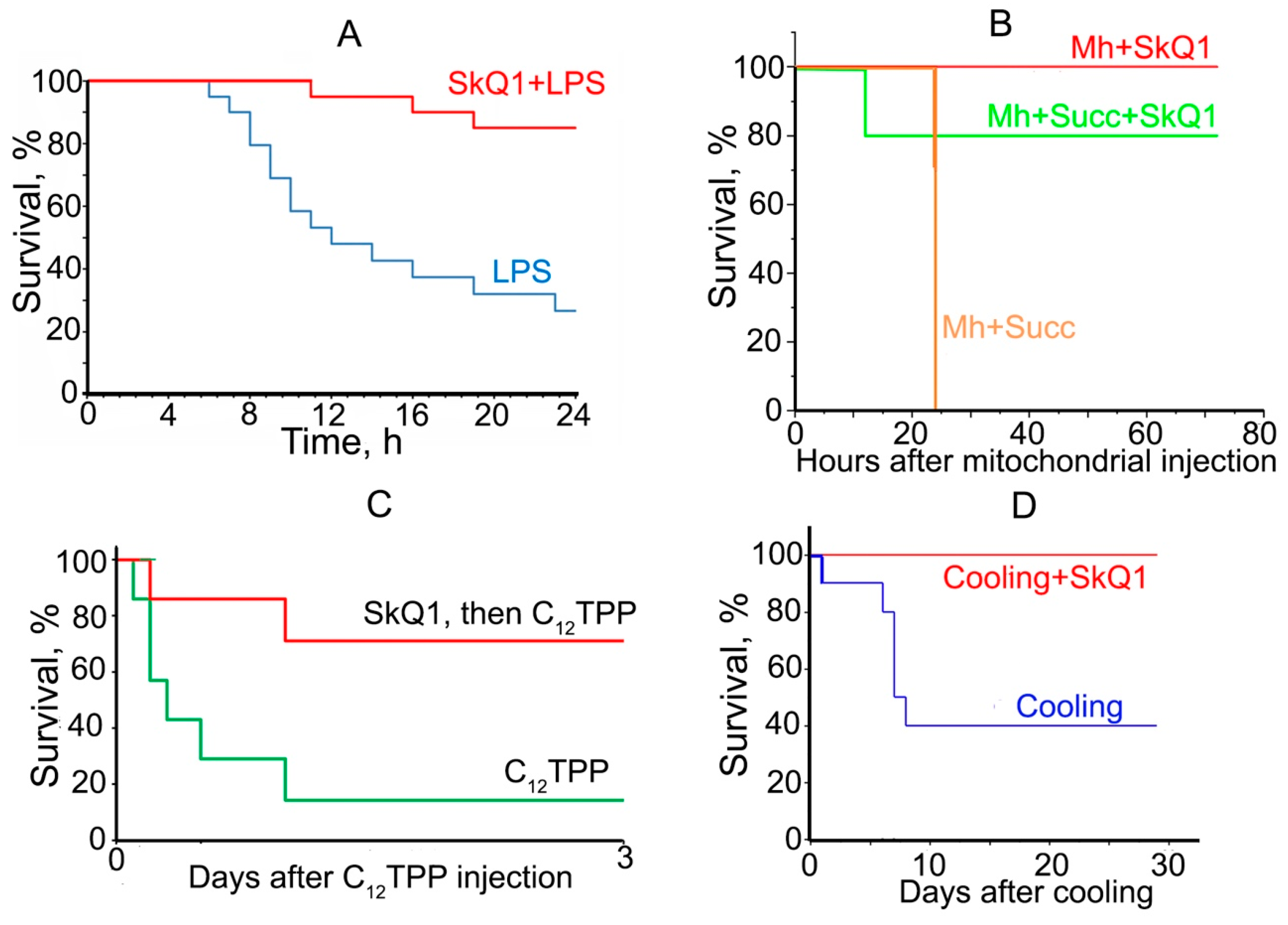
3.2. Prevention of Acute Organismic Damage
3.3. Aging (Chronic Phenoptosis) and Production of mtROS in Complex I
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skulachev, V.P. Membrane Bioenergetics; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Glass, J.B.; Elbon, C.E.; Williams, L.D. Something old, something new, something borrowed, something blue: The anaerobic microbial ancestry of aerobic respiration. Trends Microbiol. 2023, 31, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Bridges, H.R.; Blaza, J.N.; Yin, Z.; Chung, I.; Pollak, M.N.; Hirst, J. Structural basis of mammalian respiratory complex I inhibition by medicinal biguanides. Science 2023, 379, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Lai, Y.; Xu, J.; Zhang, Y.; Zhou, S.; Feng, Z.; Yu, L.; Tang, Y.; Wang, W.; et al. Structure of the human respiratory complex II. Proc. Natl. Acad. Sci. USA 2023, 120, e2216713120. [Google Scholar] [CrossRef]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef]
- Ueno, G.; Shimada, A.; Yamashita, E.; Hasegawa, K.; Kumasaka, T.; Shinzawa-Itoh, K.; Yoshikawa, S.; Tsukihara, T.; Yamamoto, M. Low-dose X-ray structure analysis of cytochrome c oxidase utilizing high-energy X-rays. J. Synchrotron Radiat. 2019, 26, 912–921. [Google Scholar] [CrossRef]
- Mirkin, N.; Jaconcic, J.; Stojanoff, V.; Moreno, A. High resolution X-ray crystallographic structure of bovine heart cytochrome c and its application to the design of an electron transfer biosensor. Proteins 2008, 70, 83–92. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trezeguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Leung, M.R.; Zenezini Chiozzi, R.; Roelofs, M.C.; Hevler, J.F.; Ravi, R.T.; Maitan, P.; Zhang, M.; Henning, H.; Bromfield, E.G.; Howes, S.C.; et al. In-cell structures of conserved supramolecular protein arrays at the mitochondria-cytoskeleton interface in mammalian sperm. Proc. Natl. Acad. Sci. USA 2021, 118, e2110996118. [Google Scholar] [CrossRef]
- Lai, Y.; Zhang, Y.; Zhou, S.; Xu, J.; Du, Z.; Feng, Z.; Yu, L.; Zhao, Z.; Wang, W.; Tang, Y.; et al. Structure of the human ATP synthase. Mol. Cell 2023, 83, 2137–2147.e2134. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.D. The ATP synthase—A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef]
- Wikstrom, M.; Krab, K.; Saraste, M. Proton-translocating cytochrome complexes. Annu. Rev. Biochem. 1981, 50, 623–655. [Google Scholar] [CrossRef] [PubMed]
- Beliakova, T.N.; Glagolev, A.N.; Skulachev, V.P. [Electrochemical gradient of H+ ions as an immediate source of energy during bacteria movement]. Biokhimiia 1976, 41, 1478–1483. [Google Scholar] [PubMed]
- Glagolev, A.N.; Skulachev, V.P. The proton pump is a molecular engine of motile bacteria. Nature 1978, 272, 280–282. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Dibrov, P.A.; Glagolev, A.N.; Sherman, M.Y.; Skulachev, V.P. A novel type of energetics in a marine alkali-tolerant bacterium. FEBS Lett. 1983, 164, 38–42. [Google Scholar] [CrossRef]
- Hoch, F.L.; Lipmann, F. The Uncoupling of Respiration and Phosphorylation by Thyroid Hormones. Proc. Natl. Acad. Sci. USA 1954, 40, 909–921. [Google Scholar] [CrossRef]
- Klingenberg, M. UCP1—A sophisticated energy valve. Biochimie 2017, 134, 19–27. [Google Scholar] [CrossRef]
- Andreyev, A.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Volkov, N.I. Carboxyatractylate inhibits the uncoupling effect of free fatty acids. FEBS Lett. 1988, 226, 265–269. [Google Scholar] [CrossRef]
- Samartsev, V.N.; Smirnov, A.V.; Zeldi, I.P.; Markova, O.V.; Mokhova, E.N.; Skulachev, V.P. Involvement of aspartate/glutamate antiporter in fatty acid-induced uncoupling of liver mitochondria. Biochim. Biophys. Acta 1997, 1319, 251–257. [Google Scholar] [CrossRef]
- Skulachev, V.P. Phenoptosis: Programmed death of an organism. Biochemistry 1999, 64, 1418–1426. [Google Scholar] [PubMed]
- Dalton, H.; Postgate, J.R. Effect of oxygen on growth of Azotobacter chroococcum in batch and continuous cultures. J. Gen. Microbiol. 1968, 54, 463–473. [Google Scholar] [CrossRef]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef]
- Kaila, V.R.; Verkhovsky, M.; Hummer, G.; Wikstrom, M. Prevention of leak in the proton pump of cytochrome c oxidase. Biochim. Biophys. Acta 2008, 1777, 890–892. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kolbe, F.; Safarian, S.; Piorek, Z.; Welsch, S.; Muller, H.; Michel, H. Cryo-EM structures of intermediates suggest an alternative catalytic reaction cycle for cytochrome c oxidase. Nat. Commun. 2021, 12, 6903. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef]
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [CrossRef]
- Cino, M.; Del Maestro, R.F. Generation of hydrogen peroxide by brain mitochondria: The effect of reoxygenation following postdecapitative ischemia. Arch. Biochem. Biophys. 1989, 269, 623–638. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial Complex I. Biochim. Biophys. Acta 2006, 1757, 553–561. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Hancock, J.T.; Desikan, R.; Neill, S.J. Role of reactive oxygen species in cell signalling pathways. Biochem. Soc. Trans. 2001, 29, 345–350. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Bogachev, A.V.; Kasparinsky, F.O. Principles of Bioenergetics; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V.; Lyamzaev, K.G.; Mulkidjanian, A.Y. Innate Immunity as an Executor of the Programmed Death of Individual Organisms for the Benefit of the Entire Population. Int. J. Mol. Sci. 2021, 22, 13480. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L. Origin of Eukaryotic Cells: Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth; Yale University Press: London, UK, 1970. [Google Scholar]
- Andersson, S.G.; Zomorodipour, A.; Andersson, J.O.; Sicheritz-Ponten, T.; Alsmark, U.C.; Podowski, R.M.; Naslund, A.K.; Eriksson, A.S.; Winkler, H.H.; Kurland, C.G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998, 396, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Moncadas, L.S.; Shabarova, T.; Kavagutti, V.S.; Bulzu, P.-A.; Chiriac, M.-C.; Park, S.-J.; Mukherjee, I.; Ghai, R.; Andrei, A.-S. Rickettsiales’ deep evolutionary history sheds light on the emergence of intracellular lifestyles. bioRxiv 2023. [Google Scholar] [CrossRef]
- Salje, J. Cells within cells: Rickettsiales and the obligate intracellular bacterial lifestyle. Nat. Rev. Microbiol. 2021, 19, 375–390. [Google Scholar] [CrossRef]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Morales-Rios, E.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 A resolution. Proc. Natl. Acad. Sci. USA 2015, 112, 13231–13236. [Google Scholar] [CrossRef]
- Srivastava, A.P.; Luo, M.; Zhou, W.; Symersky, J.; Bai, D.; Chambers, M.G.; Faraldo-Gomez, J.D.; Liao, M.; Mueller, D.M. High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science 2018, 360, eaas9699. [Google Scholar] [CrossRef]
- Heldt, H.W.; Klingenberg, M.; Milovancev, M. Differences between the ATP-ADP ratios in the mitochondrial matrix and in the extramitochondrial space. Eur. J. Biochem. 1972, 30, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y. Proton translocation by the cytochrome bc1 complexes of phototrophic bacteria: Introducing the activated Q-cycle. Photochem. Photobiol. Sci. 2007, 6, 19–34. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Khuchua, Z.A.; Vassil’eva, E.V.; Medved’eva, N.V.; Saks, V.A. Heart mitochondrial creatine kinase revisited: The outer mitochondrial membrane is not important for coupling of phosphocreatine production to oxidative phosphorylation. Arch. Biochem. Biophys. 1989, 268, 176–190. [Google Scholar] [CrossRef] [PubMed]
- da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef]
- Meyer, L.E.; Machado, L.B.; Santiago, A.P.; da-Silva, W.S.; De Felice, F.G.; Holub, O.; Oliveira, M.F.; Galina, A. Mitochondrial creatine kinase activity prevents reactive oxygen species generation: Antioxidant role of mitochondrial kinase-dependent ADP re-cycling activity. J. Biol. Chem. 2006, 281, 37361–37371. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.P.; Chaves, E.A.; Oliveira, M.F.; Galina, A. Reactive oxygen species generation is modulated by mitochondrial kinases: Correlation with mitochondrial antioxidant peroxidases in rat tissues. Biochimie 2008, 90, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef]
- Monne, M.; Palmieri, F. Antiporters of the mitochondrial carrier family. Curr. Top. Membr. 2014, 73, 289–320. [Google Scholar] [CrossRef]
- Skulachev, V.P. Anion carriers in fatty acid-mediated physiological uncoupling. J. Bioenerg. Biomembr. 1999, 31, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Korkina, O.V.; Ruuge, E.K.; Skulachev, V.P.; Starkov, A.A. Fatty acids as natural uncouplers preventing generation of O2.- and H2O2 by mitochondria in the resting state. FEBS Lett. 1998, 435, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; King, M.S.; Zogg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e415. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Nuevo, A.; Torres-Sanchez, A.; Duran, J.M.; De Guirior, C.; Martinez-Zamora, M.A.; Boke, E. Oocytes maintain ROS-free mitochondrial metabolism by suppressing complex I. Nature 2022, 607, 756–761. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Voigt, A.L.; Kondro, D.A.; Powell, D.; Valli-Pulaski, H.; Ungrin, M.; Stukenborg, J.B.; Klein, C.; Lewis, I.A.; Orwig, K.E.; Dobrinski, I. Unique metabolic phenotype and its transition during maturation of juvenile male germ cells. FASEB J. 2021, 35, e21513. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef]
- Lokhmatikov, A.V.; Voskoboynikova, N.; Cherepanov, D.A.; Skulachev, M.V.; Steinhoff, H.J.; Skulachev, V.P.; Mulkidjanian, A.Y. Impact of Antioxidants on Cardiolipin Oxidation in Liposomes: Why Mitochondrial Cardiolipin Serves as an Apoptotic Signal? Oxid. Med. Cell Longev. 2016, 2016, 8679469. [Google Scholar] [CrossRef]
- Kagan, V.E.; Chu, C.T.; Tyurina, Y.Y.; Cheikhi, A.; Bayir, H. Cardiolipin asymmetry, oxidation and signaling. Chem. Phys. Lipids 2014, 179, 64–69. [Google Scholar] [CrossRef]
- Anzmann, A.F.; Claypool, S.; Vernon, H. Mitochondrial Dysfunction and Barth Syndrome. In Handbook of Mitochondrial Dysfunction; Ahman, S.I., Ed.; CRS Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Mikel’saar, K.; Severina, I.I.; Skulachev, V.P. [Phospholipids and oxidative phosphorylation]. Usp. Sovrem. Biol. 1974, 78, 348–370. [Google Scholar] [PubMed]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 2002, 277, 43553–43556. [Google Scholar] [CrossRef] [PubMed]
- Althoff, T.; Mills, D.J.; Popot, J.L.; Kuhlbrandt, W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef]
- Muhleip, A.; Flygaard, R.K.; Baradaran, R.; Haapanen, O.; Gruhl, T.; Tobiasson, V.; Marechal, A.; Sharma, V.; Amunts, A. Structural basis of mitochondrial membrane bending by the I-II-III(2)-IV(2) supercomplex. Nature 2023, 615, 934–938. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Shalaeva, D.N.; Lyamzaev, K.G.; Chernyak, B.V. Does oxidation of mitochondrial cardiolipin trigger a chain of antiapoptotic reactions? Biochemistry 2018, 83, 1263–1278. [Google Scholar] [CrossRef]
- Koshkin, V.; Greenberg, M.L. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 2002, 364, 317–322. [Google Scholar] [CrossRef]
- Prola, A.; Blondelle, J.; Vandestienne, A.; Piquereau, J.; Denis, R.G.P.; Guyot, S.; Chauvin, H.; Mourier, A.; Maurer, M.; Henry, C.; et al. Cardiolipin content controls mitochondrial coupling and energetic efficiency in muscle. Sci. Adv. 2021, 7, eabd6322. [Google Scholar] [CrossRef]
- Nury, H.; Dahout-Gonzalez, C.; Trezeguet, V.; Lauquin, G.; Brandolin, G.; Pebay-Peyroula, E. Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 2005, 579, 6031–6036. [Google Scholar] [CrossRef]
- Tyurin, V.A.; Tyurina, Y.Y.; Kochanek, P.M.; Hamilton, R.; DeKosky, S.T.; Greenberger, J.S.; Bayir, H.; Kagan, V.E. Oxidative lipidomics of programmed cell death. Methods Enzymol. 2008, 442, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Anisimov, V.N.; Antonenko, Y.N.; Bakeeva, L.E.; Chernyak, B.V.; Erichev, V.P.; Filenko, O.F.; Kalinina, N.I.; Kapelko, V.I.; Kolosova, N.G.; et al. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Kline, A.E.; Amoscato, A.; Samhan-Arias, A.K.; Sparvero, L.J.; Tyurin, V.A.; Tyurina, Y.Y.; Fink, B.; Manole, M.D.; Puccio, A.M.; et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci. 2012, 15, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Niki, E.; Yoshida, Y.; Saito, Y.; Noguchi, N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochem. Biophys. Res. Commun. 2005, 338, 668–676. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Panteleeva, A.A.; Karpukhina, A.A.; Galkin, I.I.; Popova, E.N.; Pletjushkina, O.Y.; Rieger, B.; Busch, K.B.; Mulkidjanian, A.Y.; Chernyak, B.V. Novel Fluorescent Mitochondria-Targeted Probe MitoCLox Reports Lipid Peroxidation in Response to Oxidative Stress In Vivo. Oxid. Med. Cell Longev. 2020, 2020, 3631272. [Google Scholar] [CrossRef]
- Jain, A.; Holthuis, J.C.M. Membrane contact sites, ancient and central hubs of cellular lipid logistics. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1450–1458. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Abdrakhmanov, A.; Gogvadze, V.; Zhivotovsky, B. To Eat or to Die: Deciphering Selective Forms of Autophagy. Trends Biochem. Sci. 2020, 45, 347–364. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996, 397, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- Ten, V.S.; Starkov, A. Hypoxic-ischemic injury in the developing brain: The role of reactive oxygen species originating in mitochondria. Neurol. Res. Int. 2012, 2012, 542976. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Ryan, D.G.; O’Neill, L.A.J. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Romaschenko, V.P.; Galkin, I.I.; Zakharova, V.V.; Pletjushkina, O.Y.; Chernyak, B.V.; Popova, E.N. Role of mitochondrial reactive oxygen species in age-related inflammatory activation of endothelium. Aging 2014, 6, 661–674. [Google Scholar] [CrossRef]
- Galkin, I.I.; Pletjushkina, O.Y.; Zinovkin, R.A.; Zakharova, V.V.; Chernyak, B.V.; Popova, E.N. Mitochondria-Targeted Antioxidant SkQR1 Reduces TNF-Induced Endothelial Permeability in vitro. Biochemistry 2016, 81, 1188–1197. [Google Scholar] [CrossRef]
- Zakharova, V.V.; Pletjushkina, O.Y.; Galkin, I.I.; Zinovkin, R.A.; Chernyak, B.V.; Krysko, D.V.; Bachert, C.; Krysko, O.; Skulachev, V.P.; Popova, E.N. Low concentration of uncouplers of oxidative phosphorylation decreases the TNF-induced endothelial permeability and lethality in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 968–977. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Prikhodko, A.; Galkin, I.; Pletjushkina, O.; Zinovkin, R.; Sud’ina, G.; Chernyak, B.; Pinegin, B. Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro. Eur. J. Cell Biol. 2017, 96, 254–265. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Galkin, I.; Pletjushkina, O.; Golyshev, S.; Zinovkin, R.; Prikhodko, A.; Pinegin, V.; Kondratenko, I.; Pinegin, B.; Chernyak, B. Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165664. [Google Scholar] [CrossRef]
- Sud’ina, G.F.; Golenkina, E.A.; Prikhodko, A.S.; Kondratenko, N.D.; Gaponova, T.V.; Chernyak, B.V. Mitochondria-targeted antioxidant SkQ1 inhibits leukotriene synthesis in human neutrophils. Front. Pharmacol. 2022, 13, 1023517. [Google Scholar] [CrossRef]
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Turner, P.J. Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J. Physiol. 2013, 591, 3549–3563. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mayenco, I.; Gonzalez-Rodriguez, P.; Torres-Torrelo, H.; Gao, L.; Fernandez-Aguera, M.C.; Bonilla-Henao, V.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute O(2) Sensing: Role of Coenzyme QH(2)/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab. 2018, 28, 145–158.e144. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.; Konrad, C.; Guerrero-Castillo, S.; Manfredi, G.; Vannucci, S.; Arnold, S.; Galkin, A. Deactivation of mitochondrial complex I after hypoxia-ischemia in the immature brain. J. Cereb. Blood Flow Metab. 2019, 39, 1790–1802. [Google Scholar] [CrossRef]
- Ye, C.; Shen, Z.; Greenberg, M.L. Cardiolipin remodeling: A regulatory hub for modulating cardiolipin metabolism and function. J. Bioenerg. Biomembr. 2016, 48, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Shilovsky, G.A.; Putyatina, T.S.; Ashapkin, V.V.; Yamskova, O.V.; Lyubetsky, V.A.; Sorokina, E.V.; Shram, S.I.; Markov, A.V.; Vyssokikh, M.Y. Biological Diversity and Remodeling of Cardiolipin in Oxidative Stress and Age-Related Pathologies. Biochemistry 2019, 84, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Stoyanovsky, D.A.; Belikova, N.A.; Tyurina, Y.Y.; Zhao, Q.; Tungekar, M.A.; Kapralova, V.; Huang, Z.; Mintz, A.H.; Greenberger, J.S.; et al. A mitochondria-targeted triphenylphosphonium-conjugated nitroxide functions as a radioprotector/mitigator. Radiat. Res. 2009, 172, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Aging is a specific biological function rather than the result of a disorder in complex living systems: Biochemical evidence in support of Weismann’s hypothesis. Biochemistry 1997, 62, 1191–1195. [Google Scholar]
- Skulachev, V.P.; Shilovsky, G.A.; Putyatina, T.S.; Popov, N.A.; Markov, A.V.; Skulachev, M.V.; Sadovnichii, V.A. Perspectives of Homo sapiens lifespan extension: Focus on external or internal resources? Aging 2020, 12, 5566–5584. [Google Scholar] [CrossRef]
- Skulachev, V.P. Mitochondria in the programmed death phenomena; a principle of biology: “It is better to die than to be wrong”. IUBMB Life 2000, 49, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E. The electromechanochemical model for energy coupling in mitochondria. Biochim. Biophys. Acta 1974, 346, 27–78. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Pustovidko, A.V.; Rokitskaya, T.I.; Severina, I.I.; Simonyan, R.A.; Trendeleva, T.A.; Lyamzaev, K.G.; Antonenko, Y.N.; Rogov, A.G.; Zvyagilskaya, R.A.; Skulachev, V.P.; et al. Derivatives of the cationic plant alkaloids berberine and palmatine amplify protonophorous activity of fatty acids in model membranes and mitochondria. Mitochondrion 2013, 13, 520–525. [Google Scholar] [CrossRef]
- Severina, I.I.; Severin, F.F.; Korshunova, G.A.; Sumbatyan, N.V.; Ilyasova, T.M.; Simonyan, R.A.; Rogov, A.G.; Trendeleva, T.A.; Zvyagilskaya, R.A.; Dugina, V.B.; et al. In search of novel highly active mitochondria-targeted antioxidants: Thymoquinone and its cationic derivatives. FEBS Lett. 2013, 587, 2018–2024. [Google Scholar] [CrossRef]
- Bakeeva, L.E.; Barskov, I.V.; Egorov, M.V.; Isaev, N.K.; Kapelko, V.I.; Kazachenko, A.V.; Kirpatovsky, V.I.; Kozlovsky, S.V.; Lakomkin, V.L.; Levina, S.B.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke). Biochemistry 2008, 73, 1288–1299. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Jankauskas, S.S.; Zinovkin, R.A.; Zorova, L.D.; Zorov, S.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, D.B. A Combination of Kidney Ischemia and Injection of Isolated Mitochondria Leads to Activation of Inflammation and Increase in Mortality Rate in Rats. Bull. Exp. Biol. Med. 2020, 169, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial Damage and Mitochondria-Targeted Antioxidant Protection in LPS-Induced Acute Kidney Injury. Antioxidants 2019, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Manskikh, V.N.; Pulkova, N.V.; Galkina, S.I.; Skulachev, V.P.; Zorov, D.B. Protective effect of mitochondria-targeted antioxidants in an acute bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, E3100–E3108. [Google Scholar] [CrossRef] [PubMed]
- Kapay, N.A.; Popova, O.V.; Isaev, N.K.; Stelmashook, E.V.; Kondratenko, R.V.; Zorov, D.B.; Skrebitsky, V.G.; Skulachev, V.P. Mitochondria-targeted plastoquinone antioxidant SkQ1 prevents amyloid-beta-induced impairment of long-term potentiation in rat hippocampal slices. J. Alzheimers Dis. 2013, 36, 377–383. [Google Scholar] [CrossRef]
- Andreev-Andrievskiy, A.A.; Kolosova, N.G.; Stefanova, N.A.; Lovat, M.V.; Egorov, M.V.; Manskikh, V.N.; Zinovkin, R.A.; Galkin, I.I.; Prikhodko, A.S.; Skulachev, M.V.; et al. Efficacy of Mitochondrial Antioxidant Plastoquinonyl-decyl-triphenylphosphonium Bromide (SkQ1) in the Rat Model of Autoimmune Arthritis. Oxid. Med. Cell Longev. 2016, 2016, 8703645. [Google Scholar] [CrossRef]
- Fedorov, A.V.; Chelombitko, M.A.; Chernyavskij, D.A.; Galkin, I.I.; Pletjushkina, O.Y.; Vasilieva, T.V.; Zinovkin, R.A.; Chernyak, B.V. Mitochondria-Targeted Antioxidant SkQ1 Prevents the Development of Experimental Colitis in Mice and Impairment of the Barrier Function of the Intestinal Epithelium. Cells 2022, 11, 3441. [Google Scholar] [CrossRef]
- Fock, E.M.; Parnova, R.G. Protective Effect of Mitochondria-Targeted Antioxidants against Inflammatory Response to Lipopolysaccharide Challenge: A Review. Pharmaceutics 2021, 13, 144. [Google Scholar] [CrossRef]
- Lowes, D.A.; Thottakam, B.M.; Webster, N.R.; Murphy, M.P.; Galley, H.F. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic. Biol. Med. 2008, 45, 1559–1565. [Google Scholar] [CrossRef]
- Supinski, G.S.; Murphy, M.P.; Callahan, L.A. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1095–R1102. [Google Scholar] [CrossRef]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Renal Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Keita, A.V.; Phan, V.; McKay, C.M.; Schoultz, I.; Lee, J.; Murphy, M.P.; Fernando, M.; Ronaghan, N.; Balce, D.; et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am. J. Pathol. 2014, 184, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, A.; Mullebner, A.; Paier-Pourani, J.; Banerjee, A.; Miller, I.; Lauterbock, L.; Duvigneau, J.C.; Skulachev, V.P.; Redl, H.; Kozlov, A.V. Vicious inducible nitric oxide synthase-mitochondrial reactive oxygen species cycle accelerates inflammatory response and causes liver injury in rats. Antioxid. Redox Signal 2015, 22, 572–586. [Google Scholar] [CrossRef]
- Wang, P.F.; Xie, K.; Cao, Y.X.; Zhang, A. Hepatoprotective Effect of Mitochondria-Targeted Antioxidant Mito-TEMPO against Lipopolysaccharide-Induced Liver Injury in Mouse. Mediat. Inflamm. 2022, 2022, 6394199. [Google Scholar] [CrossRef]
- To, E.E.; Erlich, J.R.; Liong, F.; Luong, R.; Liong, S.; Esaq, F.; Oseghale, O.; Anthony, D.; McQualter, J.; Bozinovski, S.; et al. Mitochondrial Reactive Oxygen Species Contribute to Pathological Inflammation During Influenza A Virus Infection in Mice. Antioxid. Redox Signal 2020, 32, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Vyssokikh, M.Y.; Chernyak, B.V.; Averina, O.A.; Andreev-Andrievskiy, A.A.; Zinovkin, R.A.; Lyamzaev, K.G.; Marey, M.V.; Egorov, M.V.; Frolova, O.J.; et al. Mitochondrion-targeted antioxidant SkQ1 prevents rapid animal death caused by highly diverse shocks. Sci. Rep. 2023, 13, 4326. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, K.; Rica, I.; Konecna, B.; Kim, H.I.; Park, J.; Kaczmarek, E.; Hauser, C.J. Role of Mitochondria-Derived Danger Signals Released after Injury in Systemic Inflammation and Sepsis. Antioxid. Redox Signal 2021, 35, 1273–1290. [Google Scholar] [CrossRef]
- Jia, B.; Ye, J.; Gan, L.; Li, R.; Zhang, M.; Sun, D.; Weng, L.; Xiong, Y.; Xu, J.; Zhang, P.; et al. Mitochondrial antioxidant SkQ1 decreases inflammation following hemorrhagic shock by protecting myocardial mitochondria. Front. Physiol. 2022, 13, 1047909. [Google Scholar] [CrossRef]
- Wu, M.; Chen, Y.; Xia, H.; Wang, C.; Tan, C.Y.; Cai, X.; Liu, Y.; Ji, F.; Xiong, P.; Liu, R.; et al. Transcriptional and proteomic insights into the host response in fatal COVID-19 cases. Proc. Natl. Acad. Sci. USA 2020, 117, 28336–28343. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. After 62 years of regulating immunity, dexamethasone meets COVID-19. Nat. Rev. Immunol. 2020, 20, 587–588. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and Oxidative Stress. Biochemistry 2020, 85, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Sud’ina, G.F.; Chernyak, B.V. Mitochondria Are Potential Targets for the Development of New Drugs Against Neutrophilic Inflammation in Severe Pneumonia Including COVID-19. Front. Pharmacol. 2021, 12, 609508. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Popova, E.N.; Zinovkina, L.A.; Lyamzaev, K.G.; Zinovkin, R.A. Mitochondria as Targets for Endothelial Protection in COVID-19. Front. Physiol. 2020, 11, 606170. [Google Scholar] [CrossRef]
- Petcherski, A.; Sharma, M.; Satta, S.; Daskou, M.; Vasilopoulos, H.; Hugo, C.; Ritou, E.; Dillon, B.J.; Fung, E.; Garcia, G.; et al. Mitoquinone mesylate targets SARS-CoV-2 infection in preclinical models. bioRxiv 2022. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e435. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Hernandez-Camacho, J.D.; Fernandez-Ayala, D.J.M.; Navas, P. Mitochondrial dysfunction in metabolism and ageing: Shared mechanisms and outcomes? Biogerontology 2018, 19, 461–480. [Google Scholar] [CrossRef]
- Webb, M.; Sideris, D.P. Intimate Relations-Mitochondria and Ageing. Int. J. Mol. Sci. 2020, 21, 7580. [Google Scholar] [CrossRef]
- Sohal, R.S.; Mockett, R.J.; Orr, W.C. Mechanisms of aging: An appraisal of the oxidative stress hypothesis. Free Radic. Biol. Med. 2002, 33, 575–586. [Google Scholar] [CrossRef]
- Holloway, G.P.; Holwerda, A.M.; Miotto, P.M.; Dirks, M.L.; Verdijk, L.B.; van Loon, L.J.C. Age-Associated Impairments in Mitochondrial ADP Sensitivity Contribute to Redox Stress in Senescent Human Skeletal Muscle. Cell Rep. 2018, 22, 2837–2848. [Google Scholar] [CrossRef]
- Vyssokikh, M.Y.; Holtze, S.; Averina, O.A.; Lyamzaev, K.G.; Panteleeva, A.A.; Marey, M.V.; Zinovkin, R.A.; Severin, F.F.; Skulachev, M.V.; Fasel, N.; et al. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc. Natl. Acad. Sci. USA 2020, 117, 6491–6501. [Google Scholar] [CrossRef] [PubMed]
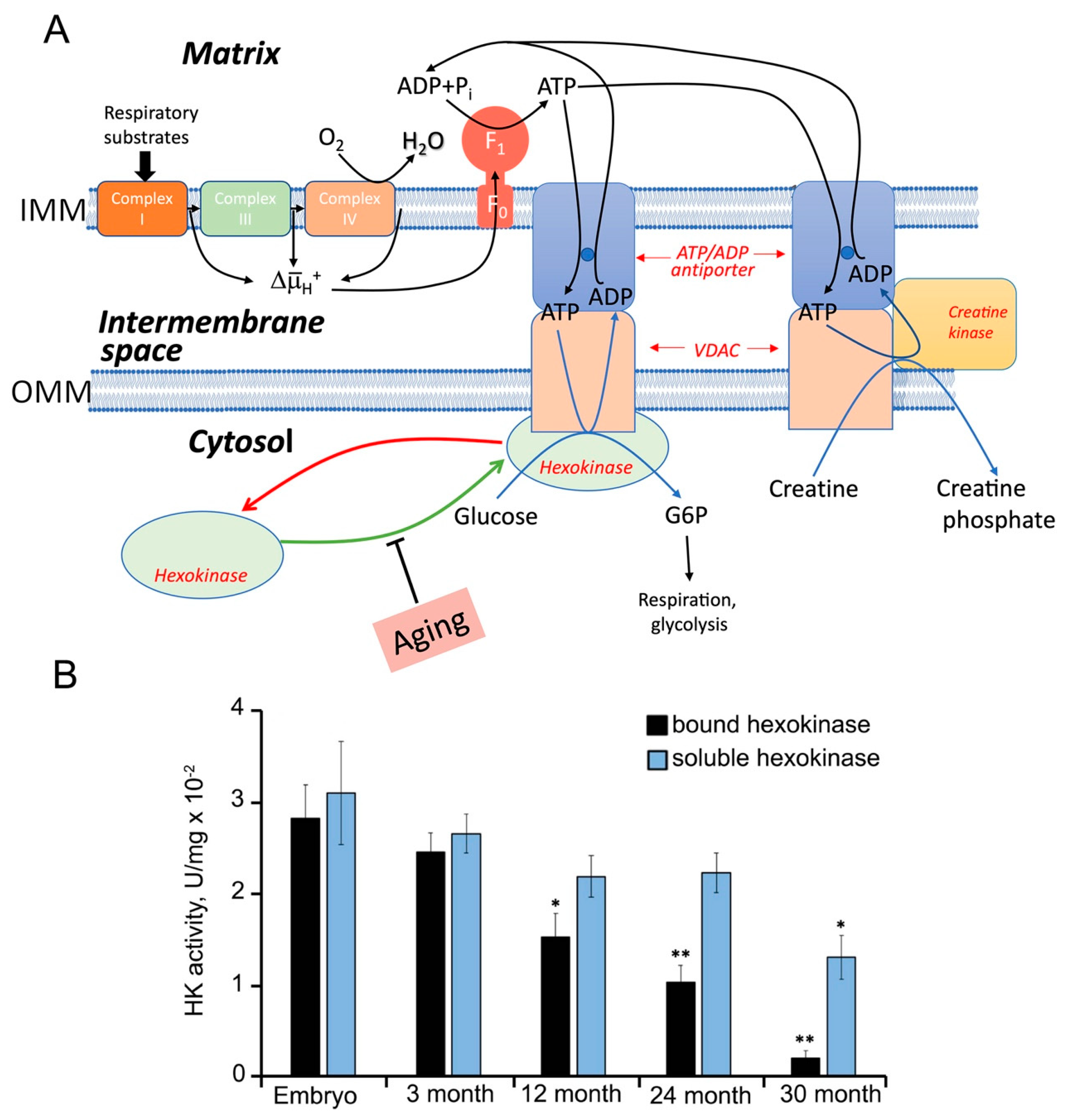
- de-Souza-Ferreira, E.; Rios-Neto, I.M.; Martins, E.L.; Galina, A. Mitochondria-coupled glucose phosphorylation develops after birth to modulate H(2) O(2) release and calcium handling in rat brain. J. Neurochem. 2019, 149, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Klingenberg, M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 1985, 24, 3821–3826. [Google Scholar] [CrossRef]
- Haloi, N.; Wen, P.C.; Cheng, Q.; Yang, M.; Natarajan, G.; Camara, A.K.S.; Kwok, W.M.; Tajkhorshid, E. Structural basis of complex formation between mitochondrial anion channel VDAC1 and Hexokinase-II. Commun. Biol. 2021, 4, 667. [Google Scholar] [CrossRef]
- Schlattner, U.; Kay, L.; Tokarska-Schlattner, M. Mitochondrial Proteolipid Complexes of Creatine Kinase. Subcell. Biochem. 2018, 87, 365–408. [Google Scholar] [CrossRef]
- Bradley, R.M.; Stark, K.D.; Duncan, R.E. Influence of tissue, diet, and enzymatic remodeling on cardiolipin fatty acyl profile. Mol. Nutr. Food Res. 2016, 60, 1804–1818. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Boengler, K.; Garcia-Dorado, D.; Hausenloy, D.J.; Kaambre, T.; Kararigas, G.; Perrino, C.; Schulz, R.; Ytrehus, K. Ageing, sex, and cardioprotection. Br. J. Pharmacol. 2020, 177, 5270–5286. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Antonenko, Y.N.; Cherepanov, D.A.; Chernyak, B.V.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; Korshunova, G.A.; Lyamzaev, K.G.; Pletjushkina, O.Y.; et al. Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs). Biochim. Biophys. Acta 2010, 1797, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef] [PubMed]
- Shilovsky, G.A.; Feniouk, B.A.; Skulachev, V.P. Thymic Involution in Ontogenesis: Role in Aging Program. Biochemistry 2015, 80, 1629–1631. [Google Scholar] [CrossRef]
- Obukhova, L.A.; Skulachev, V.P.; Kolosova, N.G. Mitochondria-targeted antioxidant SkQ1 inhibits age-dependent involution of the thymus in normal and senescence-prone rats. Aging 2009, 1, 389–401. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skulachev, V.P.; Vyssokikh, M.Y.; Chernyak, B.V.; Mulkidjanian, A.Y.; Skulachev, M.V.; Shilovsky, G.A.; Lyamzaev, K.G.; Borisov, V.B.; Severin, F.F.; Sadovnichii, V.A. Six Functions of Respiration: Isn’t It Time to Take Control over ROS Production in Mitochondria, and Aging Along with It? Int. J. Mol. Sci. 2023, 24, 12540. https://doi.org/10.3390/ijms241612540
Skulachev VP, Vyssokikh MY, Chernyak BV, Mulkidjanian AY, Skulachev MV, Shilovsky GA, Lyamzaev KG, Borisov VB, Severin FF, Sadovnichii VA. Six Functions of Respiration: Isn’t It Time to Take Control over ROS Production in Mitochondria, and Aging Along with It? International Journal of Molecular Sciences. 2023; 24(16):12540. https://doi.org/10.3390/ijms241612540
Chicago/Turabian StyleSkulachev, Vladimir P., Mikhail Yu. Vyssokikh, Boris V. Chernyak, Armen Y. Mulkidjanian, Maxim V. Skulachev, Gregory A. Shilovsky, Konstantin G. Lyamzaev, Vitaliy B. Borisov, Fedor F. Severin, and Victor A. Sadovnichii. 2023. "Six Functions of Respiration: Isn’t It Time to Take Control over ROS Production in Mitochondria, and Aging Along with It?" International Journal of Molecular Sciences 24, no. 16: 12540. https://doi.org/10.3390/ijms241612540
APA StyleSkulachev, V. P., Vyssokikh, M. Y., Chernyak, B. V., Mulkidjanian, A. Y., Skulachev, M. V., Shilovsky, G. A., Lyamzaev, K. G., Borisov, V. B., Severin, F. F., & Sadovnichii, V. A. (2023). Six Functions of Respiration: Isn’t It Time to Take Control over ROS Production in Mitochondria, and Aging Along with It? International Journal of Molecular Sciences, 24(16), 12540. https://doi.org/10.3390/ijms241612540