
Promise and Challenges of T Cell Immunotherapy for Osteosarcoma



Abstract
1. Introduction
2. Immune Checkpoint Inhibitors for Osteosarcoma
3. Adoptive T Cell Immunotherapy for Osteosarcoma
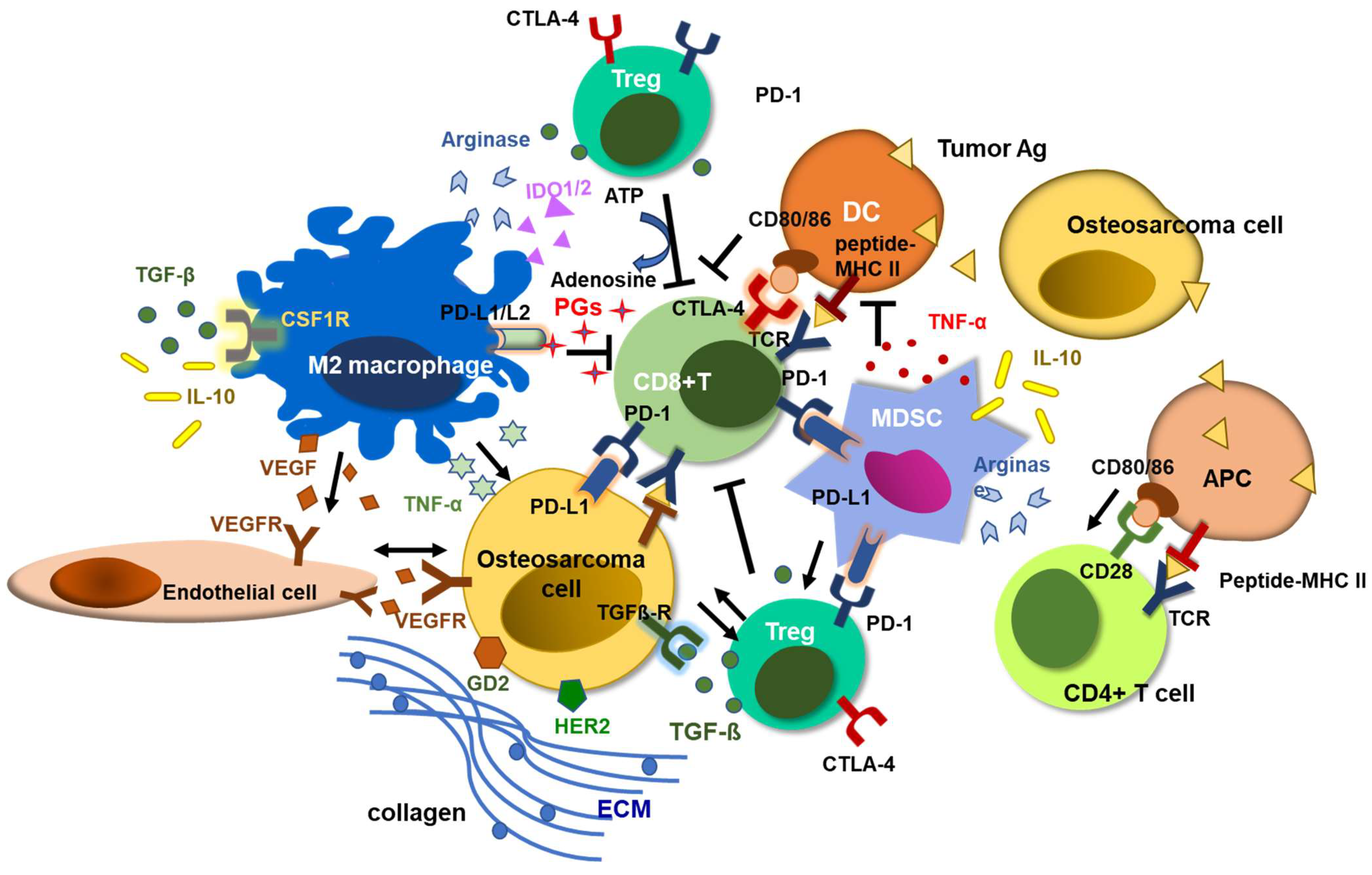
4. Tumor Microenvironment in Osteosarcoma

5. Combination Strategies to Overcome the Limitations of T Cell Immunotherapy against Osteosarcoma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luetke, A.; Meyers, P.A.; Lewis, I.; Juergens, H. Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef]
- Longhi, A.; Ferrari, S.; Tamburini, A.; Luksch, R.; Fagioli, F.; Bacci, G.; Ferrari, C. Late effects of chemotherapy and radiotherapy in osteosarcoma and Ewing sarcoma patients: The Italian Sarcoma Group Experience (1983–2006). Cancer 2012, 118, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, M.S.; Bielack, S.S.; Meltzer, P.; Gorlick, R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J. Clin. Oncol. 2015, 33, 3029–3035. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48. [Google Scholar] [CrossRef]
- Muller, C.R.; Smeland, S.; Bauer, H.C.; Saeter, G.; Strander, H. Interferon-alpha as the only adjuvant treatment in high-grade osteosarcoma: Long term results of the Karolinska Hospital series. Acta Oncol. 2005, 44, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival—A report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Bielack, S.S.; Smeland, S.; Whelan, J.S.; Marina, N.; Jovic, G.; Hook, J.M.; Krailo, M.D.; Gebhardt, M.; Papai, Z.; Meyer, J.; et al. Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b versus MAP Alone in Patients with Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. J. Clin. Oncol. 2015, 33, 2279–2287. [Google Scholar]
- Ebb, D.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef]
- Ha, H.T.; Griffith, K.A.; Zalupski, M.M.; Schuetze, S.M.; Thomas, D.G.; Lucas, D.R.; Baker, L.H.; Chugh, R. Phase II trial of cetuximab in patients with metastatic or locally advanced soft tissue or bone sarcoma. Am. J. Clin. Oncol. 2013, 36, 77–82. [Google Scholar] [CrossRef]
- Kopp, L.M.; Malempati, S.; Krailo, M.; Gao, Y.; Buxton, A.; Weigel, B.J.; Hawthorne, T.; Crowley, E.; Moscow, J.A.; Reid, J.M.; et al. Phase II trial of the glycoprotein non-metastatic B-targeted antibody-drug conjugate, glembatumumab vedotin (CDX-011), in recurrent osteosarcoma AOST1521: A report from the Children’s Oncology Group. Eur. J. Cancer 2019, 121, 177–183. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Uttenreuther-Fischer, M.M.; Huang, C.S.; Tsui, C.C.; Gillies, S.D.; Reisfeld, R.A.; Kung, F.H. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J. Clin. Oncol. 1998, 16, 2169–2180. [Google Scholar] [CrossRef]
- Edelman, M.; Dvorkin, M.; Laktionov, K.K.; Navarro, A.; Juan-Vidal, O.; Kozlov, V.; Golden, G.; Jordan, O.; Deng, C. The anti-disialoganglioside (GD2) antibody dinutuximab (D) for second-line treatment (2LT) of patients (pts) with relapsed/refractory small cell lung cancer (RR SCLC): Results from part II of the open-label, randomized, phase II/III distinct study. J. Clin. Oncol. 2020, 38, 9017. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.R.; Janeway, K.A.; Minard, C.G.; Hall, D.; Crompton, B.D.; Lazar, A.J.; Wang, W.-L.; Voss, S.D.; Militano, O.; Gorlick, R.G.; et al. PEPN1924, a phase 2 study of trastuzumab deruxtecan (DS-8201a, T-DXd) in adolescents and young adults with recurrent HER2+ osteosarcoma: A children’s oncology group pediatric early-phase clinical Trial Network study. J. Clin. Oncol. 2023, 41, 11527. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Amadei, B.; Tola, D.; Massari, M.; Schivazappa, S.; Missale, G.; Ferrari, C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 2006, 80, 11398–11403. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Hacohen, N.; Fritsch, E.F.; Carter, T.A.; Lander, E.S.; Wu, C.J. Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol. Res. 2013, 1, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Ferte, C.; Lebel-Binay, S.; Eggermont, A.; Soria, J.C. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology 2014, 3, e27817. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. CTLA-4 and PD-1 Pathway Blockade: Combinations in the Clinic. Front. Oncol. 2014, 4, 385. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Cheung, N.V. Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat. Rev. 2017, 58, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Palmerini, E.; Agostinelli, C.; Picci, P.; Pileri, S.; Marafioti, T.; Lollini, P.L.; Scotlandi, K.; Longhi, A.; Benassi, M.S.; Ferrari, S. Tumoral immune-infiltrate (IF), PD-L1 expression and role of CD8/TIA-1 lymphocytes in localized osteosarcoma patients treated within protocol ISG-OS1. Oncotarget 2017, 8, 111836–111846. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Okamoto, M.; Sasaki, J.; Kuroda, C.; Ishida, H.; Ueda, K.; Okano, S.; Ideta, H.; Kamanaka, T.; Sobajima, A.; et al. Clinical outcome of osteosarcoma and its correlation with programmed death-ligand 1 and T cell activation markers. OncoTargets Ther. 2019, 12, 2513–2518. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhou, J.; Ji, B. Evidence of Interleukin 21 Reduction in Osteosarcoma Patients Due to PD-1/PD-L1-Mediated Suppression of Follicular Helper T Cell Functionality. DNA Cell Biol. 2017, 36, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Chen, L.; Li, Y.J.; Kong, D.L. PD-L1/PD-1 axis serves an important role in natural killer cell-induced cytotoxicity in osteosarcoma. Oncol. Rep. 2019, 42, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, W.; Zhang, Z.; Shi, D.; Wu, F.; Zhong, B.; Shao, Z. Prognostic Value of Programmed Cell Death 1 Ligand-1 (PD-L1) or PD-1 Expression in Patients with Osteosarcoma: A Meta-Analysis. J. Cancer 2018, 9, 2525–2531. [Google Scholar] [CrossRef]
- Shen, J.K.; Cote, G.M.; Choy, E.; Yang, P.; Harmon, D.; Schwab, J.; Nielsen, G.P.; Chebib, I.; Ferrone, S.; Wang, X.; et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol. Res. 2014, 2, 690–698. [Google Scholar] [CrossRef]
- Liao, Y.; Chen, L.; Feng, Y.; Shen, J.; Gao, Y.; Cote, G.; Choy, E.; Harmon, D.; Mankin, H.; Hornicek, F.; et al. Targeting programmed cell death ligand 1 by CRISPR/Cas9 in osteosarcoma cells. Oncotarget 2017, 8, 30276–30287. [Google Scholar] [CrossRef]
- Lussier, D.M.; O’Neill, L.; Nieves, L.M.; McAfee, M.S.; Holechek, S.A.; Collins, A.W.; Dickman, P.; Jacobsen, J.; Hingorani, P.; Blattman, J.N. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J. Immunother. 2015, 38, 96–106. [Google Scholar] [CrossRef]
- Shimizu, T.; Fuchimoto, Y.; Fukuda, K.; Okita, H.; Kitagawa, Y.; Kuroda, T. The effect of immune checkpoint inhibitors on lung metastases of osteosarcoma. J. Pediatr. Surg. 2017, 52, 2047–2050. [Google Scholar] [CrossRef]
- Zheng, B.; Ren, T.; Huang, Y.; Sun, K.; Wang, S.; Bao, X.; Liu, K.; Guo, W. PD-1 axis expression in musculoskeletal tumors and antitumor effect of nivolumab in osteosarcoma model of humanized mouse. J. Hematol. Oncol. 2018, 11, 16. [Google Scholar] [CrossRef]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Boye, K.; Longhi, A.; Guren, T.; Lorenz, S.; Naess, S.; Pierini, M.; Taksdal, I.; Lobmaier, I.; Cesari, M.; Paioli, A.; et al. Pembrolizumab in advanced osteosarcoma: Results of a single-arm, open-label, phase 2 trial. Cancer Immunol. Immunother. 2021, 70, 2617–2624. [Google Scholar] [CrossRef]
- Lussier, D.M.; Johnson, J.L.; Hingorani, P.; Blattman, J.N. Combination immunotherapy with alpha-CTLA-4 and alpha-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J. Immunother. Cancer 2015, 3, 21. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Solis Soto, L.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Sterz, U.; Grube, M.; Herr, W.; Menhart, K.; Wendl, C.; Vogelhuber, M. Case Report: Dual Checkpoint Inhibition in Advanced Metastatic Osteosarcoma Results in Remission of All Tumor Manifestations-A Report of a Stunning Success in a 37-Year-Old Patient. Front. Oncol. 2021, 11, 684733. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, L.; Sys, G.; Creytens, D.; Lapeire, L. NGS-analysis to the rescue: Dual checkpoint inhibition in metastatic osteosarcoma—A case report and review of the literature. Acta Clin. Belg. 2021, 76, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xu, J.; Sun, X.; Guo, W.; Gu, J.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: A single-arm, open-label, phase 2 trial. J. Immunother. Cancer 2020, 8, e000798. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Lazar, A.J.; Torres, K.E.; Wang, W.L.; Cormier, J.N.; Ashleigh Guadagnolo, B.; Bishop, A.J.; Lin, H.; Hunt, K.K.; Bird, J.; et al. Phase II study of neoadjuvant checkpoint blockade in patients with surgically resectable undifferentiated pleomorphic sarcoma and dedifferentiated liposarcoma. BMC Cancer 2018, 18, 913. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Zwaan, C.M.; Marshall, L.V.; Michon, J.; Bourdeaut, F.; Casanova, M.; Corradini, N.; Rossato, G.; Farid-Kapadia, M.; Shemesh, C.S.; et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): A multicentre phase 1-2 study. Lancet Oncol. 2020, 21, 134–144. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Grignani, G.; Martinez-Trufero, J.; Redondo, A.; Valverde, C.; Stacchiotti, S.; Lopez-Pousa, A.; D’Ambrosio, L.; Gutierrez, A.; et al. Nivolumab and sunitinib combination in advanced soft tissue sarcomas: A multicenter, single-arm, phase Ib/II trial. J. Immunother. Cancer 2020, 8, e001561. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol. Ther. 2016, 166, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F. Failure at the effector phase: Immune barriers at the level of the melanoma tumor microenvironment. Clin. Cancer Res. 2007, 13, 5256–5261. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Pure, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wu, Z.; Luo, W. Chimeric Antigen Receptor T-Cell Therapy: The Light of Day for Osteosarcoma. Cancers 2021, 13, 4469. [Google Scholar] [CrossRef]
- Zhu, J.; Simayi, N.; Wan, R.; Huang, W. CAR T targets and microenvironmental barriers of osteosarcoma. Cytotherapy 2022, 24, 567–576. [Google Scholar] [CrossRef]
- Wang, H.; Jin, X.; Zhang, Y.; Wang, Z.; Zhang, T.; Xu, J.; Shen, J.; Zan, P.; Sun, M.; Wang, C.; et al. Inhibition of sphingolipid metabolism in osteosarcoma protects against CD151-mediated tumorigenicity. Cell Biosci. 2022, 12, 169. [Google Scholar] [CrossRef]
- Park, J.A.; Cheung, N.V. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J. Hematol. Oncol. 2020, 13, 172. [Google Scholar] [CrossRef]
- Koksal, H.; Muller, E.; Inderberg, E.M.; Bruland, O.; Walchli, S. Treating osteosarcoma with CAR T cells. Scand. J. Immunol. 2019, 89, e12741. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Rainusso, N.; Brawley, V.S.; Ghazi, A.; Hicks, M.J.; Gottschalk, S.; Rosen, J.M.; Ahmed, N. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 2012, 19, 212–217. [Google Scholar] [CrossRef]
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687. [Google Scholar] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Somers, G.R.; Ho, M.; Zielenska, M.; Squire, J.A.; Thorner, P.S. HER2 amplification and overexpression is not present in pediatric osteosarcoma: A tissue microarray study. Pediatr. Dev. Pathol. 2005, 8, 525–532. [Google Scholar] [CrossRef]
- Navai, S.A.; Derenzo, C.; Joseph, S.; Sanber, K.; Byrd, T.; Zhang, H.M.; Mata, M.; Gerken, C.; Shree, A.; Mathew, P.R.; et al. Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas. Cancer Res. 2019, 79, LB-147. [Google Scholar] [CrossRef]
- Roth, M.; Linkowski, M.; Tarim, J.; Piperdi, S.; Sowers, R.; Geller, D.; Gill, J.; Gorlick, R. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer 2014, 120, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Poon, V.I.; Roth, M.; Piperdi, S.; Geller, D.; Gill, J.; Rudzinski, E.R.; Hawkins, D.S.; Gorlick, R. Ganglioside GD2 expression is maintained upon recurrence in patients with osteosarcoma. Clin. Sarcoma Res. 2015, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, H.; Hamamura, K.; Hotta, H.; Matsumoto, Y.; Nishida, Y.; Hattori, H.; Furukawa, K.; Ueda, M.; Furukawa, K. Enhancement of malignant properties of human osteosarcoma cells with disialyl gangliosides GD2/GD3. Cancer Sci. 2012, 103, 1656–1664. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Q.; Chen, W.; Shan, B.; Ding, Y.; Zhang, G.; Cao, N.; Liu, L.; Zhang, Y. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS ONE 2013, 8, e70689. [Google Scholar] [CrossRef] [PubMed]
- Modak, S.; Kramer, K.; Gultekin, S.H.; Guo, H.F.; Cheung, N.K. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer Res. 2001, 61, 4048–4054. [Google Scholar]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, L.; Somovilla-Crespo, B.; Garcia-Rodriguez, P.; Morales-Molina, A.; Rodriguez-Milla, M.A.; Garcia-Castro, J. Switchable CAR T cell strategy against osteosarcoma. Cancer Immunol. Immunother. 2023, 72, 2623–2633. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Knodler, M.; Korfer, J.; Kunzmann, V.; Trojan, J.; Daum, S.; Schenk, M.; Kullmann, F.; Schroll, S.; Behringer, D.; Stahl, M.; et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM × anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Br. J. Cancer 2018, 119, 296–302. [Google Scholar] [CrossRef]
- Burges, A.; Wimberger, P.; Kumper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM × anti-CD3 antibody: A phase I/II study. Clin. Cancer Res. 2007, 13, 3899–3905. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A.; Al-Kadhimi, Z.; Colvin, G.A.; Cummings, F.J.; Legare, R.D.; Dizon, D.S.; Kouttab, N.; Maizel, A.; Colaiace, W.; et al. Targeted T-cell Therapy in Stage IV Breast Cancer: A Phase I Clinical Trial. Clin. Cancer Res. 2015, 21, 2305–2314. [Google Scholar] [CrossRef]
- Yankelevich, M.; Kondadasula, S.V.; Thakur, A.; Buck, S.; Cheung, N.K.; Lum, L.G. Anti-CD3 × anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr. Blood Cancer 2012, 59, 1198–1205. [Google Scholar] [CrossRef]
- Reusch, U.; Sundaram, M.; Davol, P.A.; Olson, S.D.; Davis, J.B.; Demel, K.; Nissim, J.; Rathore, R.; Liu, P.Y.; Lum, L.G. Anti-CD3 × anti-epidermal growth factor receptor (EGFR) bispecific antibody redirects T-cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. Clin. Cancer Res. 2006, 12, 183–190. [Google Scholar] [CrossRef]
- Vaishampayan, U.; Thakur, A.; Rathore, R.; Kouttab, N.; Lum, L.G. Phase I Study of Anti-CD3 × Anti-Her2 Bispecific Antibody in Metastatic Castrate Resistant Prostate Cancer Patients. Prostate Cancer 2015, 2015, 285193. [Google Scholar] [CrossRef] [PubMed]
- Yankelevich, M.; Modak, S.; Chu, R.; Lee, D.W.; Thakur, A.; Cheung, N.-K.V.; Lum, L.G. Phase I study of OKT3 × hu3F8 bispecific antibody (GD2Bi) armed T cells (GD2BATs) in GD2-positive tumors. J. Clin. Oncol. 2019, 37 (Suppl. S15), 2533. [Google Scholar] [CrossRef]
- Santich, B.H.; Park, J.A.; Tran, H.; Guo, H.F.; Huse, M.; Cheung, N.V. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci. Transl. Med. 2020, 12, eaax1315. [Google Scholar] [CrossRef]
- Park, J.A.; Santich, B.H.; Xu, H.; Lum, L.G.; Cheung, N.V. Potent ex vivo armed T cells using recombinant bispecific antibodies for adoptive immunotherapy with reduced cytokine release. J. Immunother. Cancer 2021, 9, e002222. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tashiro, H.; Omer, B.; Lapteva, N.; Ando, J.; Ngo, M.; Mehta, B.; Dotti, G.; Kinchington, P.R.; Leen, A.M.; et al. Vaccination Targeting Native Receptors to Enhance the Function and Proliferation of Chimeric Antigen Receptor (CAR)-Modified T Cells. Clin. Cancer Res. 2017, 23, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, X.; Jiang, Z.; Wang, H.; Yuan, H. Immune checkpoint inhibitors in osteosarcoma: A hopeful and challenging future. Front. Pharmacol. 2022, 13, 1031527. [Google Scholar] [CrossRef] [PubMed]
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex But Targetable Ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef] [PubMed]
- Alfranca, A.; Martinez-Cruzado, L.; Tornin, J.; Abarrategi, A.; Amaral, T.; de Alava, E.; Menendez, P.; Garcia-Castro, J.; Rodriguez, R. Bone microenvironment signals in osteosarcoma development. Cell. Mol. Life Sci. 2015, 72, 3097–3113. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Redini, F. Transforming Growth Factor-beta Signaling Plays a Pivotal Role in the Interplay between Osteosarcoma Cells and Their Microenvironment. Front. Oncol. 2018, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, G.; Chen, R.; Hua, Y.; Cai, Z. Mesenchymal stem cells in the osteosarcoma microenvironment: Their biological properties, influence on tumor growth, and therapeutic implications. Stem Cell Res. Ther. 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Han, J.; Yang, L.; Cai, Z.; Sun, W.; Hua, Y.; Xu, J. Immune Microenvironment in Osteosarcoma: Components, Therapeutic Strategies and Clinical Applications. Front. Immunol. 2022, 13, 907550. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.D.; Bussard, K.M. The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases. Cancers 2019, 11, 1020. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Dean, D.; Hornicek, F.J.; Chen, Z.; Duan, Z. The role of extracelluar matrix in osteosarcoma progression and metastasis. J. Exp. Clin. Cancer Res. 2020, 39, 178. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar] [CrossRef]
- Regan, D.P.; Chow, L.; Das, S.; Haines, L.; Palmer, E.; Kurihara, J.N.; Coy, J.W.; Mathias, A.; Thamm, D.H.; Gustafson, D.L.; et al. Losartan Blocks Osteosarcoma-Elicited Monocyte Recruitment, and Combined with the Kinase Inhibitor Toceranib, Exerts Significant Clinical Benefit in Canine Metastatic Osteosarcoma. Clin. Cancer Res. 2022, 28, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.Y.; Tsai, H.C.; Chou, P.Y.; Wang, S.W.; Chen, H.T.; Lin, Y.M.; Chiang, I.P.; Chang, T.M.; Hsu, S.K.; Chou, M.C.; et al. CCL3 promotes angiogenesis by dysregulation of miR-374b/ VEGF-A axis in human osteosarcoma cells. Oncotarget 2016, 7, 4310–4325. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Liu, S.C.; Sun, H.L.; Huang, T.Y.; Chan, C.H.; Yang, C.Y.; Yeh, H.I.; Huang, Y.L.; Chou, W.Y.; Lin, Y.M.; et al. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 2015, 36, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Tsai, H.C.; Cheng, Y.C.; Lin, C.Y.; Huang, Y.L.; Tsai, C.H.; Xu, G.H.; Wang, S.W.; Fong, Y.C.; Tang, C.H. CTGF promotes osteosarcoma angiogenesis by regulating miR-543/angiopoietin 2 signaling. Cancer Lett. 2017, 391, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Ohba, T.; Cates, J.M.; Cole, H.A.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Autocrine VEGF/VEGFR1 signaling in a subpopulation of cells associates with aggressive osteosarcoma. Mol. Cancer Res. 2014, 12, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, J.; Dai, R.; Xu, J.; Feng, H. Transferrin receptor-1 and VEGF are prognostic factors for osteosarcoma. J. Orthop. Surg. Res. 2019, 14, 296. [Google Scholar] [CrossRef]
- Yang, S.Y.; Yu, H.; Krygier, J.E.; Wooley, P.H.; Mott, M.P. High VEGF with rapid growth and early metastasis in a mouse osteosarcoma model. Sarcoma 2007, 2007, 95628. [Google Scholar] [CrossRef]
- Gu, J.; Ji, Z.; Li, D.; Dong, Q. Proliferation inhibition and apoptosis promotion by dual silencing of VEGF and Survivin in human osteosarcoma. Acta Biochim. Biophys. Sin. 2019, 51, 59–67. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Inagaki, Y.; Hookway, E.; Williams, K.A.; Hassan, A.B.; Oppermann, U.; Tanaka, Y.; Soilleux, E.; Athanasou, N.A. Dendritic and mast cell involvement in the inflammatory response to primary malignant bone tumours. Clin. Sarcoma Res. 2016, 6, 13. [Google Scholar] [CrossRef]
- Huang, Q.; Liang, X.; Ren, T.; Huang, Y.; Zhang, H.; Yu, Y.; Chen, C.; Wang, W.; Niu, J.; Lou, J.; et al. The role of tumor-associated macrophages in osteosarcoma progression—Therapeutic implications. Cell Oncol. 2021, 44, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Scholl, S.M.; Pallud, C.; Beuvon, F.; Hacene, K.; Stanley, E.R.; Rohrschneider, L.; Tang, R.; Pouillart, P.; Lidereau, R. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J. Natl. Cancer Inst. 1994, 86, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Xu, Y.; Fu, J.; Zhu, X.; Chen, H.; Xu, H.; Wang, G.; Song, Y.; Song, G.; Lu, J.; et al. Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci. 2020, 111, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Jakab, M.; Rostalski, T.; Lee, K.H.; Mogler, C.; Augustin, H.G. Tie2 Receptor in Tumor-Infiltrating Macrophages Is Dispensable for Tumor Angiogenesis and Tumor Relapse after Chemotherapy. Cancer Res. 2022, 82, 1353–1364. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Tian, H.; Cao, J.; Li, B.; Nice, E.C.; Mao, H.; Zhang, Y.; Huang, C. Managing the immune microenvironment of osteosarcoma: The outlook for osteosarcoma treatment. Bone Res. 2023, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef]
- Martinez-Zubiaurre, I.; Chalmers, A.J.; Hellevik, T. Radiation-Induced Transformation of Immunoregulatory Networks in the Tumor Stroma. Front. Immunol. 2018, 9, 1679. [Google Scholar] [CrossRef]
- Tsai, C.S.; Chen, F.H.; Wang, C.C.; Huang, H.L.; Jung, S.M.; Wu, C.J.; Lee, C.C.; McBride, W.H.; Chiang, C.S.; Hong, J.H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef]
- Teng, F.; Meng, X.; Kong, L.; Mu, D.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor-infiltrating lymphocytes, forkhead box P3, programmed death ligand-1, and cytotoxic T lymphocyte-associated antigen-4 expressions before and after neoadjuvant chemoradiation in rectal cancer. Transl. Res. 2015, 166, 721–732.e1. [Google Scholar] [CrossRef]
- Jagodinsky, J.C.; Jin, W.J.; Bates, A.M.; Hernandez, R.; Grudzinski, J.J.; Marsh, I.R.; Chakravarty, I.; Arthur, I.S.; Zangl, L.M.; Brown, R.J.; et al. Temporal analysis of type 1 interferon activation in tumor cells following external beam radiotherapy or targeted radionuclide therapy. Theranostics 2021, 11, 6120–6137. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.; Walker, K.L.; Grudzinski, J.J.; Aluicio-Sarduy, E.; Patel, R.; Zahm, C.D.; Pinchuk, A.N.; Massey, C.F.; Bitton, A.N.; Brown, R.J.; et al. (90)Y-NM600 targeted radionuclide therapy induces immunologic memory in syngeneic models of T-cell Non-Hodgkin’s Lymphoma. Commun. Biol. 2019, 2, 79. [Google Scholar] [CrossRef]
- Vito, A.; Rathmann, S.; Mercanti, N.; El-Sayes, N.; Mossman, K.; Valliant, J. Combined Radionuclide Therapy and Immunotherapy for Treatment of Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4843. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Vats, K.; Lohar, S.P.; Korde, A.; Samuel, G. Camptothecin Enhances Cell Death Induced by (177)Lu-EDTMP in Osteosarcoma Cells. Cancer Biother. Radiopharm. 2014, 29, 317–322. [Google Scholar]
- Alavi, M.; Omidvari, S.; Mehdizadeh, A.; Jalilian, A.R.; Bahrami-Samani, A. Metastatic Bone Pain Palliation Using (177)Lu-Ethylenediaminetetramethylene Phosphonic Acid. World J. Nucl. Med. 2015, 14, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Dias, G.; Able, S.; Skaripa-Koukelli, I.; Anderson, R.; Wilson, G.; Vallis, K.A. Evaluation of anti-tumor immunity in response to [177Lu]Lu-PSMA in a mouse model of prostate cancer. Cancer Res. 2023, 83, 5038. [Google Scholar] [CrossRef]
- Taofeek, K.; Owonikoko, J.C.M.; Yoon, J.; Slotkin, E.K.; Dowlati, A.; Ma, V.T.; Düring, M.; Sveistrup, J. Phase 1 trial of GD2-SADA:177Lu-DOTA drug complex in patients with recurrent or refractory metastatic solid tumors known to express GD2 including small cell lung cancer (SCLC), sarcoma, and malignant melanoma. J. Clin. Oncol. 2023, 41, TPS3162. [Google Scholar]
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6, 261ra151. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but Can Be Protected from Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Sam, J.; Colombetti, S.; Fauti, T.; Roller, A.; Biehl, M.; Fahrni, L.; Nicolini, V.; Perro, M.; Nayak, T.; Bommer, E.; et al. Combination of T-Cell Bispecific Antibodies with PD-L1 Checkpoint Inhibition Elicits Superior Anti-Tumor Activity. Front. Oncol. 2020, 10, 575737. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Wang, L.; Cheung, N.V. Modulating tumor infiltrating myeloid cells to enhance bispecific antibody-driven T cell infiltration and anti-tumor response. J. Hematol. Oncol. 2021, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, J.; Chen, Y.; Kang, Y.; Liao, Z.; He, Y.; Zhang, C. Novel Immunotherapies for Osteosarcoma. Front. Oncol. 2022, 12, 830546. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Guo, W.; Ren, T.; Huang, Y.; Wang, S.; Liu, K.; Zheng, B.; Yang, K.; Zhang, H.; Liang, X. Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis. Cancer Lett. 2019, 440–441, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Dennen, K.; Gordon, N.; Kleinerman, E.S. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology 2020, 9, 1747677. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Moller, H.J.; Hoyer, M.; Maniecki, M.B.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J. Clin. Oncol. 2009, 27, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Dhupkar, P.; Gordon, N.; Stewart, J.; Kleinerman, E.S. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 2018, 7, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-Trans Retinoic Acid Prevents Osteosarcoma Metastasis by Inhibiting M2 Polarization of Tumor-Associated Macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.J.; Xiang, S.F.; Chen, Y.Q.; Zhang, N.; Cao, J.; Zhu, H.; Yang, B.; Zhou, Q.; Ying, M.D.; He, Q.J. Inhibition of M2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta Pharmacol. Sin. 2019, 40, 1343–1350. [Google Scholar] [CrossRef]
- Ratti, C.; Botti, L.; Cancila, V.; Galvan, S.; Torselli, I.; Garofalo, C.; Manara, M.C.; Bongiovanni, L.; Valenti, C.F.; Burocchi, A.; et al. Trabectedin Overrides Osteosarcoma Differentiative Block and Reprograms the Tumor Immune Environment Enabling Effective Combination with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 5149–5161. [Google Scholar] [CrossRef] [PubMed]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Joseph Espat, N.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef]
- Saltz, L.B.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.S.; Rivera, F.; et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; Dieras, V.; Cortes, J.; Duenne, A.A.; Yi, J.; O’Shaughnessy, J. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: Pooled and subgroup analyses of data from 2447 patients. Ann. Oncol. 2013, 24, 2773–2780. [Google Scholar] [CrossRef]
- Upadhaya, S.; Neftelino, S.T.; Hodge, J.P.; Oliva, C.; Campbell, J.R.; Yu, J.X. Combinations take centre stage in PD1/PDL1 inhibitor clinical trials. Nat. Rev. Drug Discov. 2021, 20, 168–169. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Espinosa-Cotton, M.; Guo, H.F.; Monette, S.; Cheung, N.V. Targeting tumor vasculature to improve antitumor activity of T cells armed ex vivo with T cell engaging bispecific antibody. J. Immunother. Cancer 2023, 11, e006680. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Clinical Trial | Target | Eligibility (Age) | Phase | Status | Reference |
|---|---|---|---|---|---|
| Pembrolizumab in patients with advanced sarcomas | PD-1 inhibitor | >12 | phase 2 | completed | NCT02301039 [43,51] |
| Nivolumab with or without ipilimumab in treating recurrent or refractory solid tumors or sarcomas | PD-1 inhibitor + CTLA-4 inhibitor | >1 | phase 1 and 2 | active, not recruiting | NCT02304458 [52] |
| Nivolumab with or without ipilimumab for patients with metastatic sarcoma | PD-1 inhibitor + CTLA-4 inhibitor | >18 | phase 2 | active, not recruiting | NCT02500797 [46] |
| Atezolizumab in pediatric and young adult patients with solid tumors | PD-L1 inhibitor | <30 | phase 1 and 2 | terminated | NCT02541604 [53] |
| Durvalumab plus tremelimumab in multiple sarcoma subtypes | PD-L1 inhibitor + CTLA-4 inhibitor | >18 | phase 2 | active, not recruiting | NCT02815995 [47] |
| Avelumab in patients with recurrent or progressive osteosarcoma | PD-L1 inhibitor | 12–49 | phase 2 | active, not recruiting | NCT03006848 |
| Pembrolizumab in patients with relapsed or metastatic osteosarcoma | PD-1 inhibitor | >18 | phase 2 | terminated | NCT03013127 [44] |
| Nivolumab plus ABI-009 (nab-sirolimus) for advanced sarcoma and certain cancers | PD-1 inhibitor | >12 | phase 1 and 2 | completed | NCT03190174 |
| Sunitinib and/or nivolumab plus chemotherapy in advanced soft tissue and bone sarcomas | PD-1 inhibitor | 20–80 | phase 1 and 2 | recruiting | NCT03277924 [54] |
| Apatinib plus camrelizumab for advanced osteosarcoma | PD-1 inhibitor | phase 2 | completed | NCT03359018 [50] | |
| Nivolumab plus ipilimumab in non-resectable sarcoma and endometrial carcinoma | PD-1 inhibitor + CTLA-4 inhibitor | >18 | phase 2 | unknown | NCT03449108 |
| Nivolumab and azacitidine for recurrent, resectable osteosarcoma | PD-1 inhibitor | <39 | phase 1 and 2 | recruiting | NCT03628209 |
| Socazolimab in high-grade osteosarcoma | PD-L1 inhibitor | 18–55 | phase 1 and 2 | recruiting | NCT03676985 |
| Famitinib plus camrelizumab and famitinib alone and famitinib plus ifosfamide in advanced osteosarcoma | PD-1 inhibitor | >12 | phase 1 and 2 | withdrawn | NCT04044378 |
| Multi-antigen autoimmune cell injection (MASCT-I)combined with apatinib and/or anti-PD1 antibody in the treatment of tissue sarcoma | PD-1 inhibitor | 14–70 | phase 1 | active, not recruiting | NCT04074564 |
| Camrelizumab in combination with neoadjuvant chemotherapy in osteosarcoma | PD-1 inhibitor | 14–65 | phase 2 | recruiting | NCT04294511 |
| MAPI + camrelizumab vs. API + apatinib vs. MAPI in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma | PD-1 inhibitor | >12 | phase 2 | unknown | NCT04351308 |
| Socazolimab for maintenance therapy in patients with high-grade osteosarcoma | PD-L1 inhibitor | >12 | phase 3 | not yet recruiting | NCT04359550 |
| Niraparib and dostarlimab in pediatric participants with solid tumors | PD-1 inhibitor | 0.5–17 | phase 1 | recruiting | NCT04544995 |
| Oleclumab and durvalumab for the treatment of recurrent, refractory, or metastatic sarcoma | PD-L1 inhibitor | >12 | phase 2 | recruiting | NCT04668300 |
| Bempegaldesleukin (BEMPEG: NKTR-214) in combination with nivolumab in recurrent or treatment-resistant cancer | PD-1 inhibitor | <30 | phase 1 and 2 | terminated | NCT04730349 |
| Regorafenib and nivolumab in refractory or relapsed osteosarcoma | PD-1 inhibitor | >5 | phase 2 | active, not recruiting | NCT04803877 |
| Atezolizumab and cabozantinib for recurrent or metastatic Osteosarcoma | PD-L1 inhibitor | >12 | phase 2 | not yet recruiting | NCT05019703 |
| Combination of pembrolizumab and cabozantinib in patients with advanced sarcomas | PD-1 inhibitor | >18 | phase 2 | recruiting | NCT05182164 |
| Tislelizumab combined with chemotherapy in bone metastatic sarcoma | PD-1 inhibitor | >18 | phase 2 | recruiting | NCT05241132 |
| Neoadjuvant dual checkpoint inhibition and cryoablation in relapsed/refractory pediatric solid tumors | PD-1 inhibitor + CTLA-4 inhibitor | >1 | phase I and 2 | recruiting | NCT05302921 |
| Clinical Trial | Target | Eligibility (Age) | Phase | Status | Reference |
|---|---|---|---|---|---|
| HER2 chimeric antigen receptor expressing T cells in advanced sarcoma | HER2-CAR T cells | Child, adult, older adults | phase 1 | active, not recruiting | NCT00902044 [64] |
| Humanized 3F8 bispecific antibody (Hu3F8-BsAb) in relapsed/refractory neuroblastoma, osteosarcoma and other solid tumor cancers | Humanized 3F8 bispecific antibody | 1–17 | phase 1 and 2 | terminated | NCT03860207 |
| iC9-GD2-CAR-VZV-CTLs in refractory or metastatic GD2-positive sarcoma and neuroblastoma | GD2 T cells, VZV vaccine, CPM, fludarabine | child, adult, older adult | phase 1 | active, not recruiting | NCT01953900 [92] |
| Haploidentical transplant and donor NK cells for solid tumors | Donor NK cell | child, adult, older adult | phase 2 | active, not recruiting | NCT02100891 |
| T cells expressing an anti-GD2 CAR in GD2+ solid tumors | GD2-CAR T cell | 1–35 | phase 1 | completed | NCT02107963 |
| Activated T cells armed with GD2 bispecific antibody in neuroblastoma and osteosarcoma | GD2 BsAb armed T cell | 1–29 | phase 1 and 2 | unknown | NCT02173093 [91] |
| Aβ CD19+ depleted haploidentical transplantation plus zometa for pediatric hematologic malignancies and solid tumors | TCRαβ+/CD19+ depleted haploidentical stem cells | 0.5–21 | phase 1 | recruiting | NCT02508038 |
| Treatment of relapsed or refractory neuroblastoma and osteosarcoma with expanded haploidentical NK Cells and Hu14.18-IL2 | Ex vivo expanded and activated haploidentical donor NK cells, and Hu14.18-IL2 | 0.5–26 | phase 1 | withdrawn | NCT03209869 |
| Fourth-generation safety-engineered CAR T cells targeting sarcomas | Sarcoma-specific CAR-T cells | 1–75 | phase 1 and 2 | recruiting | NCT03356782 |
| Anti-GD2 CAR T cells in high-risk and/or relapsed/refractory neuroblastoma or other GD2-positive solid tumors | GD2-CART 01 | 1–25 | phase 1 and 2 | recruiting | NCT03373097 |
| LN-145 or LN-145-S1 in relapsed or refractory cancers | Autologous tumor infiltrating lymphocytes LN-145 or LN-145-S1 | 16–70 | phase 2 | recruiting | NCT03449108 |
| NY-ESO-1-specific T cell receptor (TCR) T cell in sarcoma | NY-ESO-1(TCR affinity enhancing T cell | 14–70 | phase 1 | recruiting | NCT03462316 |
| EGFR806 CAR T cell immunotherapy for recurrent/refractory solid tumors | EGFR 806CAR(2G)-EGFRt, CD19CAR(2G)-T2A-HER2tG | 1–30 | phase 1 | recruiting | NCT03618381 |
| C7R-GD2.CAR T cells for patients with relapsed or refractory neuroblastoma and other GD2-positive cancers | C7R-GD2.CART cells | 1–74 | phase 1 | recruiting | NCT03635632 |
| Study of CAR T cells targeting the GD2 with IL-15+i caspase 9 for relapsed/refractory neuroblastoma or relapsed/refractory osteosarcoma | iC9.GD2.CAR.IL-15 T cells | >1.6 | phase 1 | recruiting | NCT03721068 |
| Combination immunotherapy targeting sarcomas | Multiple sarcoma-specific CAR-T cells and sarcoma vaccines | 1–75 | phase 1 and 2 | recruiting | NCT04433221 |
| B7H3 CAR T cell immunotherapy for recurrent/refractory solid tumors | B7H3-specific CAR, bispecific B7H3xCD19 CAR | <26 | phase 1 | recruiting | NCT04483778 |
| GD2-targeted modified T cells (GD2CART) in relapsed/refractory osteosarcoma and neuroblastoma | GD2-CAR T cell | <35 | phase 1 | suspended | NCT04539366 |
| Clinical study of CD276 targeted CAR T cell in CD276-positive advanced solid tumor | CD276 CAR T cells | 1–70 | phase 1 | not yet recruiting | NCT04864821 |
| B7-H3-CAR T cell therapy for pediatric patients with solid tumors | B7-H3 CAR T cell | <21 | phase 1 | recruiting | NCT04897321 |
| HER2 CAR T cells in combination with checkpoint blockade in patients with advanced sarcoma | HER2-CAR T cell + pembrolizumab or nivolumab | 1–25 | Phase 1 | recruiting | NCT04995003 |
| Fluorescein-specific (FITC-E2) CAR T cells in combination with folate–fluorescein (UB-TT170) for osteogenic sarcoma | SCRI-E2CAR_EGFRtv1 + UB_TT170 | 15–30 | phase 1 | recruiting | NCT05312411 |
| T cell membrane-anchored tumor targeted IL12 (Attil12)-T cell therapy in advanced/metastatic soft tissue and bone sarcoma | autologous tumor-targeted IL12 T cells | >12 | phase 1 | not yet recruiting | NCT05621668 |
| CAR.70-engineered IL15-transduced cord blood-derived NK cells in conjunction with lymphodepleting chemotherapy for advanced renal cell carcinoma, mesothelioma and osteosarcoma | CAR.70/IL15-transduced CB-derived NK cells | 18–80 | phase 1 and 2 | not yet recruiting | NCT05703854 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.A.; Cheung, N.-K.V. Promise and Challenges of T Cell Immunotherapy for Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 12520. https://doi.org/10.3390/ijms241512520
Park JA, Cheung N-KV. Promise and Challenges of T Cell Immunotherapy for Osteosarcoma. International Journal of Molecular Sciences. 2023; 24(15):12520. https://doi.org/10.3390/ijms241512520
Chicago/Turabian StylePark, Jeong A, and Nai-Kong V. Cheung. 2023. "Promise and Challenges of T Cell Immunotherapy for Osteosarcoma" International Journal of Molecular Sciences 24, no. 15: 12520. https://doi.org/10.3390/ijms241512520
APA StylePark, J. A., & Cheung, N.-K. V. (2023). Promise and Challenges of T Cell Immunotherapy for Osteosarcoma. International Journal of Molecular Sciences, 24(15), 12520. https://doi.org/10.3390/ijms241512520