
Design, Synthesis, and Biological Evaluation of New Potential Unusual Modified Anticancer Immunomodulators for Possible Non-Teratogenic Quinazoline-Based Thalidomide Analogs

 , , , ,
, , , ,  , ,
, ,
Abstract
1. Introduction
Rationale of Molecular Design
- A flat hetero aromatic ring system that contains at least one H-bond acceptor. It occupies the phthaloyl binding pocket and can form hydrogen bonding interactions with His359 and hydrophobic stacking interactions with Pro354.
- A hydrophobic moiety interacting with the hydrophobic region of the glutarimide binding pocket forming Van der Waals interactions with Trp382, Trp388, Trp402, and Phe 404.
- A linker occupying the space region between the glutarimide binding pocket and the phthaloyl binding pocket.
- A hydrophilic head (three adjacent hydrogen-bond-forming groups) that occupies the hydrophilic region of the glutarimide binding pocket forming hydrogen bonding interactions with His380 and Trp382.
- The binding of immunomodulatory drugs (IMiDs), e.g., thalidomide, with CRBN has been associated with teratogenicity. CRBN has a mediating role in helping immunomodulatory drugs to exert their immunomodulatory and tumoricidal effects. However, the idea that cereblon modulation is responsible for the teratogenic activity of thalidomide in chicks and zebrafish was brought into doubt due to a 2013 report that pomalidomide does not cause teratogenic effects in the same animal models of thalidomide, even though it binds with CRBN more strongly than thalidomide [41,42].
- With regard to the cytotoxicity of IMiDs, CRBN likely plays an important role in the binding, ubiquitination, and degradation of factors involved in maintaining the regular function of a cell [43].
- The molecular requirement for ‘thalidomide-type’ teratogenicity is highly structure-dependent. Both the phthalimide and glutarimide groups are essential for embryopathic activity. It is also evident that the glutarimide moiety contributes to the teratogenic effect of many thalidomide analogs because they simply have R and S stereoisomers, where S is a potential teratogen [44].
- Different studies revealed that phthalimide analogs devoid of the glutarimide moiety could be involved in teratogenic effect. Other results indicate that phthalimide analogs devoid of this functional group could represent a new class of analgesic and anti-inflammatory candidates with potentially greater safety [45].



2. Results
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Anti-Proliferative Activity
2.2.2. In Vitro Protein Expression Assay
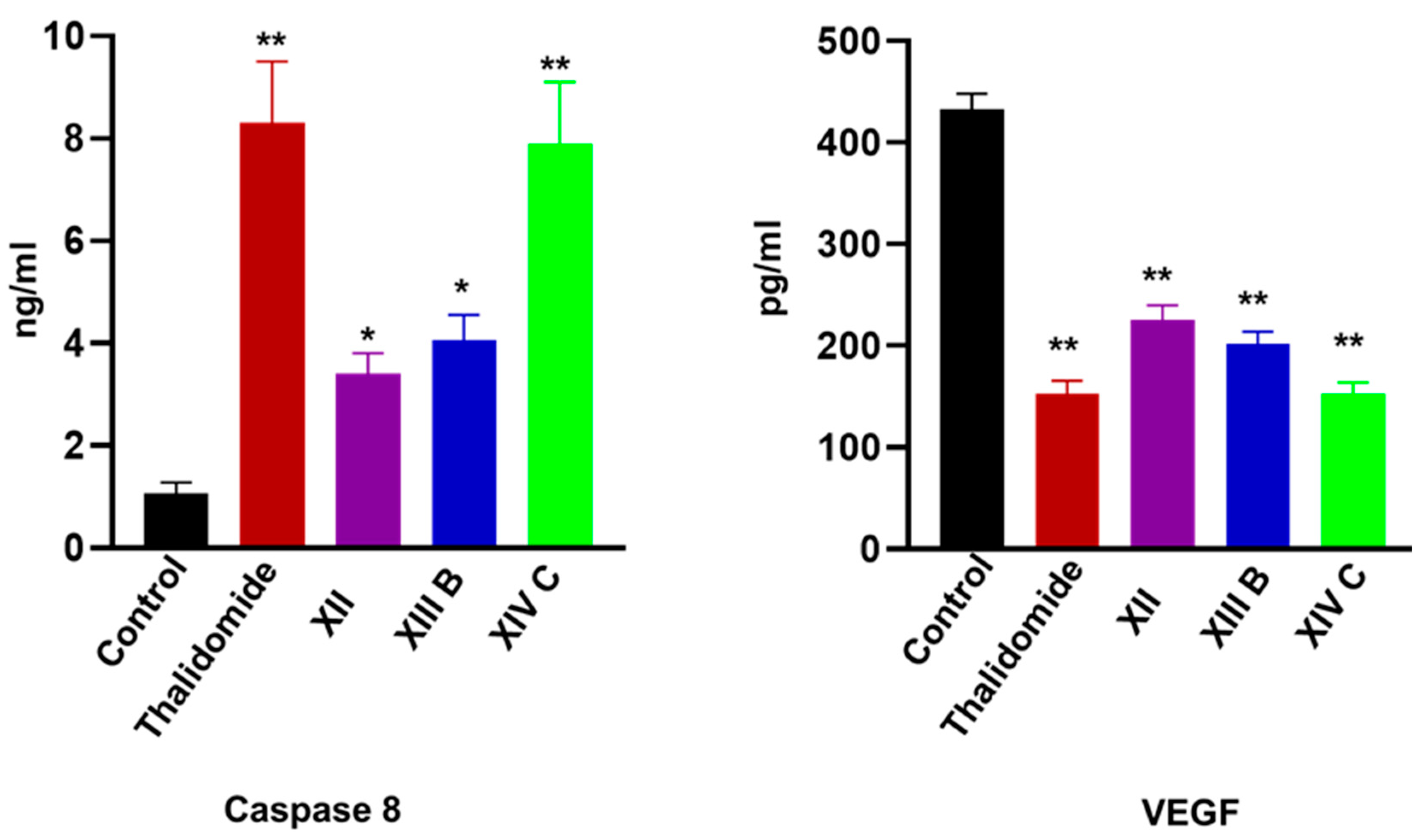
Estimation of Human Vascular Endothelial Growth Factor (VEGF) in HepG-2 Supernatant
Caspase-8 Activity Assay in HepG-2 Cell Lysate
Estimation of Nuclear Factor Kappa-B P65 (NF-κB P65) in HepG-2 Cell Lysate
The Effects on TNF-α Levels in HepG-2 Cells
Annexin V-FITC Apoptosis Assay
Cell Cycle Analysis
2.2.3. Structure–Activity Relationships (SAR)
3. Materials and Methods
3.1. Chemistry and Analysis
3.1.1. General Method for Synthesis of Compound III
3-((2-Chloroquinazolin-4-yl)amino)piperidine-2,6-dione III
3.1.2. General Procedure for Synthesis of Compounds VIIa-d
N′-(2-Chloroquinazolin-4-yl)benzohydrazide VIIa
N′-(2-Chloroquinazolin-4-yl)-2-hydroxybenzohydrazide VIIb
4-Chloro-N′-(2-chloroquinazolin-4-yl)benzohydrazide VIIc
2-Chloro-N′-(2-chloroquinazolin-4-yl)benzohydrazide VIId
3.1.3. 2,2′-(Quinazoline-2,4-diyl) bis(N-ethylhydrazine-1-carbothioamide) XI
3.1.4. 2-(2-Chloroquinazolin-4-yl)-N-phenylhydrazine-1-carboxamide XII
3.1.5. General Procedure for Synthesis of Compounds XIIIa-d
2-(2-Chloroquinazolin-4-yl)-N-propylhydrazine-1-carbothioamide XIIIa
N-Allyl-2-(2-chloroquinazolin-4-yl) hydrazine-1-carbothioamide XIIIb
2-(2-Chloroquinazolin-4-yl)-N-cyclohexylhydrazine-1-carbothioamide XIIIc
2-(2-Chloroquinazolin-4-yl)-N-phenylhydrazine-1-carbothioamide XIIId
3.1.6. General Procedure for Synthesis of Compounds XIVa-c
N′-(2-Chloroquinazolin-4-yl)benzenesulfonohydrazide XIVa
N′-(2-Chloroquinazolin-4-yl)-4-methylbenzenesulfonohydrazide XIVb
N′-(2-Chloroquinazolin-4-yl)-4-fluorobenzenesulfonohydrazide XIVc
3.1.7. Diethyl 1-(2-chloroquinazolin-4-yl)hydrazine-1,2-dicarboxylate XVI
3.1.8. General Procedure for Synthesis of Compound 4-[(2-Chloroquinazolin-4-yl)oxy]benzohydrazide XVIII
4-[(2-Chloroquinazolin-4-yl)oxy]benzohydrazide XVIII
2-(4-((2-Chloroquinazolin-4-yl)oxy)benzoyl)hydrazine-1-carboxamide XIX
4-[(2-Chloroquinazolin-4-yl)oxy]-N-(2,6-dioxopiperidin-3-yl)-benzamide XX
3.2. Biological Evaluation
3.2.1. In Vitro Antiproliferative Activities
3.2.2. In Vitro Protein Expression Assay
3.2.3. Apoptosis Assay
3.2.4. Cell Cycle Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dawoud, N.T.A.; El-Fakharany, E.M.; Abdallah, A.E.; El-Gendi, H.; Lotfy, D.R. Synthesis, and docking studies of novel heterocycles incorporating the indazolylthiazole moiety as antimicrobial and anticancer agents. Sci. Rep. 2022, 12, 3424. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Mabrouk, R.R.; Elnagar, M.R.; Farrag, A.M.; Kalaba, M.H.; Sharaf, M.H.; El-Fakharany, E.M.; Bakhotmah, D.A.; Elkaeed, E.B.; Al Ward, M.M.S. New Series of VEGFR-2 Inhibitors and Apoptosis Enhancers: Design, Synthesis and Biological Evaluation. Drug Des. Devel. Ther. 2022, 16, 587–606. [Google Scholar] [CrossRef]
- Abd-Elhamid, A.I.; El-Gendi, H.; Abdallah, A.E.; El-Fakharany, E.M. Novel nanocombinations of l-tryptophan and l-cysteine: Preparation, characterization, and their applications for antimicrobial and anticancer activities. Pharmaceutics 2021, 13, 1595. [Google Scholar] [CrossRef]
- Pecoraro, C.; Franco, M.D.; Carbone, D.; Bassani, D.; Pavan, M.; Cascioferro, S.; Parrino, B.; Cirrincione, C.; Acqua, S.D.; Moro, S.; et al. 1,2,4-Amino-triazine derivatives as pyruvate dehydrogenase kinase inhibitors: Synthesis and pharmacological evaluation, Eur. J. Med. Chem. 2023, 249, 115134. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Yousef, R.G.; Khalifa, M.M.; Ibrahim, A.; Mehany, A.B.M.; Gobaara, I.M.M.; Alsfouk, B.A.; Eldehna, W.M.; Metwaly, A.M.; Eissa, I.H.; et al. Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules 2022, 27, 6203. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Galustian, C.; Meyer, B.; Labarthe, M.C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef]
- Görgün, G.; Calabrese, E.; Soydan, E.; Hideshima, T.; Perrone, G.; Bandi, M.; Cirstea, D.; Santo, L.; Hu, Y.; Tai, Y.-T. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood, Am. J. Hematol. 2010, 116, 3227–3237. [Google Scholar]
- Anderson, K.C. Lenalidomide and thalidomide: Mechanisms of action—Similarities and differences. Semin. Hematol. 2005, 42, S3–S8. [Google Scholar] [CrossRef]
- Kumar, S.; Rajkumar, S.V. Thalidomide and lenalidomide in the treatment of multiple myeloma. Eur. J. Cancer 2006, 42, 1612–1622. [Google Scholar] [CrossRef]
- Lindner, S.; Krönke, J. The molecular mechanism of thalidomide analogs in hematologic malignancies. J. Mol. Med. 2016, 94, 1327–1334. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Reske, T.; Fulciniti, M.; Munshi, N.C. Mechanism of action of immunomodulatory agents in multiple myeloma. Med. Oncol. 2010, 27, 7–13. [Google Scholar] [CrossRef][Green Version]
- Millrine, D.; Kishimoto, T. A brighter side to thalidomide: Its potential use in immunological disorders. Trends Mol. Med. 2017, 23, 348–361. [Google Scholar] [CrossRef]
- Cortés-Hernández, J.; Torres-Salido, M.; Castro-Marrero, J.; Vilardell-Tarres, M.; Ordi-Ros, J. Thalidomide in the treatment of refractory cutaneous lupus erythematosus: Prognostic factors of clinical outcome. Br. J. Dermatol. 2012, 166, 616–623. [Google Scholar] [CrossRef]
- Chaulet, C.; Croix, C.; Alagille, D.; Normand, S.; Delwail, A.; Favot, L.; Lecron, J.-C.; Viaud-Massuard, M.-C. Design, synthesis and biological evaluation of new thalidomide analogues as TNF-α and IL-6 production inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1019–1022. [Google Scholar] [CrossRef]
- Elkady, H.; El-Adl, K.; Sakr, H.; Abdelraheem, A.S.; Eissa, S.I.; El-Zahabi, M.A. Novel promising benzoxazole/benzothiazole-derived immunomodulatory agents: Design, synthesis, anticancer evaluation, and in silico ADMET analysis. Arch. Pharm. 2023, 64, e2300097. [Google Scholar] [CrossRef]
- El-Zahabi, M. Design, molecular modeling and synthesis of new immunomodulatory agents for biological studies. Al-Azhar J. Pharm. Sci. 2021, 64, 1–20. [Google Scholar] [CrossRef]
- Sheskin, J. Thalidomide in the treatment of lepra reactions. Clin. Pharmacol. Ther. 1995, 6, 303–306. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef]
- Kotb, A.R.; Bakhotmah, D.A.; Abdallah, A.E.; Elkady, H.; Taghour, M.S.; Eissa, I.H.; El-Zahabi, M.A. Design, synthesis, and biological evaluation of novel bioactive thalidomide analogs as anticancer immunomodulatory agents. RSC Adv. 2022, 12, 33525–33539. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.B.; Dredge, K.; Dalgleish, A.G. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat. Rev. Cancer 2004, 4, 314–322. [Google Scholar] [CrossRef]
- El-Zahabi, M.A.; Elkady, H.; Sakr, H.; Abdelraheem, A.S.; Eissa, S.I.; El-Adl, K. Design, synthesis, anticancer evaluation, in silico docking and ADMET analysis of novel indole-based thalidomide analogs as promising immunomodulatory agents. J. Biomol. Struct. Dyn. 2023, 1–18. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Eissa, I.H.; Mehany, A.B.; Sakr, H.; Atwa, A.; El-Adl, K.; El-Zahabi, M. Immunomodulatory quinazoline-based thalidomide analogs: Design, synthesis, apoptosis and anticancer evaluations. J. Mol. Struct. 2023, 1281, 135164. [Google Scholar] [CrossRef]
- Abdel Razik, A.M.; El-Zahabi, M.A.; Sakr, T.M.; Eissa, I.H. Design, synthesis, and biological tracing of 99mTcDPDQA as a potential marker for tumor imaging. Al-Azhar J. Pharm. Sci. 2021, 63, 72–85. [Google Scholar]
- Hagner, P.R.; Man, H.-W.; Fontanillo, C.; Wang, M.; Couto, S.; Breider, M.; Bjorklund, C.; Havens, C.G.; Lu, G.; Rychak, E. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood Am. J. Hematol. 2015, 126, 779–789. [Google Scholar] [CrossRef]
- Hagner, P.; Wang, M.; Couto, S.; Breider, M.; Fontanillo, C.; Trotter, M.; Bjorklund, C.C.; Havens, C.G.; Raymon, H.K.; Narla, R.K. CC-122 has potent anti-lymphoma activity through destruction of the Aiolos and Ikaros transcription factors and induction of interferon response pathways. Blood 2014, 124, 3035. [Google Scholar] [CrossRef]
- Cho, N.-C.; Cha, J.H.; Kim, H.; Kwak, J.; Kim, D.; Seo, S.-H.; Shin, J.-S.; Kim, T.; Park, K.D.; Lee, J. Discovery of 2-aryloxy-4-amino-quinazoline derivatives as novel protease-activated receptor 2 (PAR2) antagonists. Bioorg. Med. Chem. 2015, 23, 7717–7727. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Y.; Dong, L.; Wang, Z.; Chen, L.; Liang, D.; Shi, D.; Shan, X.; Liang, G. Design, synthesis, and biological evaluation of novel quinazoline derivatives as anti-inflammatory agents against lipopolysaccharide-induced acute lung injury in rats. Chem. Biol. Drug Des. 2015, 85, 672–684. [Google Scholar] [CrossRef]
- Pu, Y.; Cao, D.; Xie, C.; Pei, H.; Li, D.; Tang, M.; Chen, L. Anti-arthritis effect of a novel quinazoline derivative through inhibiting production of TNF-α mediated by TNF-α converting enzyme in murine collagen-induced arthritis model. Biochem. Biophys. Res. Commun. 2015, 462, 288–293. [Google Scholar]
- Xu, L.; Russu, W.A. Molecular docking and synthesis of novel quinazoline analogues as inhibitors of transcription factors NF-κB activation and their anti-cancer activities. Bioorg. Med. Chem. 2013, 21, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.A.; Radov, L.A.; Trusso, L.A.; Georgiev, V.S. Synthesis and immunomodulating activity of 5H-thiazolo [2, 3-b] quinazolin-3 (2H)-one. J. Pharm. Sci. 1987, 76, 633–634. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.E.; Eissa, S.I.; Al Ward, M.M.S.; Mabrouk, R.R.; Mehany, A.B.; El-Zahabi, M.A. Design, synthesis and molecular modeling of new quinazolin-4 (3H)-one based VEGFR-2 kinase inhibitors for potential anticancer evaluation. Bioorg. Chem. 2021, 109, 104695. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Taghour, M.S.; Mahdy, H.A.; Eldehna, W.M.; El-Deeb, N.M.; Kenawy, A.M.; Alsfouk, B.A.; Dahab, M.A.; Metwaly, A.M.; Eissa, I.H. New quinoline and isatin derivatives as apoptotic VEGFR-2 inhibitors: Design, synthesis, anti-proliferative activity, docking, ADMET, toxicity, and MD simulation studies. J. Enzyme Inhib. Med. Chem. 2022, 37, 2191–2205. [Google Scholar] [CrossRef] [PubMed]
- Kotb, A.R.; Abdallah, A.E.; Elkady, H.; Eissa, I.H.; Taghour, M.S.; Bakhotmah, D.A.; Abdelghany, T.M.; El-Zahabi, M.A. Design, synthesis, anticancer evaluation, and in silico ADMET analysis of novel thalidomide analogs as promising immunomodulatory agents. RSC Adv. 2023, 13, 10488–10502. [Google Scholar] [CrossRef]
- Nasser, A.A.; Eissa, I.H.; Oun, M.R.; El-Zahabi, M.A.; Taghour, M.S.; Belal, A.; Saleh, A.M.; Mehany, A.B.; Luesch, H.; Mostafa, A.E. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M. Org. Biomol. Chem. 2020, 18, 7608–7634. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Mabrouk, R.R.; Al Ward, M.M.S.; Eissa, S.I.; Elkaeed, E.B.; Mehany, A.B.; Abo-Saif, M.A.; El-Feky, O.A.; Alesawy, M.S.; El-Zahabi, M.A. Synthesis, biological evaluation, and molecular docking of new series of antitumor and apoptosis inducers designed as VEGFR-2 inhibitors. J. Enzyme Inhib. Med. Chem. 2022, 37, 573–591. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; Maccoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Omar, A.M.; El-Zahabi, M.A.; Salah, G.; Abdel-Naim, A.B.; El-Araby, M.E. Substituted [1, 2, 4] Triazolo [4, 3-a] Pyrazines as Antidiabetics. U.S. Patent US 10,894,794 B1, 19 January 2021. [Google Scholar]
- Mahony, C.; Erskine, L.; Niven, J.; Greig, N.H.; Figg, W.D.; Vargesson, N. Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 12703–12708. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Chang, X.-b.; Stewart, A.K. What is the functional role of the thalidomide binding protein cereblonInt. J. Biochem. Mol. Biol. 2011, 2, 287. [Google Scholar]
- Smith, R.; Mitchell, S. Thalidomide-type teratogenicity: Structure–activity relationships for congeners. Toxicol. Res. 2018, 7, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Godin, A.M.; Araújo, D.P.; Menezes, R.R.; Brito, A.M.S.; Melo, I.S.; Coura, G.M.; Soares, D.G.; Bastos, L.F.; Amaral, F.A.; Ribeiro, L.S. Activities of 2-phthalimidethanol and 2-phthalimidethyl nitrate, phthalimide analogs devoid of the glutarimide moiety, in experimental models of inflammatory pain and edema. Pharmacol. Biochem. Behav. 2014, 122, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Zhai, X.; Zhao, Y.; Liu, Y.; Qi, B.; Tao, H.; Gong, P. Synthesis and biological evaluation of novel 2-(2-arylmethylene) hydrazinyl-4-aminoquinazoline derivatives as potent antitumor agents. Eur. J. Med. Chem. 2012, 54, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Bogert, M.T.; Scatchard, G. Researches on quinazolines XXXIII. A new and sensitive indicator for acidimetry and alkalimetry, and for the determination of hydrogen-ion concentrations between the limits of 6 and 8 on the sorensen scale. J. Am. Chem. Soc. 1916, 38, 1606–1615. [Google Scholar] [CrossRef][Green Version]
- Shah, D.; Lakum, H.; Chikhalia, K. Synthesis and in vitro antimicrobial evaluation of piperazine substituted quinazoline-based thiourea/thiazolidinone/chalcone hybrids. Russ. J. Bioorg. Chem. 2015, 41, 209–222. [Google Scholar]
- El-Zahabi, M.A.; Sakr, H.; El-Adl, K.; Zayed, M.; Abdelraheem, A.S.; Eissa, S.I.; Elkady, H.; Eissa, I.H. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg. Chem. 2020, 104, 104218. [Google Scholar] [CrossRef]
- Therese, S.K.; Geethamalika, G. Synthesis, Characterization and Anti Mycobacterial Activity of Novel Hydrazones. Orient. J. Chem. 2017, 33, 335–345. [Google Scholar] [CrossRef]
- Morgalyuk, V.; Azimov, V.; Bondarenko, V.; Denisov, A.; Yuzhakov, S.; Mashkovskii, M.; Yakhontov, L. Synthesis and cardiotonic activity of 6, 7-dimethoxyquinazoline derivatives. Pharm. Chem. J. 1991, 25, 15–21. [Google Scholar] [CrossRef]
- Prabhakar, V.; Babu, K.S.; Ravindranath, L.; Prasad, S.S.; Latha, J. Synthesis, Characterisation And Biological Evaluation Of Some Novel Quinazoline Derivatives As Potential Anti-Microbial Agents. Heterocycl. Lett. 2016, 6, 527–541. [Google Scholar]
- Kazemi, S.S.; Keivanloo, A.; Nasr-Isfahani, H.; Bamoniri, A. Synthesis of novel 1, 5-disubstituted pyrrolo [1, 2-a] quinazolines and their evaluation for anti-bacterial and anti-oxidant activities. RSC Adv. 2016, 6, 92663–92669. [Google Scholar] [CrossRef]
- El-Tombary, A.A.; Ismail, K.A.; Aboulwafa, O.M.; Omar, A.-M.M.; El-Azzouni, M.Z.; El-Mansoury, S.T. Novel triazolo [4, 3-a] quinazolinone and bis-triazolo [4, 3-a: 4, 3′-c] quinazolines: Synthesis and antitoxoplasmosis effect. Il Farm. 1999, 54, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.E.; Alesawy, M.S.; Eissa, S.I.; El-Fakharany, E.M.; Kalaba, M.H.; Sharaf, M.H.; Shama, N.M.A.; Mahmoud, S.H.; Mostafa, A.; Al-Karmalawy, A.A. Design and synthesis of new 4-(2-nitrophenoxy) benzamide derivatives as potential antiviral agents: Molecular modeling and in vitro antiviral screening. New J. Chem. 2021, 45, 16557–16571. [Google Scholar] [CrossRef]
- Elwan, A.; Abdallah, A.E.; Mahdy, H.A.; Dahab, M.A.; Taghour, M.S.; Elkaeed, E.B.; Mehany, A.B.; Nabeeh, A.; Adel, M.; Alsfouk, A.A. Modified benzoxazole-based VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, and anti-proliferative evaluation. Molecules 2022, 27, 5047. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Thabrew, M.I.; Hughes, R.D.; McFarlane, I.G. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J. Pharm. Pharmacol. 1997, 49, 1132–1135. [Google Scholar] [CrossRef]
- Paul, K.; Sharma, A.; Luxami, V. Synthesis and in vitro antitumor evaluation of primary amine substituted quinazoline linked benzimidazole. Bioorganic Med. Chem. Lett. 2014, 24, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Alesawy, M.S.; Al-Karmalawy, A.A.; Elkaeed, E.B.; Alswah, M.; Belal, A.; Taghour, M.S.; Eissa, I.H. Design and discovery of new 1, 2, 4-triazolo [4, 3-c] quinazolines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Pharm. 2021, 354, 2000237. [Google Scholar] [CrossRef]
- Zayed, M.F.; Hassan, M.H. Design, Synthesis and Biological Evaluation Studies of Novel Quinazoline Derivatives as Cytotoxic Agents. Drug Res. 2013, 63, 210–215. [Google Scholar] [CrossRef]
- Hannaford, A.; Furniss, B.; Rogers, V.; Smith, P.; Tatchell, A. Vogel’s Textbook of Practical Organic Chemistry: Including Qualitative Organic Analysis; Longman: Harlow, UK, 1978. [Google Scholar]
- Hangauer, D.; Al-Zahaby, M. Methodology for Preparing Combinatorial Libraries Based upon a Bicyclic Scaffold. WIPO (PCT) WO2005056523A3, 23 June 2005. [Google Scholar]
- Zayed, M.F.; Ahmed, S.; Ihmaid, S.; Ahmed, H.E.; Rateb, H.; Ibrahim, S. Design, Synthesis, Cytotoxic Evaluation and Molecular Docking of New Fluoroquinazolinones as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Int. J. Mol. Sci. 2018, 19, 1731. [Google Scholar] [CrossRef] [PubMed]
- Talaat, R.; El-Sayed, W.; Agwa, H.; Gamal-Eldeen, A.; Moawia, S.; Zahran, M. Novel thalidomide analogs: Anti-angiogenic and apoptotic effects on Hep-G2 and MCF-7 cancer cell lines. Biomed. Aging Pathol. 2014, 4, 179–189. [Google Scholar] [CrossRef]
- Inoue, T.; Kibata, K.; Suzuki, M.; Nakamura, S.; Motoda, R.; Orita, K. Identification of a vascular endothelial growth factor (VEGF) antagonist, sFlt-1, from a human hematopoietic cell line NALM-16. FEBS Lett. 2000, 469, 14–18. [Google Scholar] [CrossRef]
- Talaat, R.M. Soluble angiogenesis factors in sera of Egyptian patients with hepatitis C virus infection: Correlation with disease severity. Viral Immunol. 2010, 23, 151–157. [Google Scholar] [CrossRef]
- Lo, K.K.-W.; Lee, T.K.-M.; Lau, J.S.-Y.; Poon, W.-L.; Cheng, S.-H. Luminescent biological probes derived from ruthenium (II) estradiol polypyridine complexes. Inorg. Chem. 2008, 47, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lenardo, M.J. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J. Cell Sci. 2000, 113, 753–757. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | In Vitro Cytotoxicity IC50 (µg/mL) a,b | ||
|---|---|---|---|
| HepG-2 | PC3 | MCF-7 | |
| III | 9.73 ± 0.66 | 13.52 ± 0.28 | 23.14 ± 0.4 |
| VIIa | 16.16 ± 0.67 | 11.23 ± 0.54 | 8.82 ± 0.45 |
| VIIb | 12.72 ± 0.635 | 15.45 ± 0.69 | 11.29 ± 0.55 |
| VIIc | 20.35 ± 0.73 | 25.47 ± 0.79 | 30.61 ± 0.89 |
| VIId | 16.31 ± 0.69 | 19.54 ± 0.72 | 12.78 ± 0.58 |
| XI | 13.90 ± 0.87 | 12.24 ± 0.83 | 6.18 ± 0.5 |
| XII | 4.57 ± 0.12 | 5.82 ± 0.15 | 3.61 ± 0.1 |
| XIIIa | 2.03 ± 0.11 | 2.51 ± 0.2 | 0.82 ± 0.02 |
| XIIIb | 7.83 ± 0.46 | 9.36 ± 0.47 | 6.89 ± 0.24 |
| XIIIc | 9.72 ± 0.54 | 10.12 ± 0.51 | 7.19 ± 0.43 |
| XIIId | 11.24 ± 0.55 | 13.39 ± 0.64 | 10.24 ± 0.53 |
| XIVa | 6.76 ± 0.23 | 7.67 ± 0.44 | 4.81 ± 0.13 |
| XIVb | 3.41 ± 0.1 | 6.73 ± 0.23 | 2.8 ± 0.06 |
| XIVc | 2.14 ± 0.05 | 4.98 ± 0.15 | 5.57 ± 0.16 |
| XVI | 21.64 ± 0.75 | 31.01 ± 0.81 | 20.16 ± 0.74 |
| XIX | 21.32 ± 0.74 | 36.49 ± 1.25 | 21.98 ± 0.78 |
| XX | 16.62 ± 0.7 | 20.23 ± 0.73 | 13.45 ± 0.67 |
| Thalidomide | 11.26 ± 0.54 | 14.58 ± 0.57 | 16.87 ± 0.7 |
| Comp. No. | Caspase-8 (ng/mL) | VEGF (pg/mL) | NFκB P65 (pg/mL) | TNF-α (pg/mL) |
|---|---|---|---|---|
| XII | 3.4 ± 0.4 * | 225.1 ± 17.2 ** | 121.3 ± 14.6 ** | 76.4 ±14.6 ** |
| XIIIb | 4.05 ±0.5 * | 201.5 ± 19.5 ** | 118.5 ± 18.3 ** | 93.2 ± 12.5 * |
| XIVc | 7.9 ± 1.2 ** | 152.6 ± 14.5 ** | 63.1 ± 11.05 ** | 53.4 ± 11.2 ** |
| Control | 1.08 ± 0.2 | 432.5 ± 25.5 | 278.1 ± 18.5 | 162.5 ± 15.5 |
| Thalidomide | 8.3 ±1.2 ** | 153.2 ± 13.4 ** | 110.5 ± 13.2 ** | 53.1 ±12.5 ** |
| Compound | Normal Cell | Early Apoptosis | Late Apoptosis | Total Apoptosis | Necrosis |
|---|---|---|---|---|---|
| Control | 98.61 ± 5.7 | 1.19 ± 0.7 | 0.18 ± 0.01 | 1.37 ± 0.09 | 0.02 ± 0.0 |
| Thalidomide | 95.98 ± 12.3 | 3.76 ± 0.9 | 0.26 ± 0.017 | 4.02 ± 0.1 | 0.00 ± 0.0 |
| XIVc | 92.08 ± 11.23 | 7.75 ± 1.2 * | 0.15 ± 0.03 | 7.90 ± 0.1 | 0.02 ± 0.0 |
| Compound | G0/G1 | S | G2/M |
|---|---|---|---|
| Control | 10.05 | 59.55 | 29.73 |
| Thalidomide | 8.07 | 66.38 | 25.54 |
| XIVc | 10.64 | 65.05 | 24.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabrouk, R.R.; Abdallah, A.E.; Mahdy, H.A.; El-Kalyoubi, S.A.; Kamal, O.J.; Abdelghany, T.M.; Zayed, M.F.; Alshaeri, H.K.; Alasmari, M.M.; El-Zahabi, M.A. Design, Synthesis, and Biological Evaluation of New Potential Unusual Modified Anticancer Immunomodulators for Possible Non-Teratogenic Quinazoline-Based Thalidomide Analogs. Int. J. Mol. Sci. 2023, 24, 12416. https://doi.org/10.3390/ijms241512416
Mabrouk RR, Abdallah AE, Mahdy HA, El-Kalyoubi SA, Kamal OJ, Abdelghany TM, Zayed MF, Alshaeri HK, Alasmari MM, El-Zahabi MA. Design, Synthesis, and Biological Evaluation of New Potential Unusual Modified Anticancer Immunomodulators for Possible Non-Teratogenic Quinazoline-Based Thalidomide Analogs. International Journal of Molecular Sciences. 2023; 24(15):12416. https://doi.org/10.3390/ijms241512416
Chicago/Turabian StyleMabrouk, Reda R., Abdallah E. Abdallah, Hazem A. Mahdy, Samar A. El-Kalyoubi, Omar Jamal Kamal, Tamer M. Abdelghany, Mohamed F. Zayed, Heba K. Alshaeri, Moudi M. Alasmari, and Mohamed Ayman El-Zahabi. 2023. "Design, Synthesis, and Biological Evaluation of New Potential Unusual Modified Anticancer Immunomodulators for Possible Non-Teratogenic Quinazoline-Based Thalidomide Analogs" International Journal of Molecular Sciences 24, no. 15: 12416. https://doi.org/10.3390/ijms241512416
APA StyleMabrouk, R. R., Abdallah, A. E., Mahdy, H. A., El-Kalyoubi, S. A., Kamal, O. J., Abdelghany, T. M., Zayed, M. F., Alshaeri, H. K., Alasmari, M. M., & El-Zahabi, M. A. (2023). Design, Synthesis, and Biological Evaluation of New Potential Unusual Modified Anticancer Immunomodulators for Possible Non-Teratogenic Quinazoline-Based Thalidomide Analogs. International Journal of Molecular Sciences, 24(15), 12416. https://doi.org/10.3390/ijms241512416