
Prostaglandin E2 (PGE2) and Roflumilast Involvement in IPF Progression
Abstract
1. Introduction
2. Results
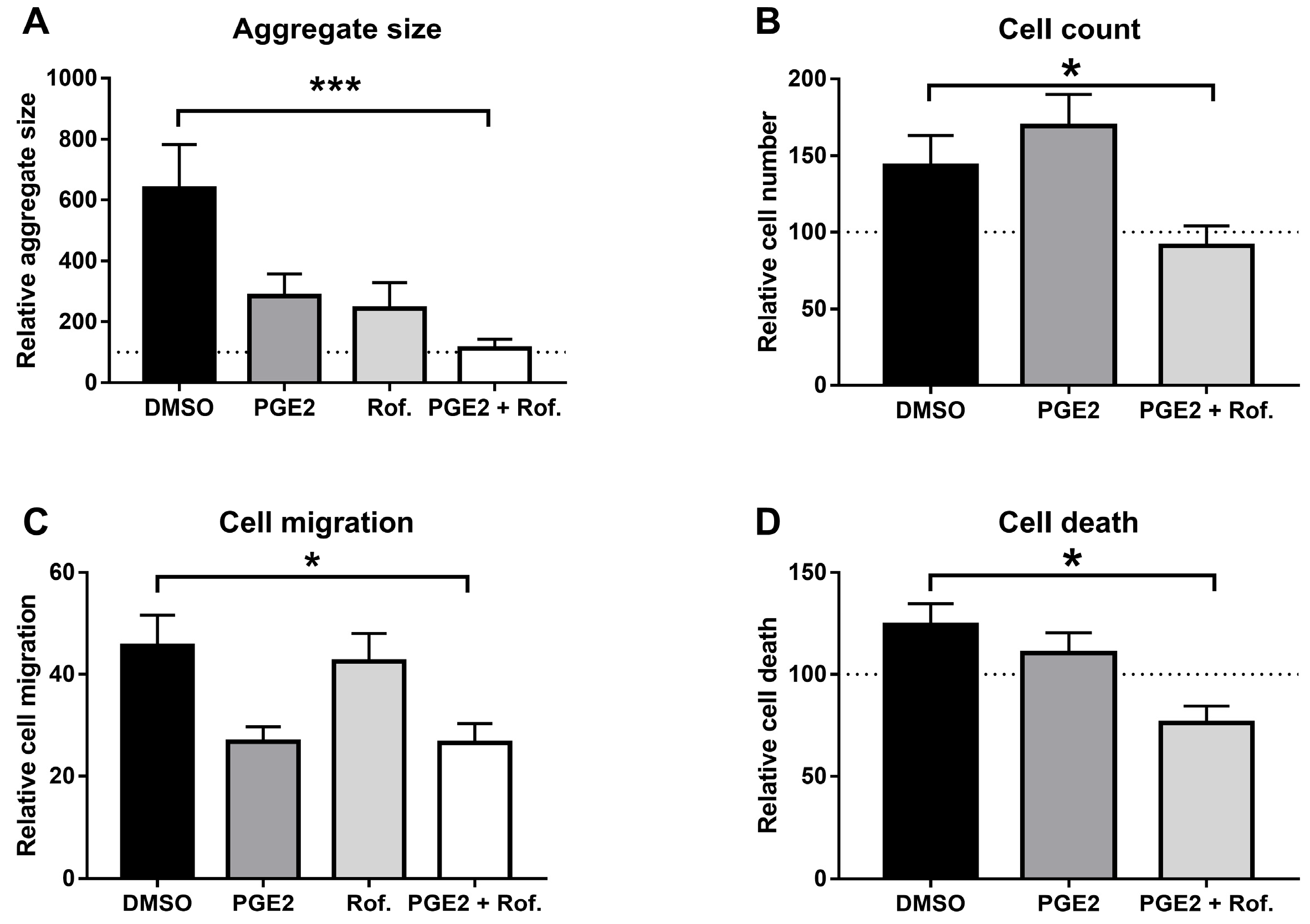
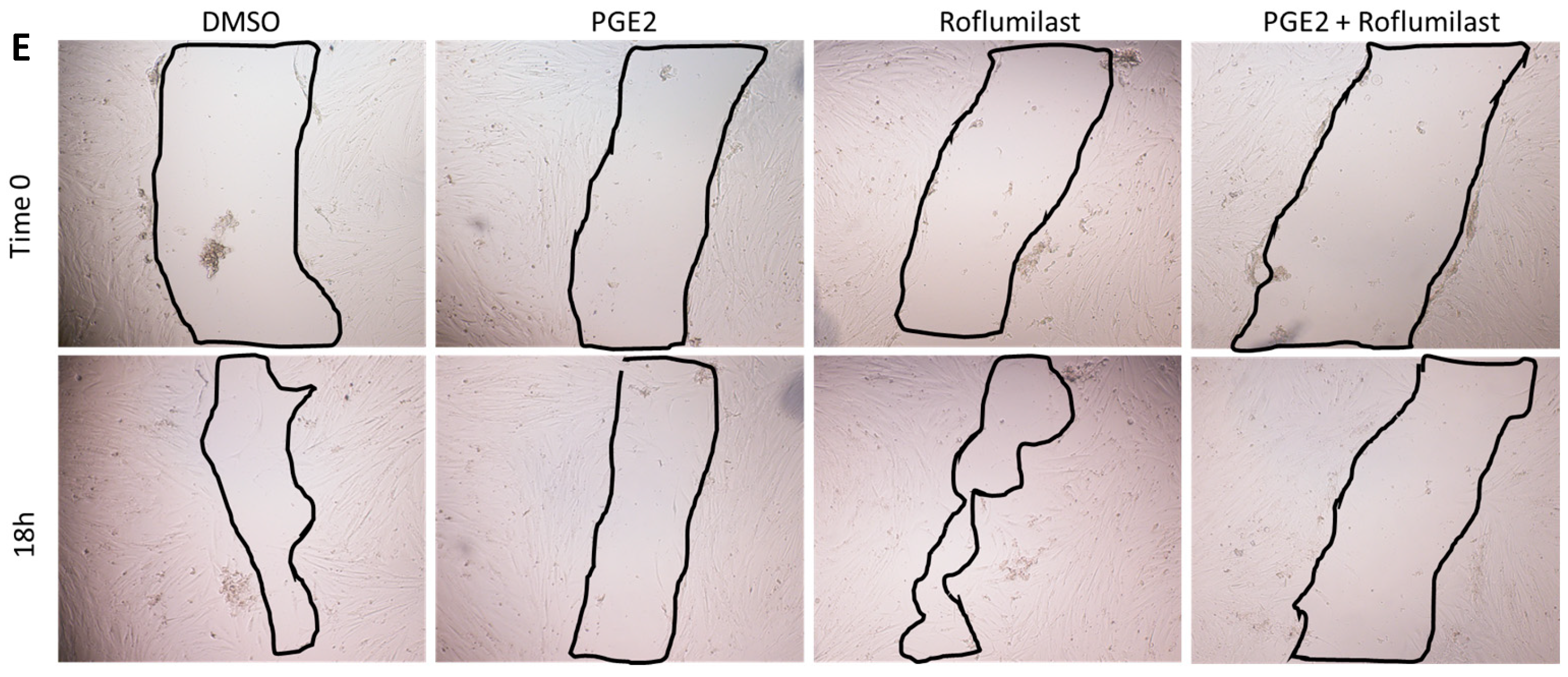
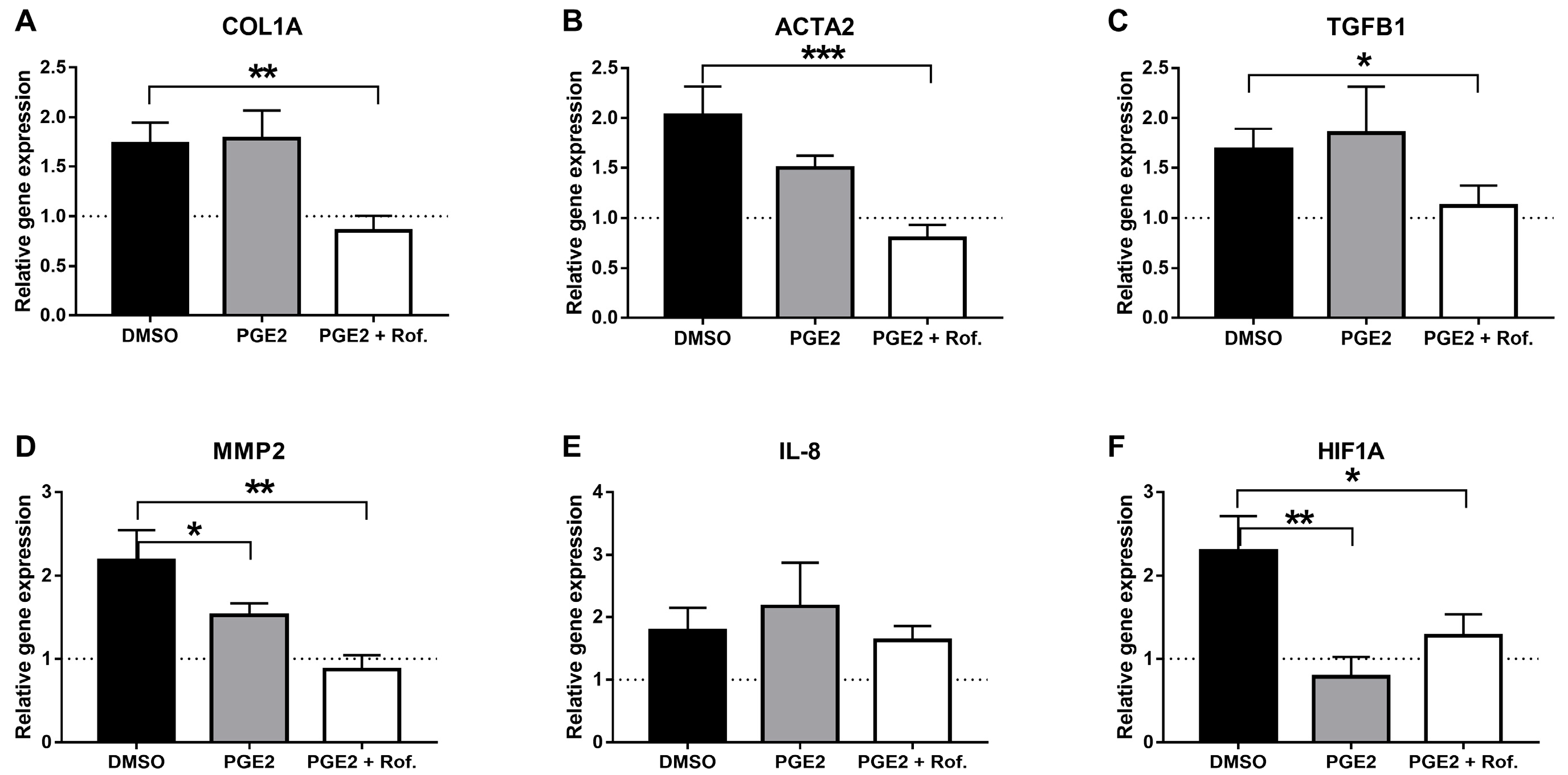
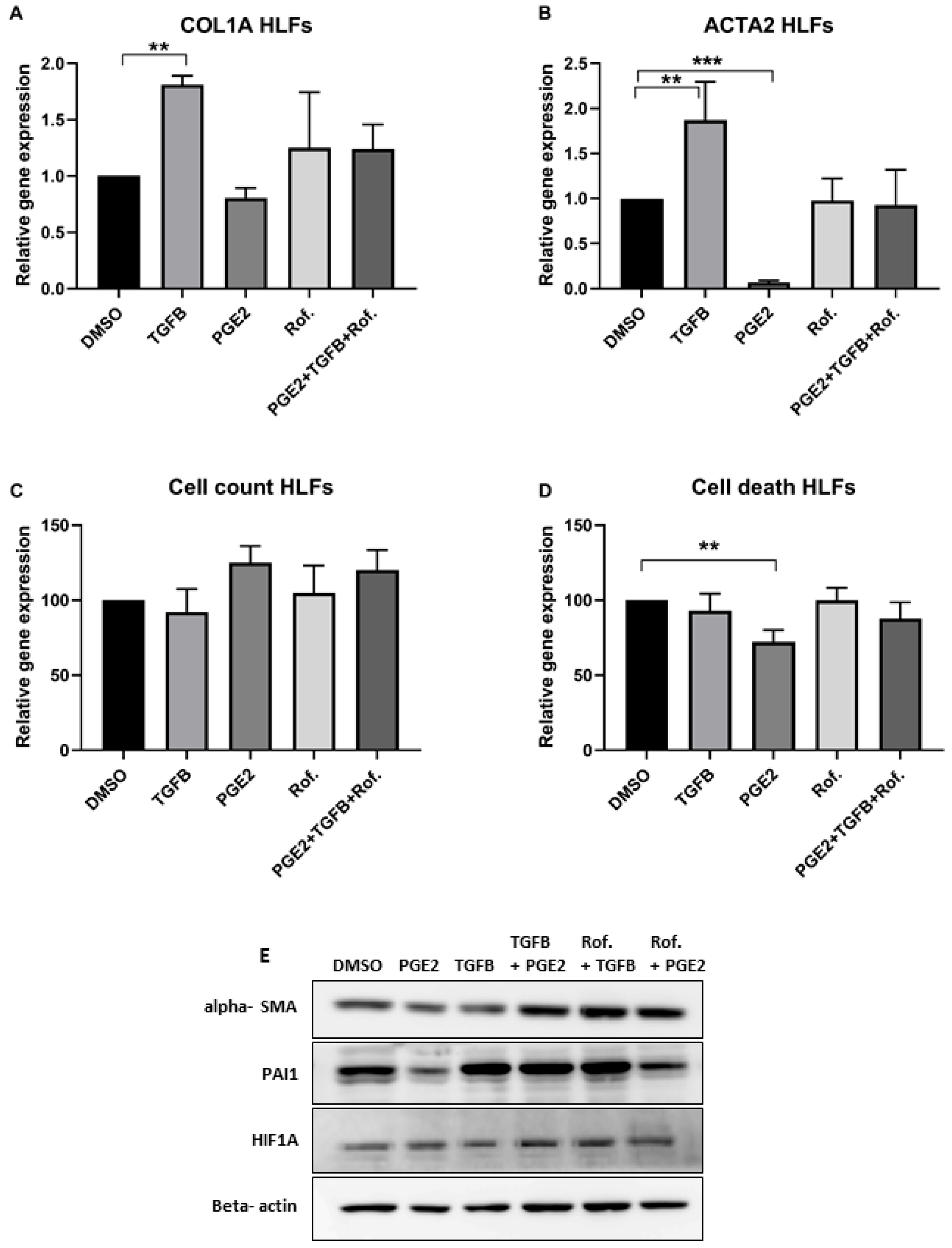
2.1. PGE2 in Combination with Roflumilast Inhibits IPF-CMs’ Pro-Fibrotic Effects on N-HLFs
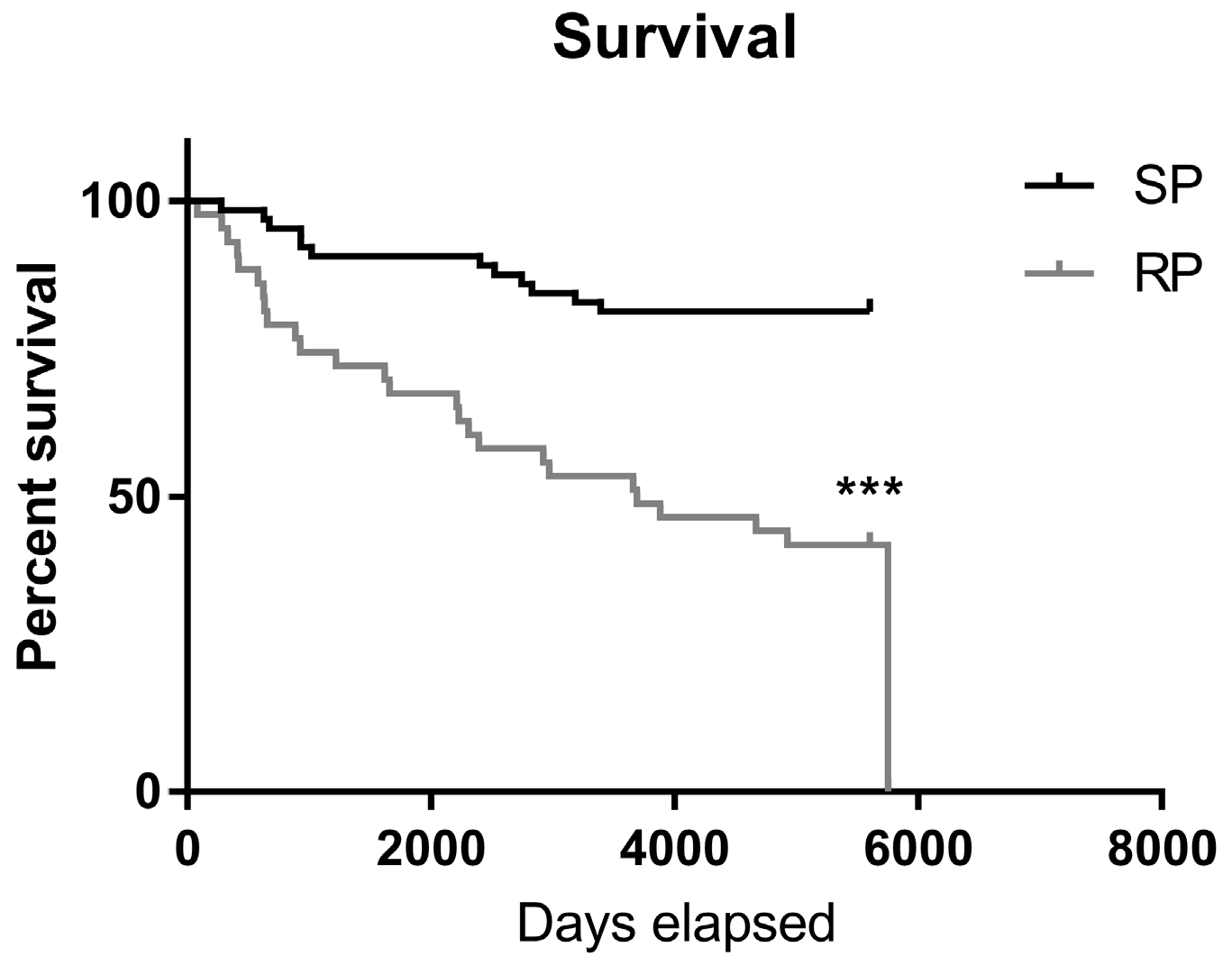
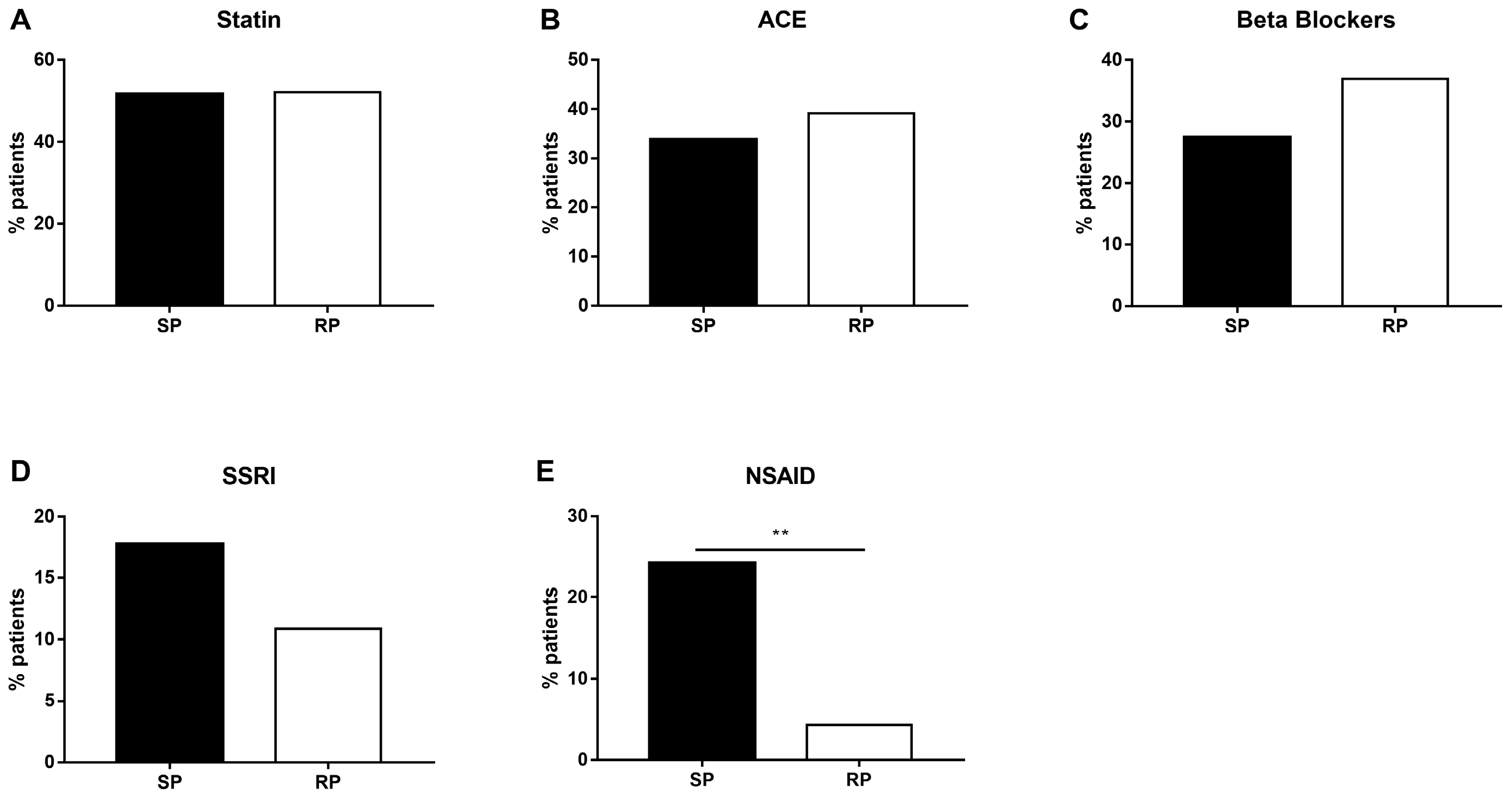
2.2. Slow-Progressing IPF Patients Were More Exposed to NSAIDs
3. Discussion
4. Materials and Methods
4.1. IPF-CM Model
4.2. Aggregate Size Measurement
4.3. Real-Time Quantitative PCR
4.4. Cell Migration
4.5. Cell Death
4.6. Western Blot
4.7. Electronic Record Retrospective Study Population
4.8. Data Collection
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Carrillo, G.; Estrada, A.; Mejia, M.; Becerril, C.; Cisneros, J.; Gaxiola, M.; Perez-Padilla, R.; Navarro, C.; Richards, T.; et al. Accelerated variant of idiopathic pulmonary fibrosis: Clinical behavior and gene expression pattern. PLoS ONE 2007, 2, e482. [Google Scholar] [CrossRef] [PubMed]
- McCormack, F.X.; King, T.E., Jr.; Bucher, B.L.; Nielsen, L.; Mason, R.J. Surfactant protein A predicts survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1995, 152, 751–759. [Google Scholar] [CrossRef]
- Martinez, F.J.; Safrin, S.; Weycker, D.; Starko, K.M.; Bradford, W.Z.; King, T.E., Jr.; Flaherty, K.R.; Schwartz, D.A.; Noble, P.W.; Raghu, G.; et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann. Intern. Med. 2005, 142, 963–967. [Google Scholar] [CrossRef]
- Ambrosini, V.; Cancellieri, A.; Chilosi, M.; Zompatori, M.; Trisolini, R.; Saragoni, L.; Poletti, V. Acute exacerbation of idiopathic pulmonary fibrosis: Report of a series. Eur. Respir. J. 2003, 22, 821–826. [Google Scholar] [CrossRef]
- Trujillo, G.; Meneghin, A.; Flaherty, K.R.; Sholl, L.M.; Myers, J.L.; Kazerooni, E.A.; Gross, B.H.; Oak, S.R.; Coelho, A.L.; Evanoff, H.; et al. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci. Transl. Med. 2010, 2, 57–82. [Google Scholar] [CrossRef]
- Balestro, E.; Calabrese, F.; Turato, G.; Lunardi, F.; Bazzan, E.; Marulli, G.; Biondini, D.; Rossi, E.; Sanduzzi, A.; Rea, F.; et al. Immune Inflammation and Disease Progression in Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0154516. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Azuma, A.; Nukiwa, T.; Tsuboi, E.; Suga, M.; Abe, S.; Nakata, K.; Taguchi, Y.; Nagai, S.; Itoh, H.; Ohi, M.; et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1040–1047. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Comment on “Idiopathic Pulmonary Fibrosis”. N. Engl. J. Med. 2018, 379, 797–798. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Wollin, L.; Shitrit, D. Fibroblast-matrix interplay: Nintedanib and pirfenidone modulate the effect of IPF fibroblast-conditioned matrix on normal fibroblast phenotype. Respirology 2018, 23, 756–763. [Google Scholar] [CrossRef]
- Epstein-Shochet, G.; Pham, S.; Beck, S.; Naiel, S.; Mekhael, O.; Revill, S.; Hayat, A.; Vierhout, M.; Bardestein-Wald, B.; Shitrit, D.; et al. Inhalation: A means to explore and optimize nintedanib’s pharmacokinetic/pharmacodynamic relationship. Pulm. Pharmacol. Ther. 2020, 63, 101933. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Bardenstein-Wald, B.; McElroy, M.; Kukuy, A.; Surber, M.; Edelstein, E.; Pertzov, B.; Kramer, M.R.; Shitrit, D. Hypoxia Inducible Factor 1A Supports a Pro-Fibrotic Phenotype Loop in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 3331. [Google Scholar] [CrossRef]
- Kawano, T.; Anrather, J.; Zhou, P.; Park, L.; Wang, G.; Frys, K.A.; Kunz, A.; Cho, S.; Orio, M.; Iadecola, C. Prostaglandin E2 EP1 receptors: Downstream effectors of COX-2 neurotoxicity. Nat. Med. 2006, 12, 225–229. [Google Scholar] [CrossRef]
- Bozyk, P.D.; Moore, B.B. Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 445–452. [Google Scholar] [CrossRef]
- Rennard, S.I.; Calverley, P.M.; Goehring, U.M.; Bredenbroker, D.; Martinez, F.J. Reduction of exacerbations by the PDE4 inhibitor roflumilast--the importance of defining different subsets of patients with COPD. Respir. Res. 2011, 12, 18. [Google Scholar] [CrossRef]
- Togo, S.; Liu, X.; Wang, X.; Sugiura, H.; Kamio, K.; Kawasaki, S.; Kobayashi, T.; Ertl, R.F.; Ahn, Y.; Holz, O.; et al. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-{beta}1-stimulated fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L959–L969. [Google Scholar] [CrossRef] [PubMed]
- Hatzelmann, A.; Morcillo, E.J.; Lungarella, G.; Adnot, S.; Sanjar, S.; Beume, R.; Schudt, C.; Tenor, H. The preclinical pharmacology of roflumilast—A selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2010, 23, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Cortijo, J.; Iranzo, A.; Milara, X.; Mata, M.; Cerda-Nicolas, M.; Ruiz-Sauri, A.; Tenor, H.; Hatzelmann, A.; Morcillo, E.J. Roflumilast, a phosphodiesterase 4 inhibitor, alleviates bleomycin-induced lung injury. Br. J. Pharmacol. 2009, 156, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Schick, M.A.; Schlegel, N. Clinical Implication of Phosphodiesterase-4-Inhibition. Int. J. Mol. Sci. 2022, 23, 1209. [Google Scholar] [CrossRef] [PubMed]
- Brunnemer, E.; Walscher, J.; Tenenbaum, S.; Hausmanns, J.; Schulze, K.; Seiter, M.; Heussel, C.P.; Warth, A.; Herth, F.J.F.; Kreuter, M. Real-World Experience with Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Respir. Int. Rev. Thorac. Dis. 2018, 95, 301–309. [Google Scholar] [CrossRef]
- Kolodsick, J.E.; Peters-Golden, M.; Larios, J.; Toews, G.B.; Thannickal, V.J.; Moore, B.B. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am. J. Respir. Cell Mol. Biol. 2003, 29, 537–544. [Google Scholar] [CrossRef]
- McAnulty, R.J.; Hernandez-Rodriguez, N.A.; Mutsaers, S.E.; Coker, R.K.; Laurent, G.J. Indomethacin suppresses the anti-proliferative effects of transforming growth factor-beta isoforms on fibroblast cell cultures. Biochem. J. 1997, 321 Pt 3, 639–643. [Google Scholar] [CrossRef]
- Thomas, P.E.; Peters-Golden, M.; White, E.S.; Thannickal, V.J.; Moore, B.B. PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L417–L428. [Google Scholar] [CrossRef]
- Moore, B.B.; Ballinger, M.N.; White, E.S.; Green, M.E.; Herrygers, A.B.; Wilke, C.A.; Toews, G.B.; Peters-Golden, M. Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J. Immunol. 2005, 174, 5644–5649. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-beta pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020, 21, 56. [Google Scholar] [CrossRef]
- Ramirez-Yanez, G.O.; Hamlet, S.; Jonarta, A.; Seymour, G.J.; Symons, A.L. Prostaglandin E2 enhances transforming growth factor-beta 1 and TGF-beta receptors synthesis: An in vivo and in vitro study. Prostaglandins Leukot. Essent. Fat. Acids 2006, 74, 183–192. [Google Scholar] [CrossRef]
- Hui, A.Y.; Dannenberg, A.J.; Sung, J.J.; Subbaramaiah, K.; Du, B.; Olinga, P.; Friedman, S.L. Prostaglandin E2 inhibits transforming growth factor beta 1-mediated induction of collagen alpha 1(I) in hepatic stellate cells. J. Hepatol. 2004, 41, 251–258. [Google Scholar] [CrossRef]
- Boak, A.M.; Roy, R.; Berk, J.; Taylor, L.; Polgar, P.; Goldstein, R.H.; Kagan, H.M. Regulation of lysyl oxidase expression in lung fibroblasts by transforming growth factor-beta 1 and prostaglandin E2. Am. J. Respir. Cell Mol. Biol. 1994, 11, 751–755. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sheng, W.; Michkov, A.; Sriarm, K.; Sun, R.; Dvorkin-Gheva, A.; Insel, P.A.; Janssen, L.J. Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca2+ signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L810–L821. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.K.; Wettlaufer, S.H.; Hogaboam, C.M.; Flaherty, K.R.; Martinez, F.J.; Myers, J.L.; Colby, T.V.; Travis, W.D.; Toews, G.B.; Peters-Golden, M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2008, 177, 66–74. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Ding, Q.; Moore, B.B.; Peters-Golden, M.; Ware, L.B.; Matthay, M.A.; Olman, M.A. Prostaglandin E2 mediates IL-1beta-related fibroblast mitogenic effects in acute lung injury through differential utilization of prostanoid receptors. J. Immunol. 2008, 180, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Berhan, A.; Harris, T.; Jaffar, J.; Jativa, F.; Langenbach, S.; Lonnstedt, I.; Alhamdoosh, M.; Ng, M.; Lee, P.; Westall, G.; et al. Cellular Microenvironment Stiffness Regulates Eicosanoid Production and Signaling Pathways. Am. J. Respir. Cell Mol. Biol. 2020, 63, 819–830. [Google Scholar] [CrossRef]
- Sisson, T.H.; Christensen, P.J.; Muraki, Y.; Dils, A.J.; Chibucos, L.; Subbotina, N.; Tohyama, K.; Horowitz, J.C.; Matsuo, T.; Bailie, M.; et al. Phosphodiesterase 4 inhibition reduces lung fibrosis following targeted type II alveolar epithelial cell injury. Physiol. Rep. 2018, 6, e13753. [Google Scholar] [CrossRef]
- McDonough, J.E.; Ahangari, F.; Li, Q.; Jain, S.; Verleden, S.E.; Herazo-Maya, J.; Vukmirovic, M.; DeIuliis, G.; Tzouvelekis, A.; Tanabe, N.; et al. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight 2019, 4, e131597. [Google Scholar] [CrossRef]
- Sabatini, F.; Petecchia, L.; Boero, S.; Silvestri, M.; Klar, J.; Tenor, H.; Beume, R.; Hatzelmann, A.; Rossi, G.A. A phosphodiesterase 4 inhibitor, roflumilast N-oxide, inhibits human lung fibroblast functions in vitro. Pulm. Pharmacol. Ther. 2010, 23, 283–291. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Brook, E.; Israeli-Shani, L.; Edelstein, E.; Shitrit, D. Fibroblast paracrine TNF-alpha signaling elevates integrin A5 expression in idiopathic pulmonary fibrosis (IPF). Respir. Res. 2017, 18, 122. [Google Scholar] [CrossRef]
- Lugnier, C. The Complexity and Multiplicity of the Specific cAMP Phosphodiesterase Family: PDE4, Open New Adapted Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 10616. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef]
- Moon, J.O.; Welch, T.P.; Gonzalez, F.J.; Copple, B.L. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G582–G592. [Google Scholar] [CrossRef]
- Ivanova, V.; Garbuzenko, O.B.; Reuhl, K.R.; Reimer, D.C.; Pozharov, V.P.; Minko, T. Inhalation treatment of pulmonary fibrosis by liposomal prostaglandin E2. Eur. J. Pharm. Biopharm. 2013, 84, 335–344. [Google Scholar] [CrossRef]
- Nio, Y.; Ookawara, M.; Yamasaki, M.; Hanauer, G.; Tohyama, K.; Shibata, S.; Sano, T.; Shimizu, F.; Anayama, H.; Hazama, M.; et al. Ameliorative effect of phosphodiesterase 4 and 5 inhibitors in deoxycorticosterone acetate-salt hypertensive uni-nephrectomized KKA(y) mice. FASEB J. 2020, 34, 14997–15014. [Google Scholar] [CrossRef]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Molteni, A.; Wolfe, L.F.; Ward, W.F.; Ts’ao, C.H.; Molteni, L.B.; Veno, P.; Fish, B.L.; Taylor, J.M.; Quintanilla, N.; Herndon, B.; et al. Effect of an angiotensin II receptor blocker and two angiotensin converting enzyme inhibitors on transforming growth factor-beta (TGF-beta) and alpha-actomyosin (alpha SMA), important mediators of radiation-induced pneumopathy and lung fibrosis. Curr. Pharm. Des. 2007, 13, 1307–1316. [Google Scholar] [CrossRef]
- Lambert, E.M.; Wuyts, W.A.; Yserbyt, J.; De Sadeleer, L.J. Statins: Cause of fibrosis or the opposite? Effect of cardiovascular drugs in idiopathic pulmonary fibrosis. Respir. Med. 2021, 176, 106259. [Google Scholar] [CrossRef]
- Rosenberg, T.; Lattimer, R.; Montgomery, P.; Wiens, C.; Levy, L. The relationship of SSRI and SNRI usage with interstitial lung disease and bronchiectasis in an elderly population: A case-control study. Clin. Interv. Aging 2017, 12, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.C.; Pini, A.; Lucarini, L.; Lanzi, C.; Veglia, E.; Thurmond, R.L.; Stark, H.; Masini, E. Prevention of bleomycin-induced lung inflammation and fibrosis in mice by naproxen and JNJ7777120 treatment. J. Pharmacol. Exp. Ther. 2014, 351, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Kankuri, E.; Cholujova, D.; Comajova, M.; Vaheri, A.; Bizik, J. Induction of hepatocyte growth factor/scatter factor by fibroblast clustering directly promotes tumor cell invasiveness. Cancer Res. 2005, 65, 9914–9922. [Google Scholar] [CrossRef]
- Bizik, J.; Kankuri, E.; Ristimaki, A.; Taieb, A.; Vapaatalo, H.; Lubitz, W.; Vaheri, A. Cell-cell contacts trigger programmed necrosis and induce cyclooxygenase-2 expression. Cell Death Differ. 2004, 11, 183–195. [Google Scholar] [CrossRef]
- Lippman, S.M.; Gibson, N.; Subbaramaiah, K.; Dannenberg, A.J. Combined targeting of the epidermal growth factor receptor and cyclooxygenase-2 pathways. Clin. Cancer Res. 2005, 11, 6097–6099. [Google Scholar] [CrossRef]
- Gilligan, M.M.; Gartung, A.; Sulciner, M.L.; Norris, P.C.; Sukhatme, V.P.; Bielenberg, D.R.; Huang, S.; Kieran, M.W.; Serhan, C.N.; Panigrahy, D. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6292–6297. [Google Scholar] [CrossRef]
- Lucotti, S.; Cerutti, C.; Soyer, M.; Gil-Bernabe, A.M.; Gomes, A.L.; Allen, P.D.; Smart, S.; Markelc, B.; Watson, K.; Armstrong, P.C.; et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J. Clin. Investig. 2019, 129, 1845–1862. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Slow Progression N = 62 | Rapid Progression N = 45 | p-Value |
|---|---|---|---|
| Age, years | 65.7 ± 12.3 | 66.3 ± 10.3 | 0.39 |
| Gender (% male) | 56% | 67% | 0.02 |
| Smoking history | 77% | 78% | 0.79 |
| Cardiovascular disease | 27% | 24% | 0.41 |
| Renal failure | 2% | 9% | <0.001 |
| Malignancy | 25% | 0 | <0.001 |
| Mortality | 19% | 54% | <0.001 |
| Parameter | Slow Progression N = 62 | Rapid Progression N = 45 | p-Value |
|---|---|---|---|
| FVC, L | 2.34 ± 0.82 | 1.85 ± 0.75 | 0.002 |
| FVC, % predicted | 77.51 ± 21 | 59.46 ± 17.9 | 0.001 |
| FRC PL % predicted | 79.24 ± 20.1 | 62.6 ± 15.4 | <0.001 |
| FRC, L | 2.46 ± 0.66 | 1.98 ± 0.57 | 0.001 |
| TLC, L | 4.16 ± 1.11 | 3.22 ± 0.88 | <0.001 |
| TLC, % predicted | 72.63 ± 16.6 | 57.23 ± 11.5 | <0.001 |
| DLCO, % predicted | 54.51 ± 15 | 42.27 ± 13.2 | <0.001 |
| 6 min walk test | |||
| SAT rest, % | 96.5 ± 2 | 95.13 ± 2.3 | 0.017 |
| SAT exercise, % | 90.90 ± 5.3 | 86.17 ± 4.8 | 0.001 |
| HR rest | 78.31 ± 10.3 | 78.08 ± 12.6 | 0.940 |
| HR exercise | 110.2 ± 18 | 107.6 ± 20 | 0.619 |
| Distance, m | 384.4 ± 150.4 | 307 ± 153.9 | 0.053 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moshkovitz, N.; Epstein Shochet, G.; Shitrit, D. Prostaglandin E2 (PGE2) and Roflumilast Involvement in IPF Progression. Int. J. Mol. Sci. 2023, 24, 12393. https://doi.org/10.3390/ijms241512393
Moshkovitz N, Epstein Shochet G, Shitrit D. Prostaglandin E2 (PGE2) and Roflumilast Involvement in IPF Progression. International Journal of Molecular Sciences. 2023; 24(15):12393. https://doi.org/10.3390/ijms241512393
Chicago/Turabian StyleMoshkovitz, Noa, Gali Epstein Shochet, and David Shitrit. 2023. "Prostaglandin E2 (PGE2) and Roflumilast Involvement in IPF Progression" International Journal of Molecular Sciences 24, no. 15: 12393. https://doi.org/10.3390/ijms241512393
APA StyleMoshkovitz, N., Epstein Shochet, G., & Shitrit, D. (2023). Prostaglandin E2 (PGE2) and Roflumilast Involvement in IPF Progression. International Journal of Molecular Sciences, 24(15), 12393. https://doi.org/10.3390/ijms241512393