
Novel Mutation Glu98Lys in Cardiac Tropomyosin Alters Its Structure and Impairs Myocardial Relaxation

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Results
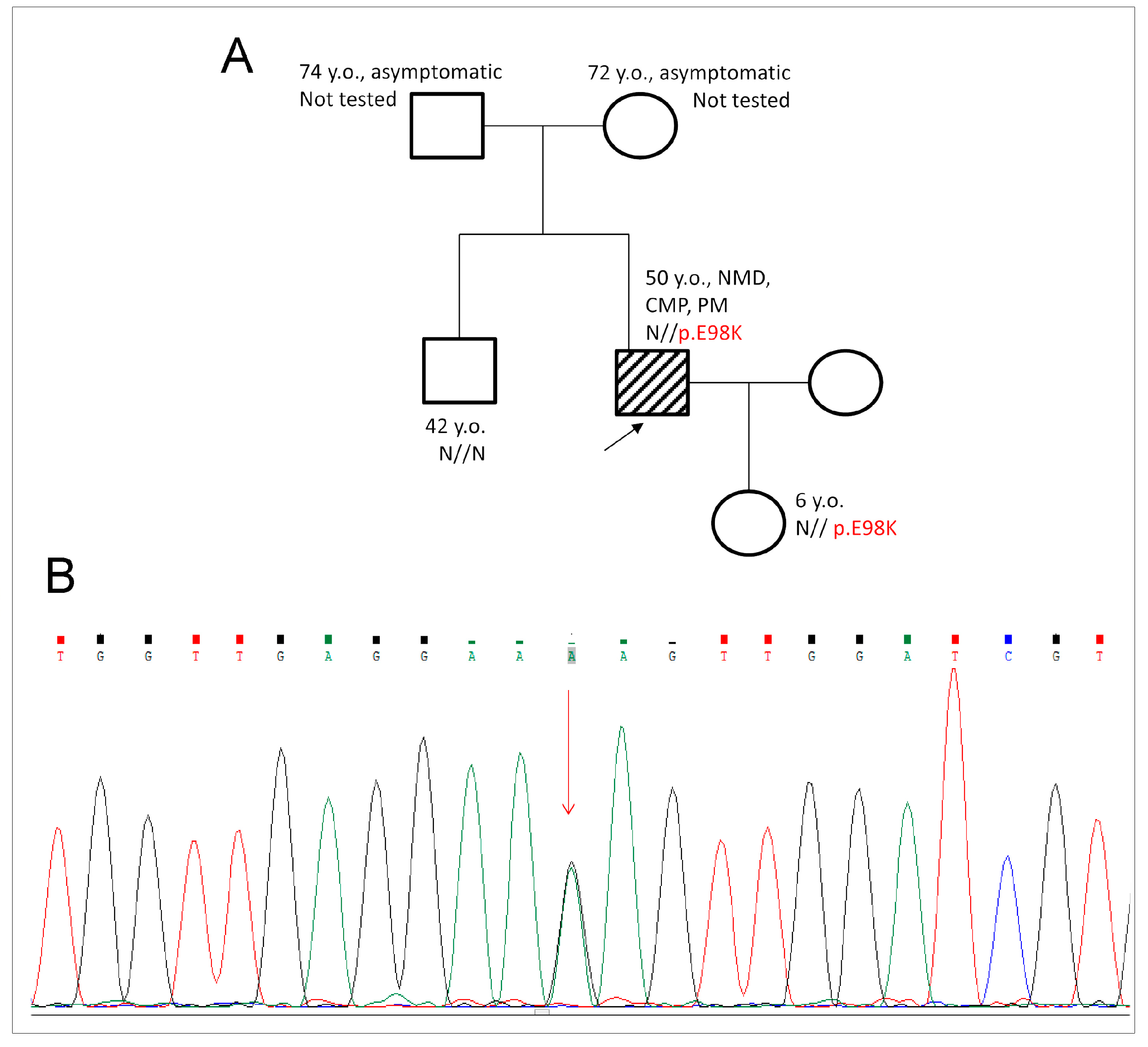
2.1. Genetic Evaluation and Detection of E98K Mutation
2.2. Effects of the E98K Mutation on the Structure of Tpm Molecule
2.2.1. Thermal Unfolding of E98K Tpm Mutant Studied with Circular Dichroism (CD) and Differential Scanning Calorimetry (DSC)
2.2.2. Solution Viscosity of E98K Tpm Mutant
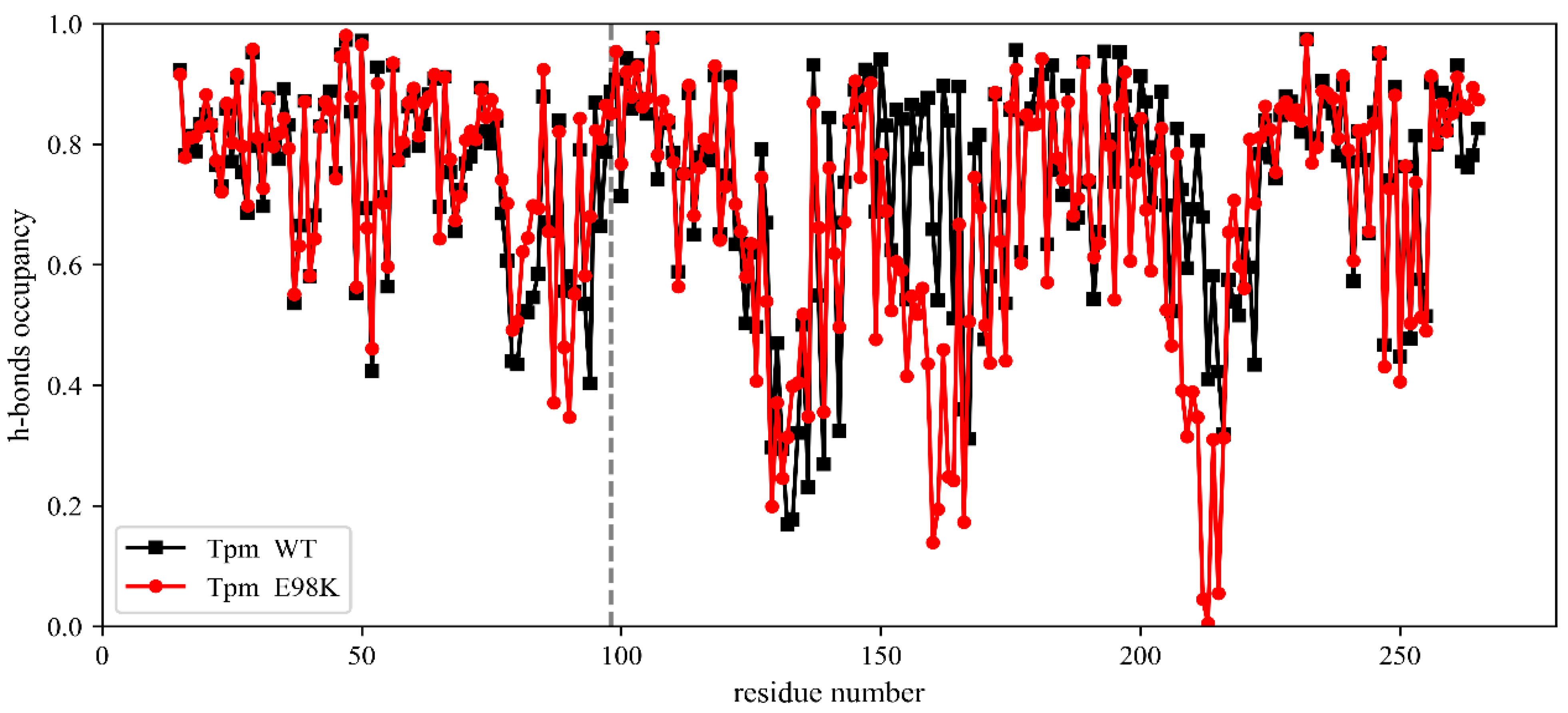
2.2.3. Molecular Dynamics (MD) Simulation
2.3. Effects of the E98K Substitution on Tpm Functional Properties
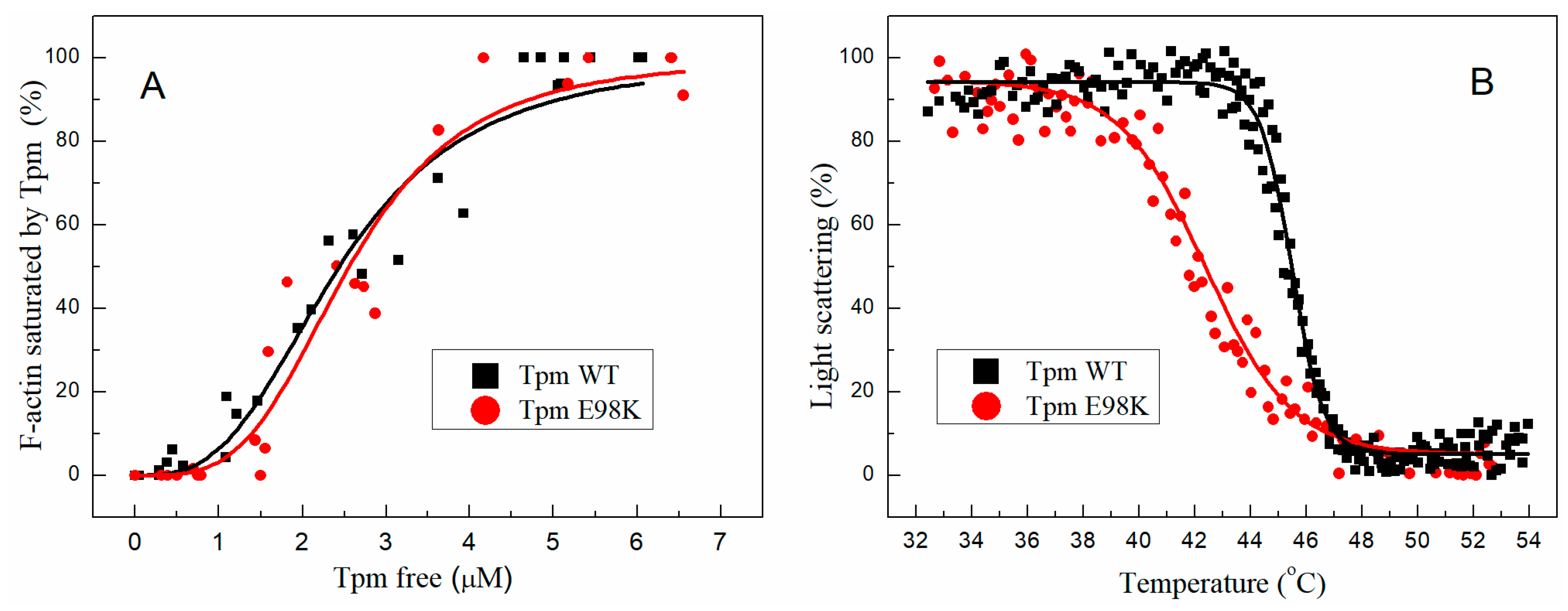
2.3.1. Affinity of E98K Tpm Mutant to F-Actin
2.3.2. Effect of E98K Substitution on the Thermal Stability of the F-Actin–Tpm Complex
2.3.3. Effect of E98K Substitution on Tpm Regulatory Properties
2.3.4. Possible Effect of E98K Substitution in Tpm on Its Interaction with TnI in the F-Actin–Tpm–Tn Complex (MD Simulation)
3. Discussion
3.1. Long-Distance Destabilization of Tpm Molecule upon Glu98Lys Substitution
3.2. Glu98Lys Tpm Substitution Destabilizes the Blocked State of the Thin Filaments and Probably Causes HCM with Restrictive Phenotype
3.3. Other HCM-Associated Mutations in the N-Terminal Part of the Tpm Molecule
3.4. Limitation of the Work
4. Materials and Methods
4.1. Genetic Investigation
4.2. Protein Preparations
4.3. Circular Dichroism (CD)
4.4. Differential Scanning Calorimetry (DSC)
4.5. Viscosimetry
4.6. Cosedimentation of Tpm Species with F-Actin
4.7. Temperature Dependences of Light Scattering
4.8. In Vitro Motility Assay
4.9. Molecular Dynamics (MD) Simulation and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Toste, A. Advances in hypertrophic cardiomyopathy: What the cardiologist needs to know. Port. J. Cardiol. 2022, 41, 499–509. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Ellsworth, E.G.; Ommen, S.R.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 2003, 108, 445–451. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Redwood, C.; Robinson, P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J. Muscle Res. Cell Motil. 2013, 34, 285–294. [Google Scholar] [CrossRef]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Keyt, L.K.; Duran, J.M.; Bui, Q.M.; Chen, C.; Miyamoto, M.I.; Silva Enciso, J.; Tardiff, J.C.; Adler, E.D. Thin filament cardiomyopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 2022, 9, 972301. [Google Scholar] [CrossRef] [PubMed]
- Moraczewska, J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1. J. Muscle Res. Cell Motil. 2020, 41, 39–53. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Levitsky, D.I. Molecular Mechanisms of Pathologies of Skeletal and Cardiac Muscles Caused by Point Mutations in the Tropomyosin Genes. Biochemistry 2020, 85 (Suppl. S1), S20–S33. [Google Scholar] [CrossRef] [PubMed]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef]
- Nevzorov, I.A.; Levitsky, D.I. Tropomyosin: Double helix from the protein world. Biochemistry 2011, 76, 1507–1527. [Google Scholar] [CrossRef]
- Kremneva, E.; Boussouf, S.; Nikolaeva, O.; Maytum, R.; Geeves, M.A.; Levitsky, D.I. Effects of two familial hypertrophic cardiomyopathy mutations in alpha-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys. J. 2004, 87, 3922–3933. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Shchepkin, D.V.; Kopylova, G.V.; Popruga, K.E.; Artemova, N.V.; Pivovarova, A.V.; Bershitsky, S.Y.; Levitsky, D.I. Structural and functional effects of cardiomyopathy-causing mutations in troponin T-binding region of cardiac tropomyosin. Biochemistry 2017, 56, 250–259. [Google Scholar] [CrossRef]
- Gupte, T.M.; Haque, F.; Gangadharan, B.; Sunitha, M.S.; Mukherjee, S.; Anandhan, S.; Rani, D.S.; Mukundan, N.; Jambekar, A.; Thangaraj, K.; et al. Mechanistic heterogeneity in contractile properties of α-tropomyosin (TPM1) mutants associated with inherited cardiomyopathies. J. Biol. Chem. 2015, 290, 7003–7015. [Google Scholar] [CrossRef] [PubMed]
- Golitsina, N.; An, Y.; Greenfield, N.J.; Thierfelder, L.; Iizuka, K.; Seidman, J.G.; Seidman, C.E.; Lehrer, S.S.; Hitchcock-DeGregori, S.E. Effects of two familial hypertrophic cardiomyopathy-causing mutations on α-tropomyosin structure and function. Biochemistry 1997, 36, 4637–4642. [Google Scholar] [CrossRef]
- Ly, S.; Lehrer, S.S. Long-range effects of familial hypertrophic cardiomyopathy mutations E180G and D175N on the properties of tropomyosin. Biochemistry 2012, 51, 6413–6420. [Google Scholar] [CrossRef][Green Version]
- Kopylova, G.V.; Shchepkin, D.V.; ·Nabiev, S.R.; Matyushenko, A.M.; ·Koubassova, N.A.; Levitsky, D.I.; Bershitsky, S.Y. Cardiomyopathy-associated mutations in tropomyosin differently affect actin–myosin interaction at single-molecule and ensemble levels. J. Muscle Res. Cell Motil. 2019, 40, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Tsaturyan, A.K.; Zaklyazminskaya, E.V.; Polyak, M.E.; Kopylova, G.V.; Shchepkin, D.V.; Kochurova, A.M.; Gonchar, A.D.; Kleymenov, S.Y.; Koubasova, N.A.; Bershitsky, S.Y.; et al. De Novo Asp219Val Mutation in Cardiac Tropomyosin Associated with Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 18. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Freire, E.; Biltonen, R.L. Statistical mechanical deconvolution of thermal transitions in macromolecules. I. Theory and application to homogeneous systems. Biopolymers 1978, 17, 463–479. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Artemova, N.V.; Sluchanko, N.N.; Levitsky, D.I. Effects of two stabilizing substitutions, D137L and G126R, in the middle part of α-tropomyosin on the domain structure of its molecule. Biophys. Chem. 2015, 196, 77–85. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Koubassova, N.A.; Shchepkin, D.V.; Kopylova, G.V.; Nabiev, S.R.; Nikitina, L.V.; Bershitsky, S.Y.; Levitsky, D.I.; Tsaturyan, A.K. The effects of cardiomyopathy-associated mutations in the head-to-tail overlap junction of α-tropomyosin on its properties and interaction with actin. Int. J. Biol. Macromol. 2019, 125, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, G.V.; Kochurova, A.M.; Yampolskaya, D.S.; Nefedova, V.V.; Tsaturyan, A.K.; Koubassova, N.A.; Kleymenov, S.Y.; Levitsky, D.I.; Bershitsky, S.Y.; Matyushenko, A.M.; et al. Structural and functional properties of kappa tropomyosin. Int. J. Mol. Sci. 2023, 24, 8340. [Google Scholar] [CrossRef]
- Marchenko, M.A.; Nefedova, V.V.; Yampolskaya, D.S.; Kopylova, G.V.; Shchepkin, D.V.; Bershitsky, S.Y.; Koubassova, N.A.; Tsaturyan, A.K.; Levitsky, D.I.; Matyushenko, A.M. Impact of A134 and E218 Amino Acid Residues of Tropomyosin on Its Flexibility and Function. Int. J. Mol. Sci. 2020, 21, 8720. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Tardiff, J.C. Thin filament mutations: Developing an integrative approach to a complex disorder. Circ. Res. 2011, 108, 765–782. [Google Scholar] [CrossRef] [PubMed]
- Shchepkin, D.V.; Nabiev, S.R.; Kopylova, G.V.; Matyushenko, A.M.; Levitsky, D.I.; Bershitsky, S.Y.; Tsaturyan, A.K. Cooperativity of myosin interaction with thin filaments is enhanced by stabilizing substitutions in tropomyosin. J. Muscle Res. Cell Motil. 2017, 38, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Koubassova, N.A.; Tsaturyan, A.K. Molecular Dynamics Assessment of Mechanical Properties of the Thin Filaments in Cardiac Muscle. Int. J. Mol. Sci. 2023, 24, 4792. [Google Scholar] [CrossRef]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.M.; Pepper, I.; Belknap, B.; Landim-Vieira, M.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. The structure of the native cardiac thin filament at systolic Ca2+ levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2024288118. [Google Scholar] [CrossRef]
- Johnson, C.A.; Brooker, H.R.; Gyamfi, I.; O’Brien, J.; Ashley, B.; Brazier, J.E.; Dean, A.; Embling, J.; Grimsey, E.; Tomlinson, A.C.; et al. Temperature sensitive point mutations in fission yeast tropomyosin have long range effects on the stability and function of the actin-tropomyosin copolymer. Biochem. Biophys. Res. Commun. 2018, 506, 339–346. [Google Scholar] [CrossRef]
- Lehman, W.; Pavadai, E.; Rynkiewicz, M.J. C-terminal troponin-I residues trap tropomyosin in the muscle thin filament blocked-state. Biochem. Biophys. Res. Commun. 2021, 551, 27–32. [Google Scholar] [CrossRef]
- Lehman, W.; Rynkiewicz, M.J. Troponin-I-induced tropomyosin pivoting defines thin-filament function in relaxed and active muscle. J. Gen. Physiol. 2023, 155, e202313387. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.S.; Geeves, M.A. The myosin-activated thin filament regulatory state, M−-open: A link to hypertrophic cardiomyopathy (HCM). J. Muscle Res. Cell Motil. 2014, 35, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Gimeno, J.R.; Bahl, A.; Steffensen, U.; Steffensen, M.; Osman, E.; Thaman, R.; Mogensen, J.; Elliott, P.M.; Doi, Y.; et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J. Am. Coll. Cardiol. 2007, 49, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Kuruba, B.; Kaczmarek, M.; Kęsik-Brodacka, M.; Fojutowska, M.; Śliwinska, M.; Kostyukova, A.S.; Moraczewska, J. Structural Effects of Disease-Related Mutations in Actin-Binding Period 3 of Tropomyosin. Molecules 2021, 26, 6980. [Google Scholar] [CrossRef] [PubMed]
- Karibe, A.; Tobacman, L.S.; Strand, J.; Butters, C.; Back, N.; Bachinski, L.L.; Arai, A.E.; Ortiz, A.; Roberts, R.; Homsher, E.; et al. Hypertrophic cardiomyopathy caused by a novel alpha-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis. Circulation 2001, 103, 65–71. [Google Scholar] [CrossRef]
- Sliwinska, M.; Robaszkiewicz, K.; Czajkowska, M.; Zheng, W.; Moraczewska, J. Functional effects of substitutions I92T and V95A in actin-binding period 3 of tropomyosin. Biochim. Biophys. Acta 2018, 1866, 558–568. [Google Scholar] [CrossRef]
- Ishii, S.; Suzuki, M.; Ishiwata, S.; Kawai, M. Functional significance of HCM mutants of tropomyosin, V95A and D175N, studied with in vitro motility assays. Biophys. Physicobiol. 2019, 16, 28–40. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef]
- Otsuka, H.; Arimura, T.; Abe, T.; Kawai, H.; Aizawa, Y.; Kubo, T.; Kitaoka, H.; Nakamura, H.; Nakamura, K.; Okamoto, H.; et al. Prevalence and distribution of sarcomeric gene mutations in Japanese patients with familial hypertrophic cardiomyopathy. Circ. J. 2012, 76, 453–461. [Google Scholar] [CrossRef]
- Ly, T.; Krieger, I.; Tolkatchev, D.; Krone, C.; Moural, T.; Samatey, F.A.; Kang, C.H.; Kostyukova, A.S. Structural destabilization of tropomyosin induced by the cardiomyopathy-linked mutation R21H. Prot. Sci. 2017, 27, 498–508. [Google Scholar] [CrossRef]
- Ly, T.; Christopher, T.; Pappas, C.T.; Johnson, D.; Schlecht, W.; Colpan, M.; Galkin, V.E.; Gregorio, C.C.; Dong, W.-J.; Kostyukova, A.S. Effects of cardiomyopathy-linked mutations K15N and R21H in tropomyosin on thin-filament regulation and pointed-end dynamics. Mol. Biol. Cell 2019, 30, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.J.; Nili, M.; Homsher, E.; Tobacman, L.S. Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivity and tropomyosin N-domain flexibility. J. Biol. Chem. 2003, 278, 41742–41748. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Hitchcock-DeGregori, S.E.; Barua, B. Investigating the effects of tropomyosin mutations on its flexibility and interactions with filamentous actin using molecular dynamics simulation. J. Muscle Res. Cell Motil. 2016, 37, 131–147. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Nefedova, V.V.; Shchepkin, D.V.; Kopylova, G.V.; Berg, V.Y.; Pivovarova, A.V.; Kleymenov, S.Y.; Bershitsky, S.Y.; Levitsky, D.I. Mechanisms of disturbance of the contractile function of slow skeletal muscles induced by myopathic mutations in the tropomyosin TPM3 gene. FASEB J. 2020, 34, 13507–13520. [Google Scholar] [CrossRef] [PubMed]
- Janco, M.; Kalyva, A.; Scellini, B.; Piroddi, N.; Tesi, C.; Poggesi, C.; Geeves, M.A. alpha-Tropomyosin with a D175N or E180G mutation in only one chain differs from tropomyosin with mutations in both chains. Biochemistry 2012, 51, 9880–9890. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Monteiro, P.B.; Lataro, R.C.; Ferro, J.A.; Reinach, F.d.C. Functional α-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 1994, 269, 10461–10466. [Google Scholar] [CrossRef]
- Nefedova, V.V.; Koubassova, N.A.; Borzova, V.A.; Kleymenov, S.Y.; Tsaturyan, A.K.; Matyushenko, A.M.; Levitsky, D.I. Tropomyosin pseudo-phosphorylation can rescue the effects of cardiomyopathy-associated mutations. Int. J. Biol. Macromol. 2021, 166, 424–434. [Google Scholar] [CrossRef]
- Margossian, S.S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1982; Volume 85, Pt B, pp. 55–71. [Google Scholar] [CrossRef]
- Pardee, J.D.; Spudich, J.A. Purification of muscle actin. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1982; Volume 85, Pt B, pp. 164–179. [Google Scholar] [CrossRef]
- Mashanov, G.I.; Molloy, J.E. Automatic detection of single fluorophores in live cells. Biophys. J. 2007, 92, 2199–2211. [Google Scholar] [CrossRef]
- Brunet, N.M.; Chase, P.B.; Mihajlović, G.; Schoffstall, B. Ca2+-regulatory function of the inhibitory peptide region of cardiac troponin I is aided by the C-terminus of cardiac troponin T: Effects of familial hypertrophic cardiomyopathy mutations cTnI R145G and cTnT R278C, alone and in combination, on filament sliding. Arch. Biochem. Biophys. 2014, 552–553, 11–20. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; Mackerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Whitby, F.G.; Phillips, G.N., Jr. Crystal structure of tropomyosin at 7 Angstroms resolution. Proteins 2000, 38, 49–59. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Pavadai, E.; Rynkiewicz, M.J.; Yang, Z.; Gould, I.R.; Marston, S.B.; Lehman, W. Modulation of cardiac thin filament structure by phosphorylated troponin-I analyzed by protein-protein docking and molecular dynamics simulation. Arch. Biochem. Biophys. 2022, 725, 109282. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tpm | Tm b (°C) | ΔHcal (kJ mol−1) | ΔHcal (% of Total) | Total ΔHcal c (kJ mol−1) |
|---|---|---|---|---|
| Tpm WT Reduced | 1330 | |||
| Domain 1 | 38.2 | 350 | 26 | |
| Domain 2 | 43.3 | 480 | 36 | |
| Domain 3 | 50.7 | 500 | 38 | |
| Tpm E98K Reduced | 950 | |||
| Domain 1 | 30.9 | 65 | 7 | |
| Domain 2 | 39.0 | 390 | 41 | |
| Domain 3 | 52.9 | 495 | 52 | |
| Tpm E98K Cross-linked | 865 | |||
| Domain 1 | 30.6 | 65 | 7 | |
| Domain 3 | 51.6 | 645 | 75 | |
| Domain 4 | 57.9 | 155 | 18 |
| Tpm | Vmax (µm/s) | V0 (µm/s) | pCa50 | n |
|---|---|---|---|---|
| WT Tpm | 1.85 ± 0.01 | 0 | 5.85 ± 0.01 | 1.7 ± 0.2 |
| E98K Tpm | 1.37 ± 0.02 * | 0.44 ± 0.04 * | 6.00 ± 0.05 * | 1.3 ± 0.1 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matyushenko, A.M.; Nefedova, V.V.; Kochurova, A.M.; Kopylova, G.V.; Koubassova, N.A.; Shestak, A.G.; Yampolskaya, D.S.; Shchepkin, D.V.; Kleymenov, S.Y.; Ryabkova, N.S.; et al. Novel Mutation Glu98Lys in Cardiac Tropomyosin Alters Its Structure and Impairs Myocardial Relaxation. Int. J. Mol. Sci. 2023, 24, 12359. https://doi.org/10.3390/ijms241512359
Matyushenko AM, Nefedova VV, Kochurova AM, Kopylova GV, Koubassova NA, Shestak AG, Yampolskaya DS, Shchepkin DV, Kleymenov SY, Ryabkova NS, et al. Novel Mutation Glu98Lys in Cardiac Tropomyosin Alters Its Structure and Impairs Myocardial Relaxation. International Journal of Molecular Sciences. 2023; 24(15):12359. https://doi.org/10.3390/ijms241512359
Chicago/Turabian StyleMatyushenko, Alexander M., Victoria V. Nefedova, Anastasia M. Kochurova, Galina V. Kopylova, Natalia A. Koubassova, Anna G. Shestak, Daria S. Yampolskaya, Daniil V. Shchepkin, Sergey Y. Kleymenov, Natalia S. Ryabkova, and et al. 2023. "Novel Mutation Glu98Lys in Cardiac Tropomyosin Alters Its Structure and Impairs Myocardial Relaxation" International Journal of Molecular Sciences 24, no. 15: 12359. https://doi.org/10.3390/ijms241512359
APA StyleMatyushenko, A. M., Nefedova, V. V., Kochurova, A. M., Kopylova, G. V., Koubassova, N. A., Shestak, A. G., Yampolskaya, D. S., Shchepkin, D. V., Kleymenov, S. Y., Ryabkova, N. S., Katrukha, I. A., Bershitsky, S. Y., Zaklyazminskaya, E. V., Tsaturyan, A. K., & Levitsky, D. I. (2023). Novel Mutation Glu98Lys in Cardiac Tropomyosin Alters Its Structure and Impairs Myocardial Relaxation. International Journal of Molecular Sciences, 24(15), 12359. https://doi.org/10.3390/ijms241512359