
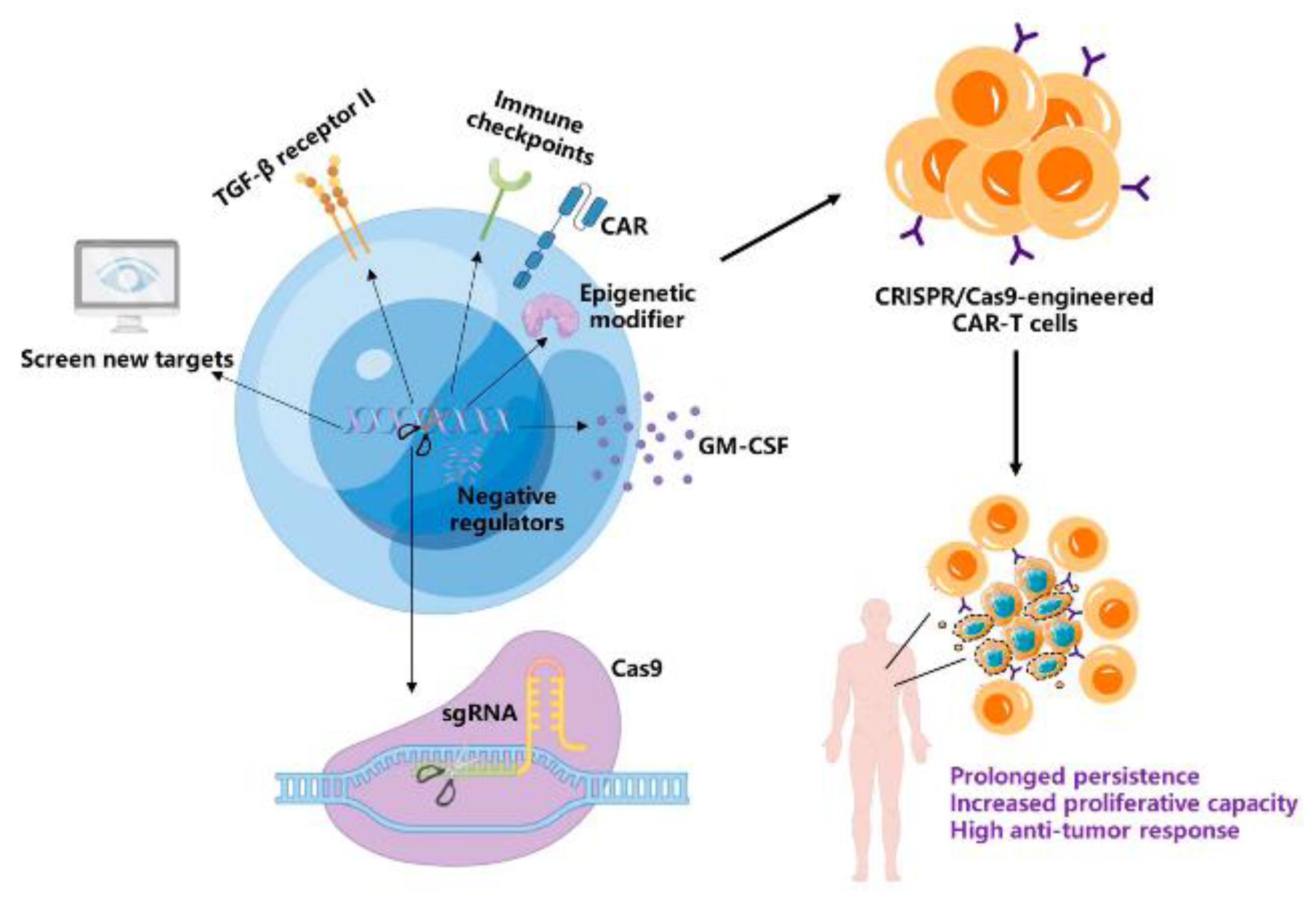
CRISPR/Cas9: A Powerful Strategy to Improve CAR-T Cell Persistence
Abstract
1. Introduction
2. Factors Limiting CAR-T Cell Persistence and Clinical Response
2.1. CAR-T Cell Differentiation Phenotype Determines Anti-Tumor Potency
2.2. CAR-T Cell Exhaustion Impairs Long-Term Anti-Tumor Activity
3. CRISPR/Cas9 Application in Improving the Persistence of CAR-T Cells
3.1. Disruption of Immune Checkpoints in CAR-T Cells
3.2. The Deletion of Negative Regulators of CAR-T Cell Function
3.3. Enhancing CAR-T Cell Resistance to Suppressive Cytokines
3.4. Epigenetic Reprogramming Strategies to Augment CAR-T Cell Effector Functions
3.5. Screening for New Targets in CAR-T Cells for Long-Time Persistence
3.5.1. Screen for Exhaustion and Memory Phenotype Targets
3.5.2. Screening for Activation, Expansion, and Effector Function Targets
3.6. CRISPR/Cas9 Technology in CAR-T Cells to Conquer Other Limitations
4. Current Clinical Application of CRISPR/Cas9 Technology in CAR-T Cell Therapy
5. Challenges of the Application of CRISPR/Cas9 in CAR-T Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zuo, S.; Deng, B.; Xu, X.; Li, C.; Zheng, Q.; Ling, Z.; Song, W.; Xu, J.; Duan, J.; et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood 2020, 135, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, M.C.; Hu, Z.H.; Curran, K.; Laetsch, T.; Locke, F.; Rouce, R.; Pulsipher, M.A.; Phillips, C.L.; Keating, A.; Frigault, M.J.; et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020, 4, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Shargian, L.; Raanani, P.; Yeshurun, M.; Gafter-Gvili, A.; Gurion, R. Chimeric antigen receptor T-cell therapy is superior to standard of care as second-line therapy for large B-cell lymphoma: A systematic review and meta-analysis. Br. J. Haematol. 2022, 198, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Canale, F.A.; Alati, C.; Vincelli, I.D.; Moscato, T.; Porto, G.; Loteta, B.; Naso, V.; Mazza, M.; Nicolini, F.; et al. CART-Cell Therapy: Recent Advances and New Evidence in Multiple Myeloma. Cancers 2021, 13, 2639. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Yang, Y.H.; Liu, J.W.; Lu, C.; Wei, J.F. CAR-T Cell Therapy for Breast Cancer: From Basic Research to Clinical Application. Int. J. Biol. Sci. 2022, 18, 2609–2626. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.; Chen, T.; Chang, L.J.; et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol. Cancer 2023, 22, 3. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. 2022, 24, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.-Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.-T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J.; et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 147, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11, 702. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Lopez-Cantillo, G.; Uruena, C.; Camacho, B.A.; Ramirez-Segura, C. CAR-T Cell Performance: How to Improve Their Persistence? Front. Immunol. 2022, 13, 878209. [Google Scholar] [CrossRef]
- Liu, Y.; An, L.; Huang, R.; Xiong, J.; Yang, H.; Wang, X.; Zhang, X. Strategies to enhance CAR-T persistence. Biomark. Res. 2022, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.F.; Yu, T. CRISPR/Cas9 therapeutics: Progress and prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef]
- Manghwar, H.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas System: Recent Advances and Future Prospects for Genome Editing. Trends Plant Sci. 2019, 24, 1102–1125. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55. [Google Scholar] [CrossRef]
- Malech, H.L. Treatment by CRISPR-Cas9 Gene Editing—A Proof of Principle. N. Engl. J. Med. 2021, 384, 286–287. [Google Scholar] [CrossRef]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021, 12, 428. [Google Scholar] [CrossRef]
- Rafii, S.; Tashkandi, E.; Bukhari, N.; Al-Shamsi, H.O. Current Status of CRISPR/Cas9 Application in Clinical Cancer Research: Opportunities and Challenges. Cancers 2022, 14, 947. [Google Scholar] [CrossRef]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.M.; Scherer, L.D.; Rouce, R.H. Replacing CAR-T cell resistance with persistence by changing a single residue. J. Clin. Investig. 2020, 130, 2806–2808. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Melief, J.; de Coana, P.Y.; Wickström, S.L.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef]
- Han, J.; Khatwani, N.; Searles, T.G.; Turk, M.J.; Angeles, C.V. Memory CD8(+) T cell responses to cancer. Semin. Immunol. 2020, 49, 101435. [Google Scholar] [CrossRef]
- Tantalo, D.G.; Oliver, A.J.; von Scheidt, B.; Harrison, A.J.; Mueller, S.N.; Kershaw, M.H.; Slaney, C.Y. Understanding T cell phenotype for the design of effective chimeric antigen receptor T cell therapies. J. Immunother. Cancer 2021, 9, e002555. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Biasco, L.; Izotova, N.; Rivat, C.; Ghorashian, S.; Richardson, R.; Guvenel, A.; Hough, R.; Wynn, R.; Popova, B.; Lopes, A.; et al. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nat. Cancer 2021, 2, 629–642. [Google Scholar] [CrossRef]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef]
- Van den Broek, T.; Borghans, J.A.M.; van Wijk, F. The full spectrum of human naive T cells. Nat. Rev. Immunol. 2018, 18, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Atsumi, T.; Stofkova, A.; Nishikawa, N.; Ohki, T.; Suzuki, H.; Katsunuma, K.; Jiang, J.J.; Bando, H.; Meng, J.; et al. Naive T Cell Homeostasis Regulated by Stress Responses and TCR Signaling. Front. Immunol. 2015, 6, 638. [Google Scholar] [CrossRef] [PubMed]
- Minato, N.; Hattori, M.; Hamazaki, Y. Physiology and pathology of T-cell aging. Int. Immunol. 2020, 32, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cheng, F.; Zheng, F. Stem cell like memory T cells: A new paradigm in cancer immunotherapy. Clin. Immunol. 2022, 241, 109078. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiu, F.; Xu, Y.; Hou, X.; Zhang, Z.; Huang, L.; Wang, H.; Xing, H.; Wu, S. Stem cell-like memory T cells: The generation and application. J. Leukoc. Biol. 2021, 110, 1209–1223. [Google Scholar] [CrossRef]
- Zhang, Q.; Lakkis, F.G. Memory T Cell Migration. Front. Immunol. 2015, 6, 504. [Google Scholar] [CrossRef]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Busch, D.H.; Frassle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin. Immunol. 2016, 28, 28–34. [Google Scholar] [CrossRef]
- Chan, J.D.; Lai, J.; Slaney, C.Y.; Kallies, A.; Beavis, P.A.; Darcy, P.K. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat. Rev. Immunol. 2021, 21, 769–784. [Google Scholar] [CrossRef]
- Meyran, D.; Terry, R.L.; Zhu, J.J.; Haber, M.; Ziegler, D.S.; Ekert, P.G.; Trapani, J.A.; Darcy, P.K.; Neeson, P.J. Early-phenotype CAR-T cells for the treatment of pediatric cancers. Ann. Oncol. 2021, 32, 1366–1380. [Google Scholar] [CrossRef]
- Arcangeli, S.; Bove, C.; Mezzanotte, C.; Camisa, B.; Falcone, L.; Manfredi, F.; Bezzecchi, E.; El Khoury, R.; Norata, R.; Sanvito, F.; et al. CAR T cell manufacturing from naive/stem memory T lymphocytes enhances antitumor responses while curtailing cytokine release syndrome. J. Clin. Investig. 2022, 132, e150807. [Google Scholar] [CrossRef] [PubMed]
- El Marabti, E.; Abdel-Wahab, O. Enhancing CD19 Chimeric Antigen Receptor T Cells Through Memory-Enriched T Cells. Clin. Cancer Res. 2023, 29, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef]
- Gallimore, A.; Glithero, A.; Godkin, A.; Tissot, A.C.; Plückthun, A.; Elliott, T.; Hengartner, H.; Zinkernagel, R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef]
- Zander, R.; Cui, W. Exhausted CD8(+) T cells face a developmental fork in the road. Trends Immunol. 2023, 44, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e828. [Google Scholar] [CrossRef] [PubMed]
- Belk, J.A.; Daniel, B.; Satpathy, A.T. Epigenetic regulation of T cell exhaustion. Nat. Immunol. 2022, 23, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Finney, O.C.; Brakke, H.M.; Rawlings-Rhea, S.; Hicks, R.; Doolittle, D.; Lopez, M.; Futrell, R.B.; Orentas, R.J.; Li, D.; Gardner, R.A.; et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Investig. 2019, 129, 2123–2132. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Q.; Zhu, X. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front. Cell Dev. Biol. 2022, 10, 1034257. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Liu, L.; Bi, E.; Ma, X.; Xiong, W.; Qian, J.; Ye, L.; Su, P.; Wang, Q.; Xiao, L.; Yang, M.; et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat. Commun. 2020, 11, 5902. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.Q.; Wei, X.W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]
- Cheng, N.; Bai, X.; Shu, Y.; Ahmad, O.; Shen, P. Targeting tumor-associated macrophages as an antitumor strategy. Biochem. Pharmacol. 2021, 183, 114354. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Niu, Y.R.; Wang, Z.H.; Ye, L.L.; Peng, W.B.; Zhou, Q. Cancer-associated fibroblasts: Vital suppressors of the immune response in the tumor microenvironment. Cytokine Growth Factor. Rev. 2022, 67, 35–48. [Google Scholar] [CrossRef]
- Dwivedi, M.; Tiwari, S.; Kemp, E.H.; Begum, R. Implications of regulatory T cells in anti-cancer immunity: From pathogenesis to therapeutics. Heliyon 2022, 8, e10450. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Hossain, M.A.; Liu, G.; Dai, B.; Si, Y.; Yang, Q.; Wazir, J.; Birnbaumer, L.; Yang, Y. Reinvigorating exhausted CD8(+) cytotoxic T lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med. Res. Rev. 2021, 41, 156–201. [Google Scholar] [CrossRef]
- Chaput, N.; Louafi, S.; Bardier, A.; Charlotte, F.; Vaillant, J.C.; Menegaux, F.; Rosenzwajg, M.; Lemoine, F.; Klatzmann, D.; Taieb, J. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut 2009, 58, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.J.; et al. TGFbeta1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Mosaheb, M.M.; Flavell, R.A. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 2009, 31, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, S.K.; Rana, B.; Rana, A. Tumor-infiltrating CD8(+) T cell antitumor efficacy and exhaustion: Molecular insights. Drug Discov. Today 2021, 26, 951–967. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef]
- Gleditzsch, D.; Pausch, P.; Muller-Esparza, H.; Ozcan, A.; Guo, X.; Bange, G.; Randau, L. PAM identification by CRISPR-Cas effector complexes: Diversified mechanisms and structures. RNA Biol. 2019, 16, 504–517. [Google Scholar] [CrossRef]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Wing, J.B.; Ise, W.; Kurosaki, T.; Sakaguchi, S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity 2014, 41, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy—Current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Nakazawa, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Motoyama, Y.; Park, Y.S.; et al. Effect of CRISPR/Cas9-Mediated PD-1-Disrupted Primary Human Third-Generation CAR-T Cells Targeting EGFRvIII on In Vitro Human Glioblastoma Cell Growth. Cells 2020, 9, 998. [Google Scholar] [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]
- Dotsch, S.; Svec, M.; Schober, K.; Hammel, M.; Wanisch, A.; Gokmen, F.; Jarosch, S.; Warmuth, L.; Barton, J.; Cicin-Sain, L.; et al. Long-term persistence and functionality of adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression. Proc. Natl. Acad. Sci. USA 2023, 120, e2200626120. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Kumar, R.; Kumar Singh, A.; Tsakem, E.L.; Kathania, M.; Riese, M.J.; Theiss, A.L.; Davila, M.L.; Venuprasad, K. Deletion of Cbl-b inhibits CD8+ T-cell exhaustion and promotes CAR T-cell function. J. Immunother. Cancer 2021, 9, e001688. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Lu, K.H.; Du, X.; Zeissig, M.N.; Xu, R.; Goh, P.K.; Xirouchaki, C.E.; Hogarth, S.J.; Greatorex, S.; Sek, K.; et al. PTP1B Is an Intracellular Checkpoint that Limits T-cell and CAR T-cell Antitumor Immunity. Cancer Discov. 2022, 12, 752–773. [Google Scholar] [CrossRef]
- Good, C.R.; Aznar, M.A.; Kuramitsu, S.; Samareh, P.; Agarwal, S.; Donahue, G.; Ishiyama, K.; Wellhausen, N.; Rennels, A.K.; Ma, Y.; et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 2021, 184, 6081–6100.e26. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef]
- Moller, S.H.; Hsueh, P.C.; Yu, Y.R.; Zhang, L.; Ho, P.C. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab. 2022, 34, 378–395. [Google Scholar] [CrossRef]
- Riese, M.J.; Moon, E.K.; Johnson, B.D.; Albelda, S.M. Diacylglycerol Kinases (DGKs): Novel Targets for Improving T Cell Activity in Cancer. Front. Cell Dev. Biol. 2016, 4, 108. [Google Scholar] [CrossRef]
- Jung, I.Y.; Kim, Y.Y.; Yu, H.S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef]
- Leone, R.D.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Decombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derre, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.Y.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Molck, C.; et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2021, 11, 622509. [Google Scholar] [CrossRef]
- Alishah, K.; Birtel, M.; Masoumi, E.; Jafarzadeh, L.; Mirzaee, H.R.; Hadjati, J.; Voss, R.H.; Diken, M.; Asad, S. CRISPR/Cas9-mediated TGFbetaRII disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells in vitro. J. Transl. Med. 2021, 19, 482. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. J. Clin. Investig. 2020, 5, e133977. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.J.; Richardson, R.T.; Alexander, W.S.; Viney, E.M.; Willson, T.A.; Sprigg, N.S.; Starr, R.; Nicholson, S.E.; Metcalf, D.; Nicola, N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA 1998, 95, 114–119. [Google Scholar] [CrossRef]
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Qin, L.; Zhao, R.; Wu, D.; Wu, Z.; Zheng, D.; Li, S.; Luo, M.; Wu, Q.; Long, Y.; et al. Disruption of CISH promotes the antitumor activity of human T cells and decreases PD-1 expression levels. Mol. Ther. Oncolytics 2023, 28, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef]
- Cong, B.; Zhang, Q.; Cao, X. The function and regulation of TET2 in innate immunity and inflammation. Protein Cell 2021, 12, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Zhao, Z.; Feucht, J.; Koche, R.; Iyer, A.; Dobrin, A.; Mansilla-Soto, J.; Yang, J.; Zhan, Y.; Lopez, M.; et al. TET2 guards against unchecked BATF3-induced CAR T cell expansion. Nature 2023, 615, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Wu, Z.; Inoue, S.; Kasuya, H.; Matsushita, H.; Takahashi, Y.; Kuroda, H.; Hosoda, W.; Suzuki, S.; Kagoya, Y. Genetic ablation of PRDM1 in antitumor T cells enhances therapeutic efficacy of adoptive immunotherapy. Blood 2022, 139, 2156–2172. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e119. [Google Scholar] [CrossRef]
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med. 2021, 13, eabh0272. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Liu, D.; Zhao, X.; Tang, A.; Xu, X.; Liu, S.; Zha, L.; Ma, W.; Zheng, J.; Shi, M. CRISPR screen in mechanism and target discovery for cancer immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188378. [Google Scholar] [CrossRef]
- Carnevale, J.; Shifrut, E.; Kale, N.; Nyberg, W.A.; Blaeschke, F.; Chen, Y.Y.; Li, Z.; Bapat, S.P.; Diolaiti, M.E.; O’Leary, P.; et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature 2022, 609, 174–182. [Google Scholar] [CrossRef]
- Freitas, K.A.; Belk, J.A.; Sotillo, E.; Quinn, P.J.; Ramello, M.C.; Malipatlolla, M.; Daniel, B.; Sandor, K.; Klysz, D.; Bjelajac, J.; et al. Enhanced T cell effector activity by targeting the Mediator kinase module. Science 2022, 378, eabn5647. [Google Scholar] [CrossRef]
- Belk, J.A.; Yao, W.; Ly, N.; Freitas, K.A.; Chen, Y.T.; Shi, Q.; Valencia, A.M.; Shifrut, E.; Kale, N.; Yost, K.E.; et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 2022, 40, 768–786.e7. [Google Scholar] [CrossRef]
- Trefny, M.P.; Kirchhammer, N.; Auf der Maur, P.; Natoli, M.; Schmid, D.; Germann, M.; Fernandez Rodriguez, L.; Herzig, P.; Lotscher, J.; Akrami, M.; et al. Deletion of SNX9 alleviates CD8 T cell exhaustion for effective cellular cancer immunotherapy. Nat. Commun. 2023, 14, 86. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, C.; Qiao, M.; Cheng, C.; Tang, N.; Lu, S.; Sun, W.; Xu, B.; Cao, Y.; Wei, X.; et al. Depletion of BATF in CAR-T cells enhances antitumor activity by inducing resistance against exhaustion and formation of central memory cells. Cancer Cell 2022, 40, 1407–1422.e7. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Gajic, Z.; Guarino, M.; Daniloski, Z.; Rahman, J.A.; Xue, X.; Lu, C.; Lu, L.; Mimitou, E.P.; Hao, S.; et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nature 2022, 603, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Prager, B.C.; Gimple, R.C.; Aguilar, B.; Alizadeh, D.; Tang, H.; Lv, D.; Starr, R.; Brito, A.; Wu, Q.; et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021, 11, 1192–1211. [Google Scholar] [CrossRef] [PubMed]
- Gurusamy, D.; Henning, A.N.; Yamamoto, T.N.; Yu, Z.; Zacharakis, N.; Krishna, S.; Kishton, R.J.; Vodnala, S.K.; Eidizadeh, A.; Jia, L.; et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 2020, 37, 818–833.e9. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Park, J.J.; Dong, M.B.; Yang, Q.; Chow, R.D.; Peng, L.; Du, Y.; Guo, J.; Dai, X.; Wang, G.; et al. In vivo CRISPR screening in CD8 T cells with AAV–Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat. Biotechnol. 2019, 37, 1302–1313. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef]
- Ye, L.; Park, J.J.; Peng, L.; Yang, Q.; Chow, R.D.; Dong, M.B.; Lam, S.Z.; Guo, J.; Tang, E.; Zhang, Y.; et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022, 34, 595–614.e14. [Google Scholar] [CrossRef]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An "off-the-shelf" fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef]
- Mailankody, S.; Matous, J.V.; Chhabra, S.; Liedtke, M.; Sidana, S.; Oluwole, O.O.; Malik, S.; Nath, R.; Anwer, F.; Cruz, J.C.; et al. Allogeneic BCMA-targeting CAR T cells in relapsed/refractory multiple myeloma: Phase 1 UNIVERSAL trial interim results. Nat. Med. 2023, 29, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Khan, J.F.; Godfrey, W.D.; Lopez, A.V.; Ciampricotti, M.; Rudin, C.M.; Brentjens, R.J. IL-18-secreting CAR T cells targeting DLL3 are highly effective in small cell lung cancer models. J. Clin. Investig. 2023, 133, e166028. [Google Scholar] [CrossRef]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Ottaviano, G.; Georgiadis, C.; Gkazi, S.A.; Syed, F.; Zhan, H.; Etuk, A.; Preece, R.; Chu, J.; Kubat, A.; Adams, S.; et al. TT52 CRISPR-CAR group. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci. Transl. Med. 2022, 14, eabq3010. [Google Scholar] [CrossRef]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Hoijer, I.; Emmanouilidou, A.; Ostlund, R.; van Schendel, R.; Bozorgpana, S.; Tijsterman, M.; Feuk, L.; Gyllensten, U.; den Hoed, M.; Ameur, A. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun. 2022, 13, 627. [Google Scholar] [CrossRef]
- Kang, S.-H.; Lee, W.-J.; An, J.-H.; Lee, J.-H.; Kim, Y.-H.; Kim, H.; Oh, Y.; Park, Y.-H.; Jin, Y.B.; Jun, B.-H.; et al. Prediction-based highly sensitive CRISPR off-target validation using target-specific DNA enrichment. Nat. Commun. 2020, 11, 3596. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.; Khalili, N.; Rezaei, N. CRISPR/Cas9 revitalizes adoptive T-cell therapy for cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 269. [Google Scholar] [CrossRef]
- Rasul, M.F.; Hussen, B.M.; Salihi, A.; Ismael, B.S.; Jalal, P.J.; Zanichelli, A.; Jamali, E.; Baniahmad, A.; Ghafouri-Fard, S.; Basiri, A.; et al. Strategies to overcome the main challenges of the use of CRISPR/Cas9 as a replacement for cancer therapy. Mol. Cancer 2022, 21, 64. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Merkel, O.M. Immunogenicity of Cas9 Protein. J. Pharm. Sci. 2020, 109, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 3497. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L. Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1408. [Google Scholar] [CrossRef]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef]


{kind=link}
| NCT Number | Phase | Target Gene | CAR-T Cell | Cancer | Location | Status |
|---|---|---|---|---|---|---|
| NCT03545815 | I | TRAC and PDCD1 | Mesothelin- CART | Mesothelin-positive multiple solid tumors | China | Completed |
| NCT04557436 | I | TRAC and CD52 | CD19-CART | Refractory B-cell leukemia | The United Kingdom | Active; not recruiting |
| NCT03166878 | I/II | B2M | CD19-CART | B-cell leukemia | China | Recruiting |
| NCT04244656 | I | TCR and MHC class I | BCMA-CART | Multiple myeloma | The United States | Active, Not Recruiting |
| NCT04438083 | I | TRAC, B2M and CD70 | CD70-CART | Relapsed or refractory renal cell carcinoma | The United States | Active; not recruiting |
| NCT05643742 | I/II | TRAC and B2M | CD19-CART | Relapsed or refractory B-cell malignancies | The United States | Recruiting |
| NCT03747965 | I | PDCD1 | Mesothelin-CART | Mesothelin-positive multiple solid tumors | China | Completed |
| NCT04035434 | I | TRAC and B2M | CD19-CART | Relapsed or refractory B-cell malignancies | The United States | Recruiting |
| NCT03166878 | I/II | TRAC and B2M | CD19-CART | Relapsed or refractory CD19+ leukemia and lymphoma | China | Completed |
| NCT03398967 | I/II | TRAC and CD52 | CD19/20- or CD19/22-UCART | Relapsed or refractory hematological malignancies | China | Unknown |
| NCT04976218 | I | TGF-β receptor II | EGFR-CART | Advanced EGFR-positive solid tumors | China | Recruiting |
| NCT05812326 | I/II | PDCD1 | MUC1-CART | MUC1-positive advanced breast cancer | China | Completed |
| NCT04037566 | I | HPK1 | CD19-CART | Hematopoietic malignancies | China | Recruiting |
| NCT03203369 | I | TCR | CD123-CART | Blastic plasmacytoid dendritic cell neoplasm | The United States | Discontinued |
| NCT04637763 | I | Unknown | CD19-CART | Relapsed/refractory B-cell non-Hodgkin lymphoma | The United States | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, W.; Chen, Z.-N.; Wang, K. CRISPR/Cas9: A Powerful Strategy to Improve CAR-T Cell Persistence. Int. J. Mol. Sci. 2023, 24, 12317. https://doi.org/10.3390/ijms241512317
Wei W, Chen Z-N, Wang K. CRISPR/Cas9: A Powerful Strategy to Improve CAR-T Cell Persistence. International Journal of Molecular Sciences. 2023; 24(15):12317. https://doi.org/10.3390/ijms241512317
Chicago/Turabian StyleWei, Wei, Zhi-Nan Chen, and Ke Wang. 2023. "CRISPR/Cas9: A Powerful Strategy to Improve CAR-T Cell Persistence" International Journal of Molecular Sciences 24, no. 15: 12317. https://doi.org/10.3390/ijms241512317
APA StyleWei, W., Chen, Z.-N., & Wang, K. (2023). CRISPR/Cas9: A Powerful Strategy to Improve CAR-T Cell Persistence. International Journal of Molecular Sciences, 24(15), 12317. https://doi.org/10.3390/ijms241512317