
The Role of Intracellular Ca2+ and Mitochondrial ROS in Small Aβ1-42 Oligomer-Induced Microglial Death


Abstract
1. Introduction
2. Results
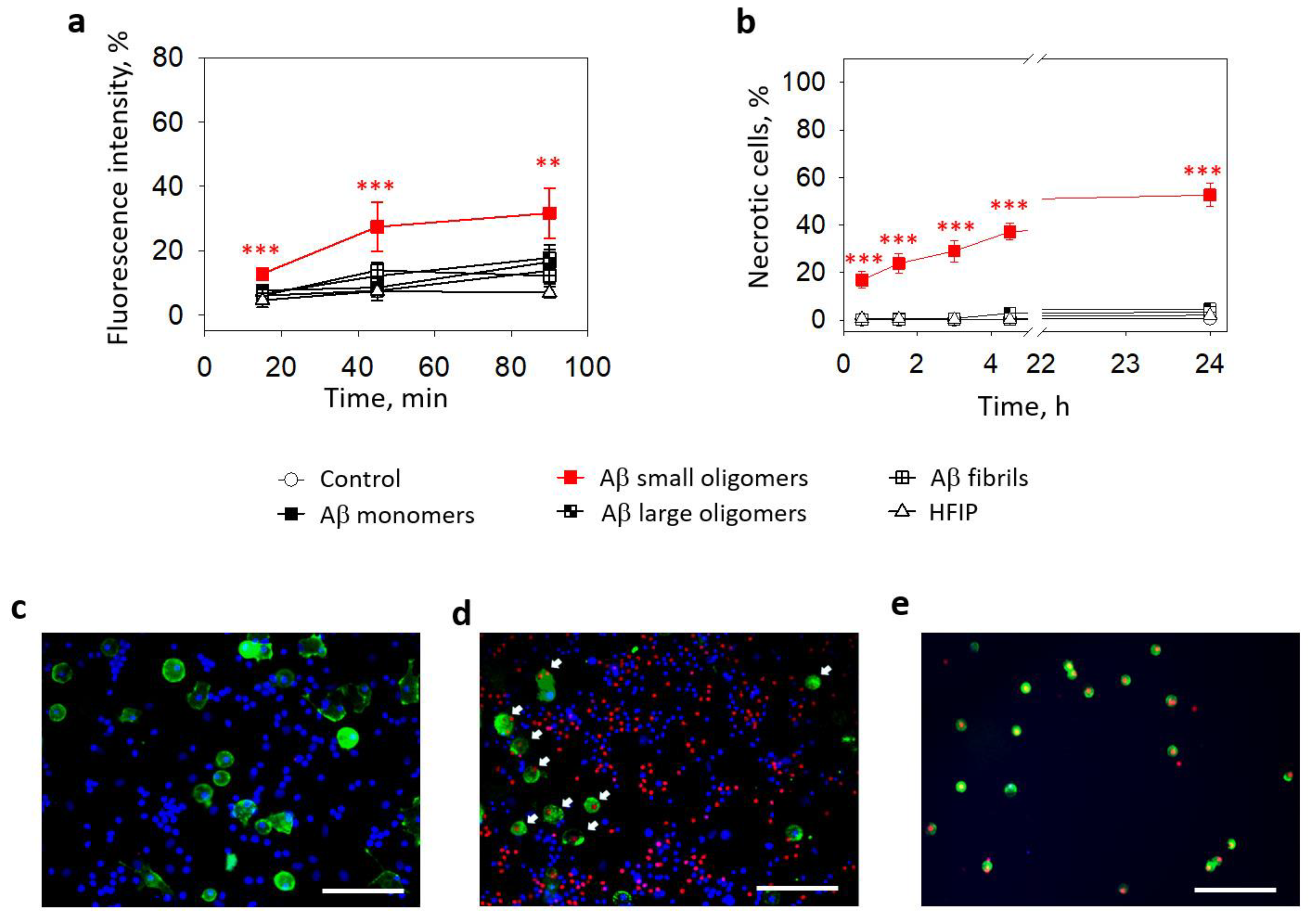
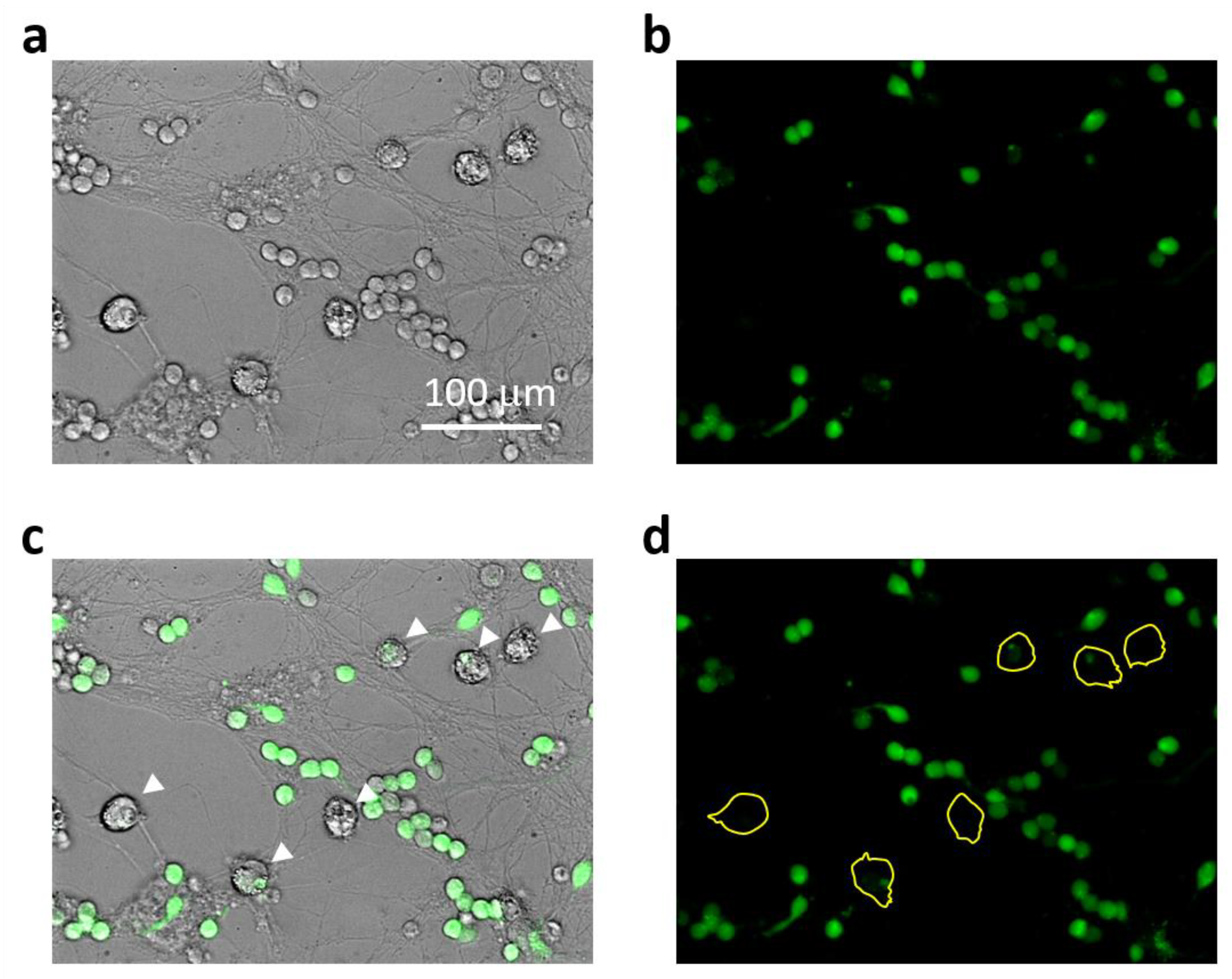
2.1. The Role of NMDARs in Aβ1-42-Induced Microglial Cytoplasmic Calcium Increase and Death
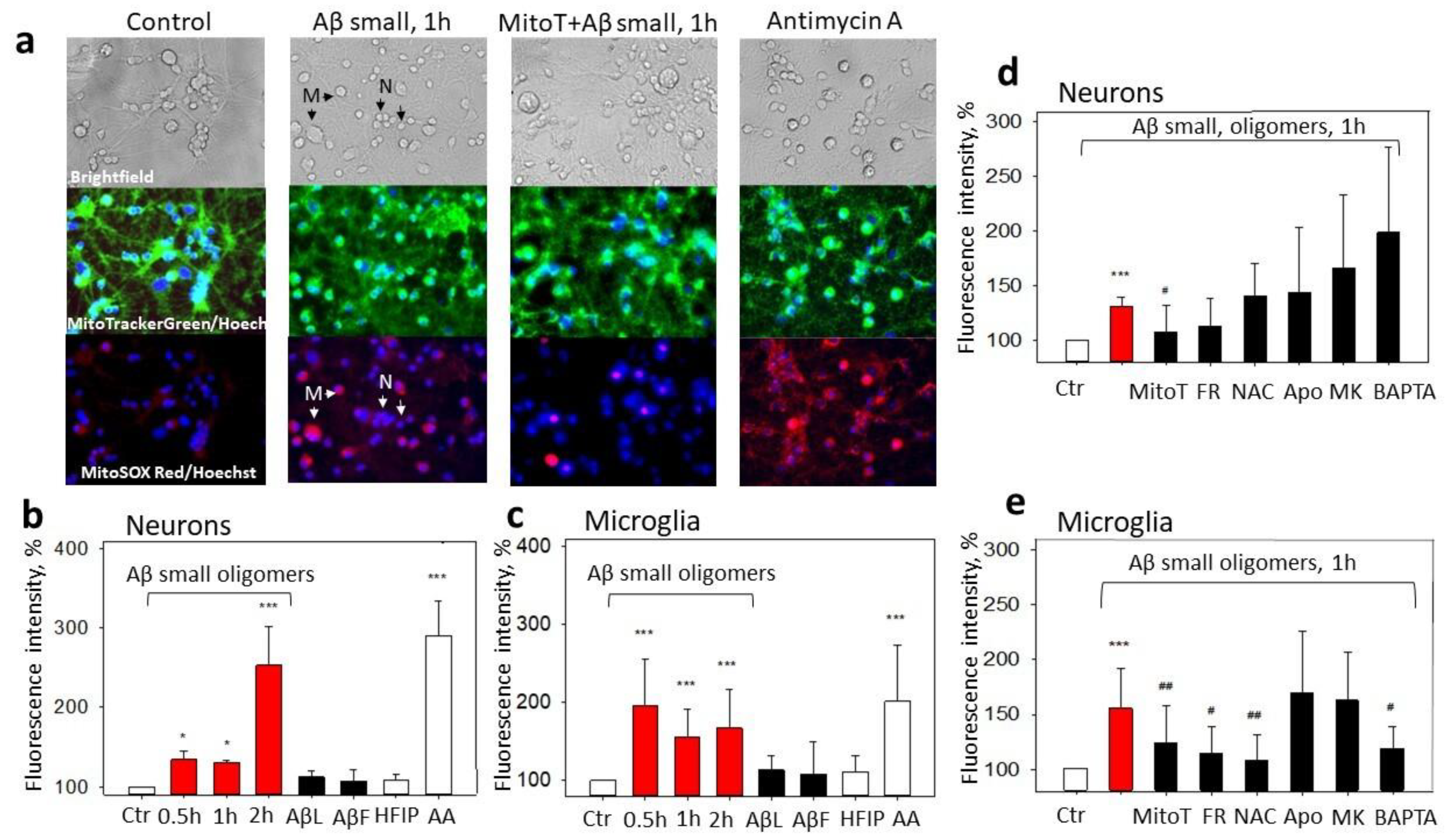
2.2. The Effect of Aβ1-42 Species on Superoxide Production by Mitochondria
2.3. The Role of Mitochondrial ROS in Small Aβ1-42 Oligomer-Induced Neuronal and Microglial Death
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of Aβ1-42 Aggregates
4.3. Cell Cultures and Treatments
4.4. Measurement of Intracellular Calcium Concentration
4.5. Cell Viability Assay
4.6. Mitochondrial Superoxide Measurements
4.7. Mitochondrial Membrane Potential Assessment
4.8. Measurement of Glutamate Concentration
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: London, UK, 2021. [Google Scholar]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed]
- Fani, G.; Mannini, B.; Vecchi, G.; Cascella, R.; Cecchi, C.; Dobson, C.M.; Vendruscolo, M.; Chiti, F. Aβ Oligomers Dysregulate Calcium Homeostasis by Mechanosensitive Activation of AMPA and NMDA Receptors. ACS Chem. Neurosci. 2021, 12, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’reilly, J.-A.; Mills, K.H.G.; Lynch, M.A. IFN-γ Production by Amyloid β–Specific Th1 Cells Promotes Microglial Activation and Increases Plaque Burden in a Mouse Model of Alzheimer’s Disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S.D. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Morkuniene, R.; Cizas, P.; Jankeviciute, S.; Petrolis, R.; Arandarcikaite, O.; Krisciukaitis, A.; Borutaite, V. Small Aβ1-42oligomer-induced membrane depolarization of neuronal and microglial cells: Role of N-methyl-D-aspartate receptors. J. Neurosci. Res. 2015, 93, 475–486. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; Le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef]
- Murugan, M.; Sivakumar, V.; Lu, J.; Ling, E.-A.; Kaur, C. Expression of N-methyl D-aspartate receptor subunits in amoeboid microglia mediates production of nitric oxide via NF-κB signaling pathway and oligodendrocyte cell death in hypoxic postnatal rats. Glia 2011, 59, 521–539. [Google Scholar] [CrossRef]
- Perkins, D.O.; Jeffries, C.D.; Do, K.Q. Potential Roles of Redox Dysregulation in the Development of Schizophrenia. Biol. Psychiatry 2020, 88, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Bodhinathan, K.; Kumar, A.; Foster, T.C. Intracellular Redox State Alters NMDA Receptor Response during Aging through Ca2+/Calmodulin-Dependent Protein Kinase II. J. Neurosci. 2010, 30, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three Distinct Mechanisms Generate Oxygen Free Radicals in Neurons and Contribute to Cell Death during Anoxia and Reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef]
- Esteras, N.; Kopach, O.; Maiolino, M.; Lariccia, V.; Amoroso, S.; Qamar, S.; Wray, S.; Rusakov, D.A.; Jaganjac, M.; Abramov, A.Y. Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimer’s Dement. 2022, 18, 318–338. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.-C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD enhances Aβ-induced cell stress via mitochondrial dysfunction. FASEB J. 2005, 19, 597–598. [Google Scholar] [CrossRef]
- Morsy, A.; Trippier, P.C. Amyloid-Binding Alcohol Dehydrogenase (ABAD) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2019, 62, 4252–4264. [Google Scholar] [CrossRef]
- Pan, R.-Y.; Ma, J.; Kong, X.-X.; Wang, X.-F.; Li, S.-S.; Qi, X.-L.; Yan, Y.-H.; Cheng, J.; Liu, Q.; Jin, W.; et al. Sodium rutin ameliorates Alzheimer’s disease–like pathology by enhancing microglial amyloid-β clearance. Sci. Adv. 2019, 5, eaau6328. [Google Scholar] [CrossRef]
- Traber, J.; Suter, M.; Walter, P.; Richter, C. In vivo modulation of total and mitochondrial glutathione in rat liver: Depletion by phorone and rescue by N-acetylcysteine. Biochem. Pharmacol. 1992, 43, 961–964. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001, 495, 12–15. [Google Scholar] [CrossRef]
- Cizas, P.; Budvytyte, R.; Morkuniene, R.; Moldovan, R.; Broccio, M.; Lösche, M.; Niaura, G.; Valincius, G.; Borutaite, V. Size-dependent neurotoxicity of β-amyloid oligomers. Arch. Biochem. Biophys. 2010, 496, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef]
- Woollacott, I.O.C.; Toomey, C.E.; Strand, C.; Courtney, R.; Benson, B.C.; Rohrer, J.D.; Lashley, T. Microglial burden, activation and dystrophy patterns in frontotemporal lobar degeneration. J. Neuroinflam. 2020, 17, 234. [Google Scholar] [CrossRef] [PubMed]
- Shahidehpour, R.K.; Higdon, R.E.; Crawford, N.G.; Neltner, J.H.; Ighodaro, E.T.; Patel, E.; Price, D.; Nelson, P.T.; Bachstetter, A.D. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 2021, 99, 19–27. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Brawek, B.; Schwendele, B.; Riester, K.; Kohsaka, S.; Lerdkrai, C.; Liang, Y.; Garaschuk, O. Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 2014, 127, 495–505. [Google Scholar] [CrossRef]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.-F.; Guo, L.; Chen, D.; Stern, D.M.; Moore, F.J.G.; et al. Inhibition of Amyloid-β (Aβ) Peptide-Binding Alcohol Dehydrogenase-Aβ Interaction Reduces Aβ Accumulation and Improves Mitochondrial Function in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; López, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox Control of Microglial Function: Molecular Mechanisms and Functional Significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef]
- Nesci, S. The mitochondrial permeability transition pore in cell death: A promising drug binding bioarchitecture. Med. Res. Rev. 2020, 40, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ Oligomers Induce Neuronal Oxidative Stress through an N-Methyl-D-aspartate Receptor-dependent Mechanism That Is Blocked by the Alzheimer Drug Memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Eun, S.-Y.; Hong, Y.H.; Kim, E.H.; Jeon, H.; Suh, Y.H.; Lee, J.E.; Jo, C.; Jo, S.A.; Kim, J. Glutamate receptor-mediated regulation of c-fos expression in cultured microglia. Biochem. Biophys. Res. Commun. 2004, 325, 320–327. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Estrada-Sánchez, A.M.; Montiel, T.; Páramo, B.; Massieu, L.; Morán, J. Activation of NOX2 by the Stimulation of Ionotropic and Metabotropic Glutamate Receptors Contributes to Glutamate Neurotoxicity In Vivo Through the Production of Reactive Oxygen Species and Calpain Activation. J. Neuropathol. Exp. Neurol. 2011, 70, 1020–1035. [Google Scholar] [CrossRef]
- Liang, J.; Takeuchi, H.; Jin, S.; Noda, M.; Li, H.; Doi, Y.; Kawanokuchi, J.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Glutamate induces neurotrophic factor production from microglia via protein kinase C pathway. Brain Res. 2010, 1322, 8–23. [Google Scholar] [CrossRef]
- Yeckel, M.F.; Kapur, A.; Johnston, D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat. Neurosci. 1999, 2, 625–633. [Google Scholar] [CrossRef]
- Verkhratsky, A. Physiology and Pathophysiology of the Calcium Store in the Endoplasmic Reticulum of Neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.L.; Ferreiro, E.; Schmidt, J.; Cardoso, J.M.; Pereira, C.M.; Carvalho, A.L.; Oliveira, C.R.; Rego, A.C. Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol. Aging 2015, 36, 680–692. [Google Scholar] [CrossRef]
- Jekabsone, A.; Mander, P.K.; Tickler, A.; Sharpe, M.; Brown, G.C. Fibrillar beta-amyloid peptide Aβ1–40 activates microglial proliferation via stimulating TNF-α release and H2O2 derived from NADPH oxidase: A cell culture study. J. Neuroinflam. 2006, 3, 24. [Google Scholar] [CrossRef]
- Bal-Price, A.; Brown, G.C. Inflammatory Neurodegeneration Mediated by Nitric Oxide from Activated Glia-Inhibiting Neuronal Respiration, Causing Glutamate Release and Excitotoxicity. J. Neurosci. 2001, 21, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Tricklebank, M.D.; Singh, L.; Oles, R.J.; Preston, C.; Iversen, S.D. The behavioural effects of MK-801: A comparison with antagonists acting non-competitively and competitively at the NMDA receptor. Eur. J. Pharmacol. 1989, 167, 127–135. [Google Scholar] [CrossRef]
- Lipton, S. The Molecular Basis of Memantine Action in Alzheimers Disease and Other Neurologic Disorders: Low-affinity, Uncompetitive Antagonism. Curr. Alzheimer Res. 2005, 2, 155–165. [Google Scholar] [CrossRef]
- Pedre, B.; Barayeu, U.; Ezeriņa, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Stefanska, J.; Pawliczak, R. Apocynin: Molecular Aptitudes. Mediat. Inflamm. 2008, 2008, 106507. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Mechanism of Action |
|---|---|
| MK801 | A potent noncompetitive NMDAR antagonist [47]. |
| Memantine | A low-affinity noncompetitive NMDAR antagonist [48]. |
| D-2-Amino-5-phosphopentanoic acid | A competitive NMDAR antagonist [47]. |
| BAPTA | A selective cell-permeable Ca2+ chelator (information provided by manufacturer). |
| MitoTEMPO | A mitochondrially targeted antioxidant and specific scavenger of mitochondrial superoxide (information provided by manufacturer). |
| Frentizole | An inhibitor of amyloid beta peptide binding alcohol dehydrogenase (ABAD)–Aβ interaction (information provided by manufacturer). |
| N-Acetyl L-Cysteine | A disulfide reductant, a direct scavenger of oxidants, and a driver of glutathione synthesis [20,49]. |
| Apocynin | A potent and selective inhibitor of NADPH oxidase [50]. |
| Antimycin A | An inhibitor of mitochondrial cytochrome bc1 (complex III), often used as a positive control for superoxide generation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jekabsone, A.; Jankeviciute, S.; Pampuscenko, K.; Borutaite, V.; Morkuniene, R. The Role of Intracellular Ca2+ and Mitochondrial ROS in Small Aβ1-42 Oligomer-Induced Microglial Death. Int. J. Mol. Sci. 2023, 24, 12315. https://doi.org/10.3390/ijms241512315
Jekabsone A, Jankeviciute S, Pampuscenko K, Borutaite V, Morkuniene R. The Role of Intracellular Ca2+ and Mitochondrial ROS in Small Aβ1-42 Oligomer-Induced Microglial Death. International Journal of Molecular Sciences. 2023; 24(15):12315. https://doi.org/10.3390/ijms241512315
Chicago/Turabian StyleJekabsone, Aiste, Silvija Jankeviciute, Katryna Pampuscenko, Vilmante Borutaite, and Ramune Morkuniene. 2023. "The Role of Intracellular Ca2+ and Mitochondrial ROS in Small Aβ1-42 Oligomer-Induced Microglial Death" International Journal of Molecular Sciences 24, no. 15: 12315. https://doi.org/10.3390/ijms241512315
APA StyleJekabsone, A., Jankeviciute, S., Pampuscenko, K., Borutaite, V., & Morkuniene, R. (2023). The Role of Intracellular Ca2+ and Mitochondrial ROS in Small Aβ1-42 Oligomer-Induced Microglial Death. International Journal of Molecular Sciences, 24(15), 12315. https://doi.org/10.3390/ijms241512315