

EP300 as a Molecular Integrator of Fibrotic Transcriptional Programs

 ,
,  , ,
, ,
Abstract
1. Introduction
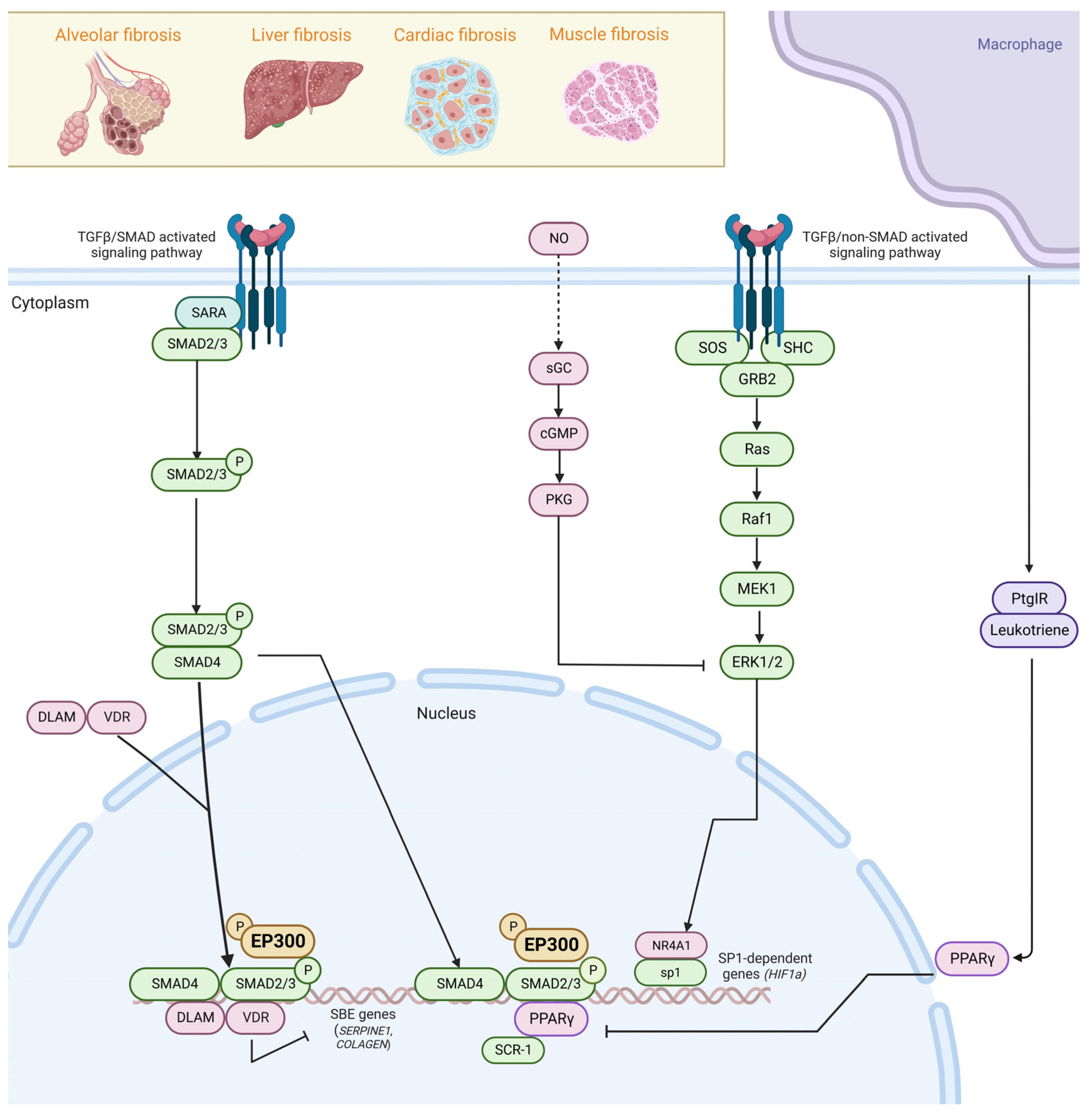
2. Pathogenesis of Fibrosis
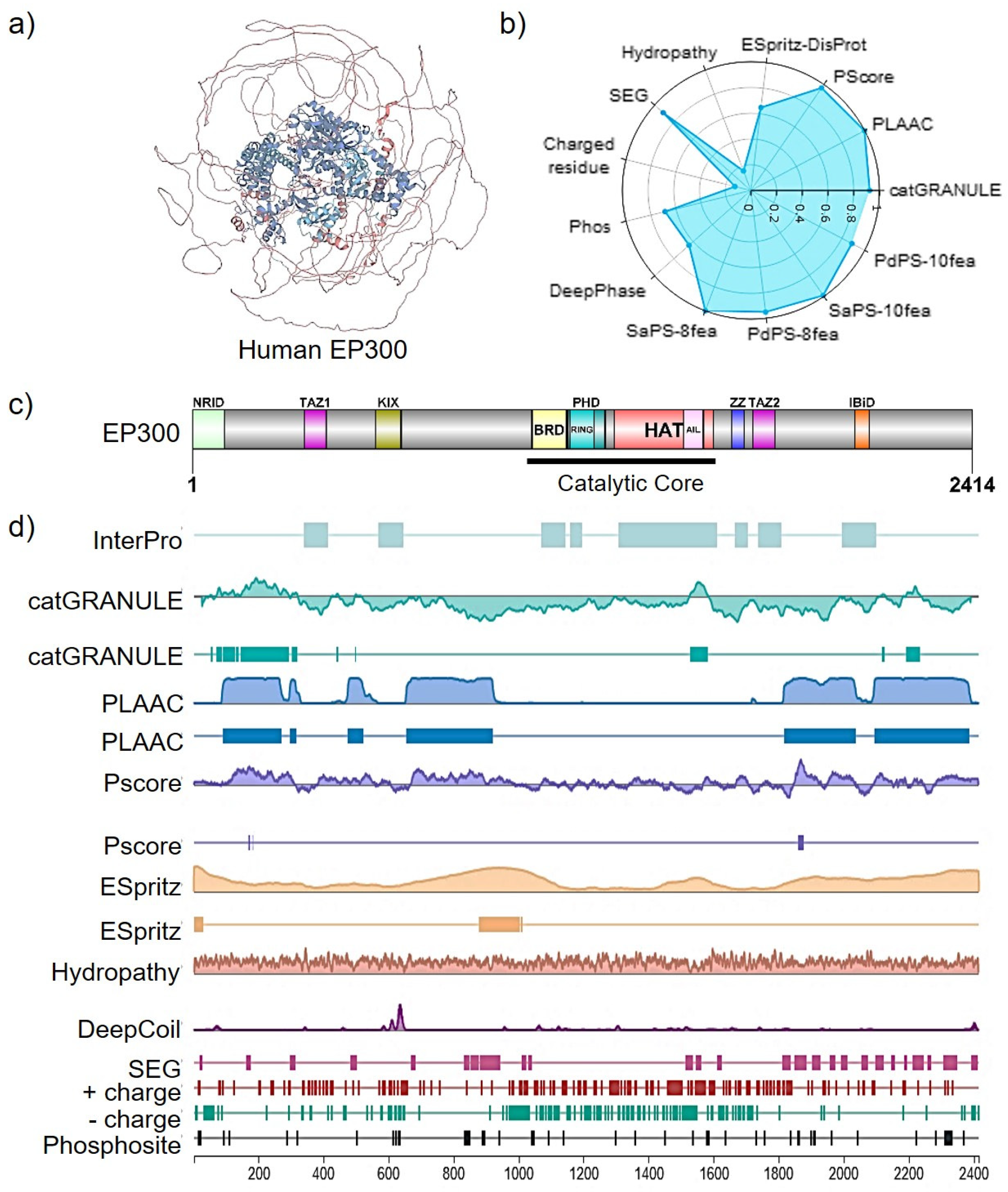
3. EP300 Functional Domains, Enzymatic Activity, and Cellular Distribution
4. Multiorgan EP300 Dysregulation
4.1. Renal Fibrosis
4.2. Cardiac Fibrosis
4.3. Lung Fibrosis
4.4. Liver Fibrosis
5. Epithelial-to-Mesenchymal Transition and TGFβ Crosstalk
6. EP300 Inhibitors
6.1. Pharmacological EP300 HAT Inhibitors
6.1.1. C646
6.1.2. A485
6.1.3. (E)-3-(3-(4-((3-Carbamoylbenzyl) oxy)-3-iodo-5-methoxyphenyl)acryloyl)benzamide(A6)
6.1.4. L002
6.2. Natural Products as EP300 HAT Inhibitors
6.2.1. Plumbagin
6.2.2. Curcumin
6.3. Pharmacological EP300 Bromodomain Inhibitors
6.3.1. SGC-CBP30 and I-CBP112
6.3.2. Clinical Trials with CBP/EP300 Bromodomain Inhibitors
6.4. EP300 Degraders
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dou, C.; Liu, Z.; Tu, K.; Zhang, H.; Chen, C.; Yaqoob, U.; Wang, Y.; Wen, J.; van Deursen, J.; Sicard, D.; et al. P300 Acetyltransferase Mediates Stiffness-Induced Activation of Hepatic Stellate Cells Into Tumor-Promoting Myofibroblasts. Gastroenterology 2018, 154, 2209–2221.e14. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wei, B.; Liu, M.; Hirsova, P.; Sehrawat, T.S.; Cao, S.; Hu, X.; Xue, F.; Yaqoob, U.; Kang, N.; et al. Endothelial p300 Promotes Portal Hypertension and Hepatic Fibrosis Through C-C Motif Chemokine Ligand 2–Mediated Angiocrine Signaling. Hepatology 2021, 73, 2468–2483. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bhattacharyya, S.; Lafyatis, R.; Farina, G.; Yu, J.; Thimmapaya, B.; Wei, J.; Varga, J. p300 Is Elevated in Systemic Sclerosis and Its Expression Is Positively Regulated by TGF-β: Epigenetic Feed-Forward Amplification of Fibrosis. J. Investig. Dermatol. 2013, 133, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Niu, Y.; Liang, K.; Du, Y. Mechanical communication in fibrosis progression. Trends Cell Biol. 2022, 32, 70–90. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Günther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.-M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 2229. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Lyv, X.; Zhou, Q.J.; Xiang, Z.; Stanford, D.; Bodduluri, S.; Rowe, S.M.; Thannickal, V.J. Brd4-p300 inhibition downregulates Nox4 and ac-celerates lung fibrosis resolution in aged mice. JCI Insight 2020, 5, e137127. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Xue, T.; Qiu, X.; Liu, H.; Gan, C.; Tan, Z.; Xie, Y.; Wang, Y.; Ye, T. Epigenetic regulation in fibrosis progress. Pharmacol. Res. 2021, 173, 105910. [Google Scholar] [CrossRef]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.-F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Przyborowski, M.; Berthiaume, F. Stem Cells for Skin Tissue Engineering and Wound Healing. Crit. Rev. Biomed. Eng. 2009, 37, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, E.; Watt, F.M. Skin Cell Heterogeneity in Development, Wound Healing, and Cancer. Trends Cell Biol. 2018, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, H.P.; Longaker, M.T.; Perkocha, L.A.; Jennings, R.W.; Harrison, M.R.; Adzick, N.S. Scarless wound repair: A human fetal skin model. Development 1992, 114, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.; Liu, Y. Targeting Hypoxia Inducible Factors-1α As a Novel Therapy in Fibrosis. Front. Pharmacol. 2017, 8, 326. [Google Scholar] [CrossRef]
- Jiménez-Uribe, A.P.; Gómez-Sierra, T.; Aparicio-Trejo, O.E.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Backstage players of fi-brosis: NOX4, mTOR, HDAC, and S1P; companions of TGF-β. Cell. Signal. 2021, 87, 110123. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, K.; Li, Q.; Pang, H.; Pan, Z.; Huang, X.; Wang, L.; Wu, F.; He, G. Recent Advances on Small-Molecule Bromodomain-Containing Histone Acetyltransferase Inhibitors. J. Med. Chem. 2023, 66, 1678–1699. [Google Scholar] [CrossRef]
- Arndt, L.; Lindhorst, A.; Neugebauer, J.; Hoffmann, A.; Hobusch, C.; Alexaki, V.-I.; Ghosh, A.; Blüher, M.; Wolfrum, C.; Gla ß, M.; et al. The Role of IL-13 and IL-4 in Adipose Tissue Fibrosis. Int. J. Mol. Sci. 2023, 24, 5672. [Google Scholar] [CrossRef]
- Vierhout, M.; Ayoub, A.; Naiel, S.; Yazdanshenas, P.; Revill, S.D.; Reihani, A.; Dvorkin-Gheva, A.; Shi, S.; Ask, K. Monocyte and macrophage derived myofi-broblasts: Is it fate? A review of the current evidence. Wound Repair Regen. 2021, 29, 548–562. [Google Scholar] [CrossRef]
- Schuster, R.; Younesi, F.; Ezzo, M.; Hinz, B. The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harb. Perspect. Biol. 2023, 15, a041231. [Google Scholar] [CrossRef]
- Mehal, W.Z.; Iredale, J.; Friedman, S.L. Scraping fibrosis: Expressway to the core of fibrosis. Nat. Med. 2011, 17, 552–553. [Google Scholar] [CrossRef]
- Jeong, H.-H.; Jia, J.; Dai, Y.; Simon, L.M.; Zhao, Z. Investigating Cellular Trajectories in the Severity of COVID-19 and Their Transcriptional Programs Using Machine Learning Approaches. Genes 2021, 12, 635. [Google Scholar] [CrossRef]
- Dobersch, S.; Rubio, K.; Singh, I.; Günther, S.; Graumann, J.; Cordero, J.; Castillo-Negrete, R.; Huynh, M.B.; Mehta, A.; Braubach, P. Positioning of nucleosomes containing γ-H2AX precedes active DNA demethylation and transcription initiation. Nat. Commun. 2021, 12, 1072. [Google Scholar] [CrossRef]
- Amos, L.A.; Ma, F.Y.; Tesch, G.H.; Liles, J.T.; Breckenridge, D.G.; Nikolic-Paterson, D.J.; Han, Y. ASK1 inhibitor treatment suppresses p38/JNK signalling with reduced kidney inflammation and fibrosis in rat crescentic glomerulonephritis. J. Cell. Mol. Med. 2018, 22, 4522–4533. [Google Scholar] [CrossRef]
- Liu, L.; Song, W.; Zeng, J.; Peng, Z. Evaluating a Specific Dual ROCK Inhibitor against Bleomycin-Induced Idiopathic Pulmonary Fibrosis in Rats. ACS Pharmacol. Transl. Sci. 2022, 5, 819–828. [Google Scholar] [CrossRef]
- Antognelli, C.; Moretti, S.; Frosini, R.; Puxeddu, E.; Sidoni, A.; Talesa, V.N. Methylglyoxal Acts as a Tumor-Promoting Factor in Anaplastic Thyroid Cancer. Cells 2019, 8, 547. [Google Scholar] [CrossRef]
- Liu, Z.; Li, S.; Qian, X.; Li, L.; Zhang, H. RhoA/ROCK-YAP/TAZ Axis Regulates the Fibrotic Activity in Dexamethasone-Treated Human Trabecular Meshwork Cells. Front. Mol. Biosci. 2021, 8, 728932. [Google Scholar] [CrossRef]
- Mikhailova, E.V.; Romanova, I.V.; Bagrov, A.Y.; Agalakova, N.I. Fli1 and Tissue Fibrosis in Various Diseases. Int. J. Mol. Sci. 2023, 24, 1881. [Google Scholar] [CrossRef]
- Dees, C.; Pötter, S.; Zhang, Y.; Bergmann, C.; Zhou, X.; Luber, M.; Wohlfahrt, T.; Karouzakis, E.; Ramming, A.; Gelse, K.; et al. TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Investig. 2020, 130, 2347–2363. [Google Scholar] [CrossRef]
- Schinner, E.; Wetzl, V.; Schramm, A.; Kees, F.; Sandner, P.; Stasch, J.P.; Hofmann, F.; Schlossmann, J. Inhibition of the TGFβ signalling pathway by cGMP and cGMP-dependent kinase I in renal fibrosis. FEBS Open Bio 2017, 7, 550–561. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bhattacharyya, S.; Wei, J.; Kim, S.; Barak, Y.; Mori, Y.; Varga, J. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. 2009, 23, 2968–2977. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.L.; Liu, Z.; Shah, V.H.; Dou, C.; et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor β-Stimulated Hepatic Stellate Cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.-P.; Branton, P.E. Detection of cellular proteins associated with human adenovirus type 5 early region 1A polypeptides. Virology 1985, 147, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Schölz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018, 174, 231–244.e12. [Google Scholar] [CrossRef]
- Martire, S.; Nguyen, J.; Sundaresan, A.; Banaszynski, L.A. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol. Cell Biol. 2020, 21, 55. [Google Scholar] [CrossRef]
- Delvecchio, M.; Gaucher, J.; Aguilar-Gurrieri, C.; Ortega, E.; Panne, D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 2013, 20, 1040–1046. [Google Scholar] [CrossRef]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, T.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef]
- Thompson, P.R.; Wang, D.; Wang, L.; Fulco, M.; Pediconi, N.; Zhang, D.; An, W.; Ge, Q.; Roeder, R.G.; Wong, J.; et al. Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol. 2004, 11, 308–315. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J. Biol. Chem. 2016, 291, 6714–6722. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, Y.; Shi, J.; Ahn, J.; Mi, W.; Ali, M.; Wang, X.; Klein, B.J.; Wen, H.; Li, W.; et al. The ZZ domain of p300 mediates specificity of the adjacent HAT domain for histone H3. Nat. Struct. Mol. Biol. 2018, 25, 841–849. [Google Scholar] [CrossRef]
- Ibrahim, Z.; Wang, T.; Destaing, O.; Salvi, N.; Hoghoughi, N.; Chabert, C.; Rusu, A.; Gao, J.; Feletto, L.; Reynoird, N.; et al. Structural insights into p300 regulation and acetylation-dependent genome organisation. Nat. Commun. 2022, 13, 7759. [Google Scholar] [CrossRef]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2552. [Google Scholar] [CrossRef]
- Grossman, S.R.; Deato, M.E.; Brignone, C.; Chan, H.M.; Kung, A.L.; Tagami, H.; Nakatani, Y.; Livingston, D.M. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003, 300, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Pop, M.S.; Kulikov, R.; Love, I.M.; Kung, A.L.; Grossman, S.R. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc. Natl. Acad. Sci. USA 2009, 106, 16275–16280. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’agnese, A.; Young, R.A. Biomolecular Condensates in the Nucleus. Trends Biochem. Sci. 2020, 45, 961–977. [Google Scholar] [CrossRef]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2021, 22, 215–235. [Google Scholar] [CrossRef]
- Ma, L.; Gao, Z.; Wu, J.; Zhong, B.; Xie, Y.; Huang, W.; Lin, Y. Co-condensation between transcription factor and coactivator p300 modulates transcriptional bursting kinetics. Mol. Cell 2021, 81, 1682–1697.e7. [Google Scholar] [CrossRef]
- Efstratiadis, G.; Divani, M.; Katsioulis, E.; Vergoulas, G. Renal fibrosis. Hippokratia 2009, 13, 224–229. [Google Scholar]
- Chun, P. Chapter 34—Histone Deacetylase Inhibitors in Medical Therapeutics. In Medical Epigenetics; Academic Press: Cambridge, MA, USA, 2016; pp. 633–655. ISBN 9780128032398. [Google Scholar]
- Gong, Y.; Dou, Y.; Wang, L.; Wang, X.; Zhao, Z. EP300 promotes renal tubular epithelial cell fibrosis by increasing HIF2α expression in diabetic nephropathy. Cell. Signal. 2022, 98, 110407. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, W.; Niu, Y.; Zhu, S.; Zhang, Y.; Li, X.; Yu, C. Lysyl oxidase promotes renal fibrosis via accelerating collagen cross-link driving by β-arrestin/ERK/STAT3 pathway. FASEB J. 2022, 36, e22427. [Google Scholar] [CrossRef] [PubMed]
- Cippà, P.E.; Sun, B.; Liu, J.; Chen, L.; Naesens, M.; McMahon, A.P. Transcriptional trajectories of human kidney injury progression. J. Clin. Investig. 2018, 3, e123151. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, L.; Yang, H.; Chen, X.; Zheng, H.; Liao, X. SIK2 protects against renal tubular injury and the progression of diabetic kidney disease. Transl. Res. 2023, 253, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.Y.; Tin, A.; Schlosser, P.; Ko, Y.-A.; Qiu, C.; Yao, C.; Joehanes, R.; Grams, M.E.; Liang, L.; Gluck, C.A.; et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 2017, 8, 1286. [Google Scholar] [CrossRef]
- Stanton, T.; Leano, R.; Marwick, T.H. Prediction of all-cause mortality from global longitudinal speckle strain: Comparison with ejection fraction and wall motion scoring. Circ. Cardiovasc. Imaging 2009, 2, 356–364. [Google Scholar] [CrossRef]
- Moukette, B.; Barupala, N.P.; Aonuma, T.; Sepulveda, M.; Kawaguchi, S.; Kim, I.-M. Interactions between noncoding RNAs as epigenetic regulatory mechanisms in cardiovascular diseases. Methods Cell Biol. 2021, 166, 309–348. [Google Scholar] [CrossRef]
- Brønnum, H.; Kalluri, R. Chapter 29—Cardiac Fibrosis: Cellular and Molecular Determinants. In Muscle; Academic Press: Cambridge, MA, USA, 2012; pp. 389–404. ISBN 9780123815101. [Google Scholar] [CrossRef]
- Gao, X.Y.; Lai, Y.Y.; Luo, X.S.; Peng, D.W.; Li, Q.Q.; Zhou, H.S.; Xue, Y.M.; Guo, H.M.; Zhao, J.F.; Yang, H.; et al. Acetyltransferase p300 regulates atrial fibroblast senescence and age-related atrial fibrosis through p53/Smad3 axis. Aging Cell 2023, 22, e13743. [Google Scholar] [CrossRef]
- Fan, M.; Yang, K.; Wang, X.; Chen, L.; Gill, P.S.; Ha, T.; Liu, L.; Lewis, N.H.; Williams, D.L.; Li, C. Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci. Adv. 2023, 9, eadc9465. [Google Scholar] [CrossRef]
- Lim, Y.; Jeong, A.; Kwon, D.H.; Lee, Y.U.; Kim, Y.K.; Ahn, Y.; Kook, T.; Park, W.J.; Kook, H. P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. Int. J. Mol. Sci. 2021, 22, 9944. [Google Scholar] [CrossRef]
- Chunyu, Z.; Zaicheng, X.; Cong, L.; Caiyu, C.; Yang, Y.; Cao, N.; Yu, J.; Luo, H.; Chen, K.; Guo, L.; et al. Heart-Enriched Long Noncoding RNA NPPA-AS1 Regulates Pathological Cardiac Hypertrophy by Initiating a EP300-GATA4 Axis. Res. Sq. 2022, preprint. [Google Scholar] [CrossRef]
- Gambelunghe, A.; Giovagnoli, S.; Di Michele, A.; Boncompagni, S.; Dell’omo, M.; Leopold, K.; Iavicoli, I.; Talesa, V.N.; Antognelli, C. Redox-Sensitive Glyoxalase 1 Up-Regulation Is Crucial for Protecting Human Lung Cells from Gold Nanoparticles Toxicity. Antioxidants 2020, 9, 697. [Google Scholar] [CrossRef]
- George, G.; Ramirez, M.I.; Roman, J. Chapter—Lung Mesenchyme. In Murray & Nadel’s Textbook of Respiratory Medicine; Elsevier Health Sciences: Maryland Heights, MO, USA, 2021; Volume 5, pp. 66–75.e4. [Google Scholar]
- Murray, L.; Homer, R.J.; Gulati, M.; Herzog, E. Chapter: Pulmonary Fibrosis. In Pathobiology of Human Disease; Academic Press: Cambridge, MA, USA, 2014; pp. 2636–2653. ISBN 9780123864574. [Google Scholar]
- Hua, H.-S.; Wen, H.-C.; Weng, C.-M.; Lee, H.-S.; Chen, B.-C.; Lin, C.-H. Histone deacetylase 7 mediates endothelin-1-induced connective tissue growth factor expression in human lung fibroblasts through p300 and activator protein-1 activation. J. Biomed. Sci. 2021, 28, 38. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Hasegawa, D.; Wallace, M.C.; Friedman, S.L. Chapter 4—Stellate Cells and Hepatic Fibrosis. In Stellate Cells in Health and Disease; Academic Press: Cambridge, MA, USA, 2015; pp. 41–62. ISBN 9780128001349. [Google Scholar]
- Declerck, P.J.; Gils, A.; De Taeye, B. Chapter five—Use of Mouse Models to Study Plasminogen Activator Inhibitor-1. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2011; Volume 499, pp. 77–104. ISBN 9780123864710. [Google Scholar]
- Wang, Z.-Y.; Keogh, A.; Waldt, A.; Cuttat, R.; Neri, M.; Zhu, S.; Schuierer, S.; Ruchti, A.; Crochemore, C.; Knehr, J.; et al. Single-cell and bulk transcriptomics of the liver reveals potential targets of NASH with fibrosis. Sci. Rep. 2021, 11, 19396. [Google Scholar] [CrossRef]
- Zhao, J.; Peng, L.; Cui, R.; Guo, X.; Yan, M. Dimethyl α-ketoglutarate reduces CCl. Biochem. Biophys. Res. Commun. 2016, 481, 90–96. [Google Scholar] [CrossRef]
- Sampieri, C.L.; Orozco-Ortega, R.A. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in chronic kidney disease and acute kidney injury: A systematic review of the literature. Hippokratia 2018, 22, 99–104. [Google Scholar]
- Pinto, S.; Hoek, M.; Huang, Y.; Costet, P.; Ma, L.; Imbriglio, J.E. Drug Discovery in Tissue Fibrosis. In Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 7.19; pp. 694–713. ISBN 9780128032015. [Google Scholar]
- Stanciu, A.E.; Vatasescu, R.G.; Stanciu, M.M.; Serdarevic, N.; Dorobantu, M. The role of pro-fibrotic biomarkers in paroxysmal and persistent atrial fibrillation. Cytokine 2018, 103, 63–68. [Google Scholar] [CrossRef]
- Goldmann, T.; Zissel, G.; Watz, H.; Drömann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human alveolar epithelial cells type II are capable of TGFβ-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef]
- Gao, L.; Wang, L.Y.; Liu, Z.Q.; Jiang, D.; Wu, S.Y.; Guo, Y.Q.; Tao, H.M.; Sun, M.; You, L.N.; Qin, S.; et al. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-β1/Smads and activating P53 signaling pathways. Cell Death Dis. 2020, 11, 44. [Google Scholar] [CrossRef]
- Dees, C.; Schlottmann, I.; Funke, R.; Distler, A.; Palumbo-Zerr, K.; Zerr, P.; Lin, N.-Y.; Beyer, C.; Distler, O.; Schett, G.; et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann. Rheum. Dis. 2013, 73, 1232–1239. [Google Scholar] [CrossRef]
- Zeng, Z.; Cheng, S.; Chen, H.; Li, Q.; Hu, Y.; Wang, Q.; Zhu, X.; Wang, J. Activation and overexpression of Sirt1 attenuates lung fibrosis via P300. Biochem. Biophys. Res. Commun. 2017, 486, 1021–1026. [Google Scholar] [CrossRef]
- Xiao, Y.; Ye, J.; Zhou, Y.; Huang, J.; Liu, X.; Huang, B.; Zhu, L.; Wu, B.; Zhang, G.; Cai, Y. Baicalin inhibits pressure overload-induced cardiac fibrosis through regulating AMPK/TGF-β/Smads signaling pathway. Arch. Biochem. Biophys. 2018, 640, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Cecchetti, R.; Riuzzi, F.; Peirce, M.J.; Talesa, V.N. Glyoxalase 1 sustains the metastatic phenotype of prostate cancer cells via EMT control. J. Cell. Mol. Med. 2018, 22, 2865–2883. [Google Scholar] [CrossRef]
- Sunagawa, Y.; Shimizu, K.; Katayama, A.; Funamoto, M.; Shimizu, K.; Nurmila, S.; Shimizu, S.; Miyazaki, Y.; Katanasaka, Y.; Hasegawa, K.; et al. Metformin suppresses phenylephrine-induced hypertrophic responses by inhibiting p300-HAT activity in cardiomyocytes. J. Pharmacol. Sci. 2021, 147, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Han, J.; Wang, X.; Wu, T.; Zhang, H.; An, H.; Qin, L.; Sun, Y.; Zhong, W.; Yang, C.; et al. Twist1-YY1-p300 complex promotes the malignant progression of HCC through activation of miR-9 by forming phase-separated condensates at super-enhancers and relieved by metformin. Pharmacol. Res. 2023, 188, 106661. [Google Scholar] [CrossRef] [PubMed]
- Szymczak-Pajor, I.; Drzewoski, J.; Świderska, E.; Strycharz, J.; Gabryanczyk, A.; Kasznicki, J.; Bogdańska, M.; Śliwińska, A. Metformin Induces Apoptosis in Human Pancreatic Cancer (PC) Cells Accompanied by Changes in the Levels of Histone Acetyltransferases (Particularly, p300/CBP-Associated Factor (PCAF) Protein Levels). Pharmaceuticals 2023, 16, 115. [Google Scholar] [CrossRef]
- Yang, H.; Pinello, C.E.; Luo, J.; Li, D.; Wang, Y.; Zhao, L.Y.; Jahn, S.C.; Saldanha, S.A.; Chase, P.; Planck, J.; et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol. Cancer Ther. 2013, 12, 610–620. [Google Scholar] [CrossRef]
- Rai, R.; Verma, S.K.; Kim, D.; Ramirez, V.; Lux, E.; Li, C.; Sahoo, S.; Wilsbacher, L.D.; Vaughan, D.E.; Quaggin, S.E.; et al. A novel acetyltransferase p300 inhibitor ameliorates hypertension-associated cardio-renal fibrosis. Epigenetics 2017, 12, 1004–1013. [Google Scholar] [CrossRef]
- Hwang, S.-Y.; Park, S.-Y.; Hong, J.Y.; Lee, S.Y.; Shin, J.-H.; Na, Y.; Sohn, M.H.; Yoon, H.-G.; Kwon, Y. Field-based rational design of p300 histone acetyltransferase inhibitor and systematic evaluation as an anti-fibrotic agent. Chem. Commun. 2020, 56, 9795–9798. [Google Scholar] [CrossRef]
- Kim, H.; Park, S.-Y.; Lee, S.Y.; Kwon, J.-H.; Byun, S.; Kim, M.J.; Yu, S.; Yoo, J.-Y.; Yoon, H.-G. Therapeutic effects of selective p300 histone acetyl-transferase inhibitor on liver fibrosis. BMB Rep. 2023, 56, 114–119. [Google Scholar] [CrossRef]
- Su, H.; Zeng, H.; He, X.; Zhu, S.-H.; Chen, J.-X. Histone Acetyltransferase p300 Inhibitor Improves Coronary Flow Reserve in SIRT3 (Sirtuin 3) Knockout Mice. J. Am. Heart Assoc. 2020, 9, e017176. [Google Scholar] [CrossRef]
- Lazar, A.G.; Vlad, M.L.; Manea, A.; Simionescu, M.; Manea, S.A. Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney. Antioxidants 2021, 10, 1356. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Williams, L.M.; McCann, F.E.; Cabrita, M.A.; Layton, T.; Cribbs, A.; Knezevic, B.; Fang, H.; Knight, J.; Zhang, M.; Fischer, R.; et al. Identifying collagen VI as a target of fibrotic diseases regulated by CREBBP/EP300. Proc. Natl. Acad. Sci. USA 2020, 117, 20753–20763. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef]
- Sunagawa, Y.; Funamoto, M.; Shimizu, K.; Shimizu, S.; Sari, N.; Katanasaka, Y.; Miyazaki, Y.; Kakeya, H.; Hasegawa, K.; Morimoto, T. Curcumin, an Inhibitor of p300-HAT Activity, Suppresses the Development of Hypertension-Induced Left Ventricular Hypertrophy with Preserved Ejection Fraction in Dahl Rats. Nutrients 2021, 13, 2608. [Google Scholar] [CrossRef]
- Ravindra, K.C.; Selvi, B.R.; Arif, M.; Reddy, B.A.A.; Thanuja, G.R.; Agrawal, S.; Pradhan, S.K.; Nagashayana, N.; Dasgupta, D.; Kundu, T.K. Inhibition of Lysine Acetyltransferase KAT3B/p300 Activity by a Naturally Occurring Hydroxynaphthoquinone, Plumbagin. J. Biol. Chem. 2009, 284, 24453–24464. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, M.J.; Jang, S.; Lee, G.-E.; Hwang, S.-Y.; Kwon, Y.; Hong, J.Y.; Sohn, M.H.; Park, S.-Y.; Yoon, H.-G. Plumbagin Suppresses Pulmonary Fibrosis via Inhibition of p300 Histone Acetyltransferase Activity. J. Med. Food 2020, 23, 633–640. [Google Scholar] [CrossRef]
- Hay, D.A.; Fedorov, O.; Martin, S.; Singleton, D.C.; Tallant, C.; Wells, C.; Picaud, S.; Philpott, M.; Monteiro, O.P.; Rogers, C.M.; et al. Discovery and Optimization of Small-Molecule Ligands for the CBP/p300 Bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319. [Google Scholar] [CrossRef]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef]
- Strachowska, M.; Gronkowska, K.; Michlewska, S.; Robaszkiewicz, A. CBP/p300 Bromodomain Inhibitor–I–CBP112 Declines Transcription of the Key ABC Transporters and Sensitizes Cancer Cells to Chemotherapy Drugs. Cancers 2021, 13, 4614. [Google Scholar] [CrossRef] [PubMed]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, G.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Sorum, A.W.; Shrimp, J.H.; Roberts, A.M.; Montgomery, D.C.; Tiwari, N.K.; Lal-Nag, M.; Simeonov, A.; Jadhav, A.; Meier, J.L. Microfluidic Mobility Shift Profiling of Lysine Acetyltransferases Enables Screening and Mechanistic Analysis of Cellular Acetylation Inhibitors. ACS Chem. Biol. 2015, 11, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Nelson, K.M.; Strasser, J.M.; Barsyte-Lovejoy, D.; Szewczyk, M.M.; Organ, S.; Cuellar, M.; Singh, G.; Shrimp, J.H.; Nguyen, N.; et al. Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat. Commun. 2017, 8, 1527. [Google Scholar] [CrossRef]
- Thakor, N.; Janathia, B. Plumbagin: A potential candidate for future research and development. Curr. Pharm. Biotechnol. 2021, 23, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Theret, M.; Low, M.; Rempel, L.; Li, F.F.; Tung, L.W.; Contreras, O.; Chang, C.-K.; Wu, A.; Soliman, H.; Rossi, F.M. In vitro assessment of anti-fibrotic drug activity does not predict in vivo efficacy in murine models of Duchenne muscular dystrophy. Life Sci. 2021, 279, 119482. [Google Scholar] [CrossRef]
- Hammitzsch, A.; Tallant, C.; Fedorov, O.; O’Mahony, A.; Brennan, P.E.; Hay, D.A.; Martinez, F.O.; Al-Mossawi, M.H.; de Wit, J.; Vecellio, M.; et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. USA 2015, 112, 10768–10773. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, D.; Wang, L.; Wang, S.; Roden, A.C.; Zhao, H.; Li, X.; Prakash, Y.S.; Matteson, E.L.; Tschumperlin, D.J.; et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2019, 316, L487–L497. [Google Scholar] [CrossRef]
- Zucconi, B.E.; Makofske, J.L.; Meyers, D.J.; Hwang, Y.; Wu, M.; Kuroda, M.I.; Cole, P.A. Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP. Biochemistry 2019, 58, 2133–2143. [Google Scholar] [CrossRef]
- Navarro-Corcuera, A.; Sehrawat, T.S.; Jalan-Sakrikar, N.; Gibbons, H.R.; Pirius, N.E.; Khanal, S.; Hamdan, F.H.; Aseem, S.O.; Cao, S.; Banales, J.M.; et al. Long non-coding RNA ACTA2-AS1 promotes ductular reaction by interacting with the p300/ELK1 complex. J. Hepatol. 2022, 76, 921–933. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Gordon, M.S.; Reimers, M.A.; Hussain, A.; Patel, V.G.; Lam, E.T.; Sedkov, A.; Potter, V.; Shore, N. Abstract P202: Initial findings from an ongoing first-in-human phase 1 study of the CBP/p300 inhibitor FT-7051 in men with metastatic castration-resistant prostate cancer. Mol. Cancer Ther. 2021, 20, P202. [Google Scholar] [CrossRef]
- Wei, W.; Song, Z.; Chiba, M.; Wu, W.; Jeong, S.; Zhang, J.-P.; Kadin, M.E.; Nakagawa, M.; Yang, Y. Analysis and therapeutic targeting of the EP300 and CREBBP acetyltransferases in anaplastic large cell lymphoma and Hodgkin lymphoma. Leukemia 2023, 37, 396–407. [Google Scholar] [CrossRef]
- Pan, Z.; Zhao, Y.; Wang, X.; Xie, X.; Liu, M.; Zhang, K.; Wang, L.; Bai, D.; Foster, L.J.; Shu, R.; et al. Targeting bromodomain-containing proteins: Research advances of drug discovery. Mol. Biomed. 2023, 4, 13. [Google Scholar] [CrossRef]
- Vannam, R.; Sayilgan, J.; Ojeda, S.; Karakyriakou, B.; Hu, E.; Kreuzer, J.; Morris, R.; Lopez, X.I.H.; Rai, S.; Haas, W.; et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem. Biol. 2021, 28, 503–514.e12. [Google Scholar] [CrossRef]
- Durbin, A.D.; Wang, T.; Wimalasena, V.K.; Zimmerman, M.W.; Li, D.; Dharia, N.V.; Mariani, L.; Shendy, N.A.M.; Nance, S.; Patel, A.G.; et al. EP300 Selectively Controls the Enhancer Landscape of MYCN-Amplified Neuroblastoma. Cancer Discov. 2022, 12, 730–751. [Google Scholar] [CrossRef]
- Modell, A.E.; Lim, D.; Nguyen, T.M.; Sreekanth, V.; Choudhary, A. CRISPR-based therapeutics: Current challenges and future applications. Trends Pharmacol. Sci. 2022, 43, 151–161. [Google Scholar] [CrossRef]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40, BSR20200127. [Google Scholar] [CrossRef]
- Kesavan, G. Innovations in CRISPR-Based Therapies. Mol. Biotechnol. 2023, 65, 138–145. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound | IC50/Kd | Selectivity | Model | Molecular Mechanism | Fibrotic Phenotype | Ref. |
|---|---|---|---|---|---|---|
| Inhibitors of HAT Activity | ||||||
| L002 | 1.98 µm a | 3.7-fold more selective for PCALF and GCN5 than for EP300 | Ang II-induced hypertense mice | Reduction in cardiac fibrosis, hypertrophy, and renal fibrosis; Reverses hypertension-induced myofibroblast differentiation in murine ventricles. | [5,84,85] | |
| Human and rat cardiac fibroblasts, human podocytes, human mesangial cells + TGFβ | Inhibits profibrogenic AT1 receptor and partially rescues suppression of antifibrogenic AT2 receptor in human cardiac fibroblasts; Suppresses ERK1/2 MAPK pathway in human mesangial cells; Suppresses phosphorylation of pSMAD2 and pERK1/2 (canonical and non-canonical TGFβ signaling) in rat cardiac fibroblasts. | Reduces TGFβ-mediated cellular migration, proliferation, ECM protein synthesis (collagen, α-SMA), and CBP/EP300 upregulation. Blocks TGFβ-induced fibroblast-to-myofibroblast differentiation (reduction in αSMA); Suppresses pro-fibrogenic responses in cardiac fibroblasts, podocytes, and mesangial cells. | ||||
| A6 | 0.87 µM a | Bleomycin-induced mouse model of lung fibrosis | Reduced the level of activated TGFβ1 in the bronchoalveolar lavage fluid. | Reduced collagen deposition in the lung parenchyma. | [86,87] | |
| Liver fibrosis mouse models (choline-deficient/high-fat diet and thioacetamide models) | Reduced liver fibrosis; Reduced fibrosis marker genes (smooth muscle actin (αSMA), tenascin C, collagen type 1, collagen type 3, connective tissue growth factor). | |||||
| Hepatic stellate cells + TGFβ | Dissociation of EP300 from AKT. | Decreasing levels of TGFβ1-induced αSMA and fibrosis markers COL1A, CTGF, fibronectin (FN), TNC, and periostin. | ||||
| Mouse lung fibroblast + TGFβ1 | Epithelial-to-mesenchymal transition (EMT)-inducible transcription factors (SNAI1, SNAI2) and decrease in the endogenous and trichostatin A-induced histone acetylation level. | Reduced expression of COL1A1, COL1A2, FN and ACTA2. | ||||
| C646 | 1.6 µM b | Human cardiac fibroblasts + TGFβ | Blocks collagen synthesis. | [85,88,89] | ||
| Streptozotocin-induced diabetic mice | Reduced glomerular hypertrophy by down-regulating diabetes-induced pro-fibrotic molecules (collagen IV, fibronectin, and laminin). | |||||
| Coronary microvascular dysfunction mice model (SIRT3KO) | Suppressed TLR-4/IRAK-4/MyD88–mediated NF-κB | Attenuated cardiac remodeling (cardiac fibrosis, hypertrophy). | ||||
| A485 | 9.8 nM c | Myofibroblasts from patients Dupuytren’s disease | Inhibited profibrotic genes ACTA2 and COL1A1 expression. | [90,91] | ||
| Curcumin | ∼25 μM b | Dahl salt-sensitive hypertensive rats | Attenuated acetylation levels of GATA4 | Inhibited left ventricular hypertrophy development; Reduced increases in myocardial cell diameter, perivascular fibrosis and transcription of the hypertrophy-response genes, including Anf and Myh7. | [59,92,93] | |
| Aged mice | Inhibited TP53/SMAD3 axis activity | Rescued senescence and fibrosis in the atrial tissue, and atrial fibrillation vulnerability. | ||||
| Senescent human atrial fibroblasts | Inhibited TP53/SMAD3 axis activity | Fibrosis proteins COL1A1, MMP2/9, and TGFβ levels were reduced. | ||||
| Plumbagin | 20 µM b | Fibroblast + TGFβ | Inhibited fibroblast proliferation and fibrotic targets transcription: COL1A1, COL3A1, ACTA2 FN, SNAIL, and SERPINE1. | [94,95] | ||
| Bleomycin-induced mouse model of lung fibrosis | Inhibited pulmonary fibrosis | |||||
| Inhibitors of bromodomain (Non-BET) | ||||||
| SGC-CBP30 | 32 nM d | 40-fold e | Myofibroblasts from patients Dupuytren’s disease | Specifically, SGC-CBP30 identified collagen VI (Col VI) as a prominent downstream regulator of myofibroblast activity. | Inhibited profibrotic genes ACTA2 and COL1A1 expression. | [2,7,91,96] |
| Liver sinusoidal endothelial cells (LSECs) + TNFα | Prevented recruitment of EP300/BRD4/NFκB complex to the CCL2 enhancer and promoter regions. | Abrogated CBP/EP300-dependent CCL2 transcription and associated macrophage chemotaxis in vitro. | ||||
| Idiopathic pulmonary fibrosis models: patient-derived primary fibroblast, bleomycin mouse model, and ex vivo (precision-cut lung slices). | Reconstituted HDAC activity and MiCEE function in IPF fibroblasts | Reduced fibrotic hallmarks: matrix protein deposition, levels of fibrotic markers, such as FN1, COL1A1, and ACTA2, and reduced cell migration and proliferation. | ||||
| I-CBP112 | 170 nM for CPB f | 37 fold e | Myofibroblasts from patients Dupuytren’s disease. | Inhibited profibrotic genes ACTA2 and COL1A1 expression. | [91,97,98] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio, K.; Molina-Herrera, A.; Pérez-González, A.; Hernández-Galdámez, H.V.; Piña-Vázquez, C.; Araujo-Ramos, T.; Singh, I. EP300 as a Molecular Integrator of Fibrotic Transcriptional Programs. Int. J. Mol. Sci. 2023, 24, 12302. https://doi.org/10.3390/ijms241512302
Rubio K, Molina-Herrera A, Pérez-González A, Hernández-Galdámez HV, Piña-Vázquez C, Araujo-Ramos T, Singh I. EP300 as a Molecular Integrator of Fibrotic Transcriptional Programs. International Journal of Molecular Sciences. 2023; 24(15):12302. https://doi.org/10.3390/ijms241512302
Chicago/Turabian StyleRubio, Karla, Alejandro Molina-Herrera, Andrea Pérez-González, Hury Viridiana Hernández-Galdámez, Carolina Piña-Vázquez, Tania Araujo-Ramos, and Indrabahadur Singh. 2023. "EP300 as a Molecular Integrator of Fibrotic Transcriptional Programs" International Journal of Molecular Sciences 24, no. 15: 12302. https://doi.org/10.3390/ijms241512302
APA StyleRubio, K., Molina-Herrera, A., Pérez-González, A., Hernández-Galdámez, H. V., Piña-Vázquez, C., Araujo-Ramos, T., & Singh, I. (2023). EP300 as a Molecular Integrator of Fibrotic Transcriptional Programs. International Journal of Molecular Sciences, 24(15), 12302. https://doi.org/10.3390/ijms241512302