
Raman Spectroscopy Combined with Malaria Protein for Early Capture and Recognition of Broad-Spectrum Circulating Tumor Cells
 and
and
Abstract
1. Introduction
2. Results
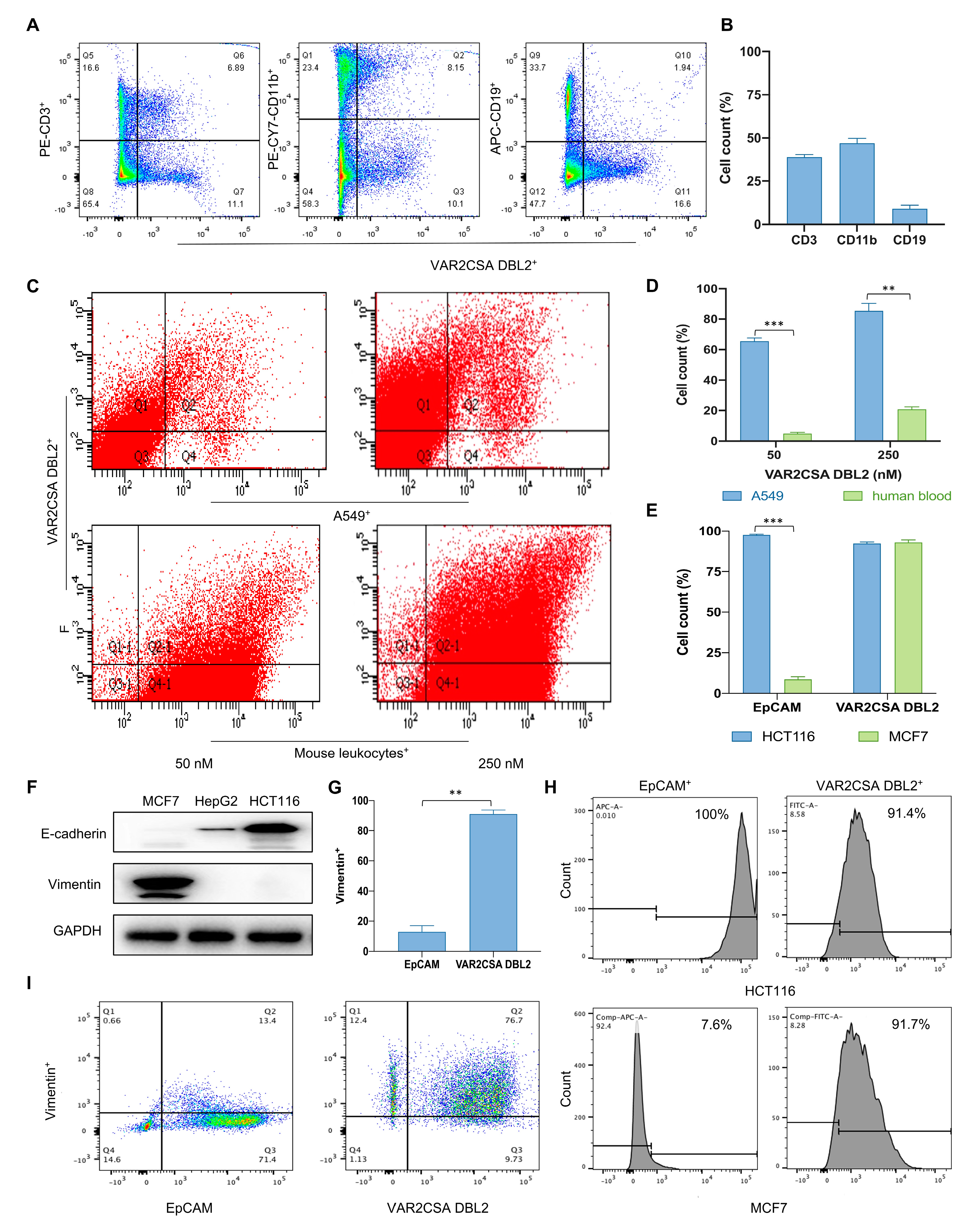
2.1. VAR2CSA DBL2 Can Efficiently Bind to Cancer Cells
2.2. Taking Only PBMC Layer Cells Can Greatly Reduce the Non-Specific Binding of Malaria Proteins to Blood Cells
2.3. Malaria Protein Is a More Broad-Spectrum Tumor Surface Marker than EpCAM
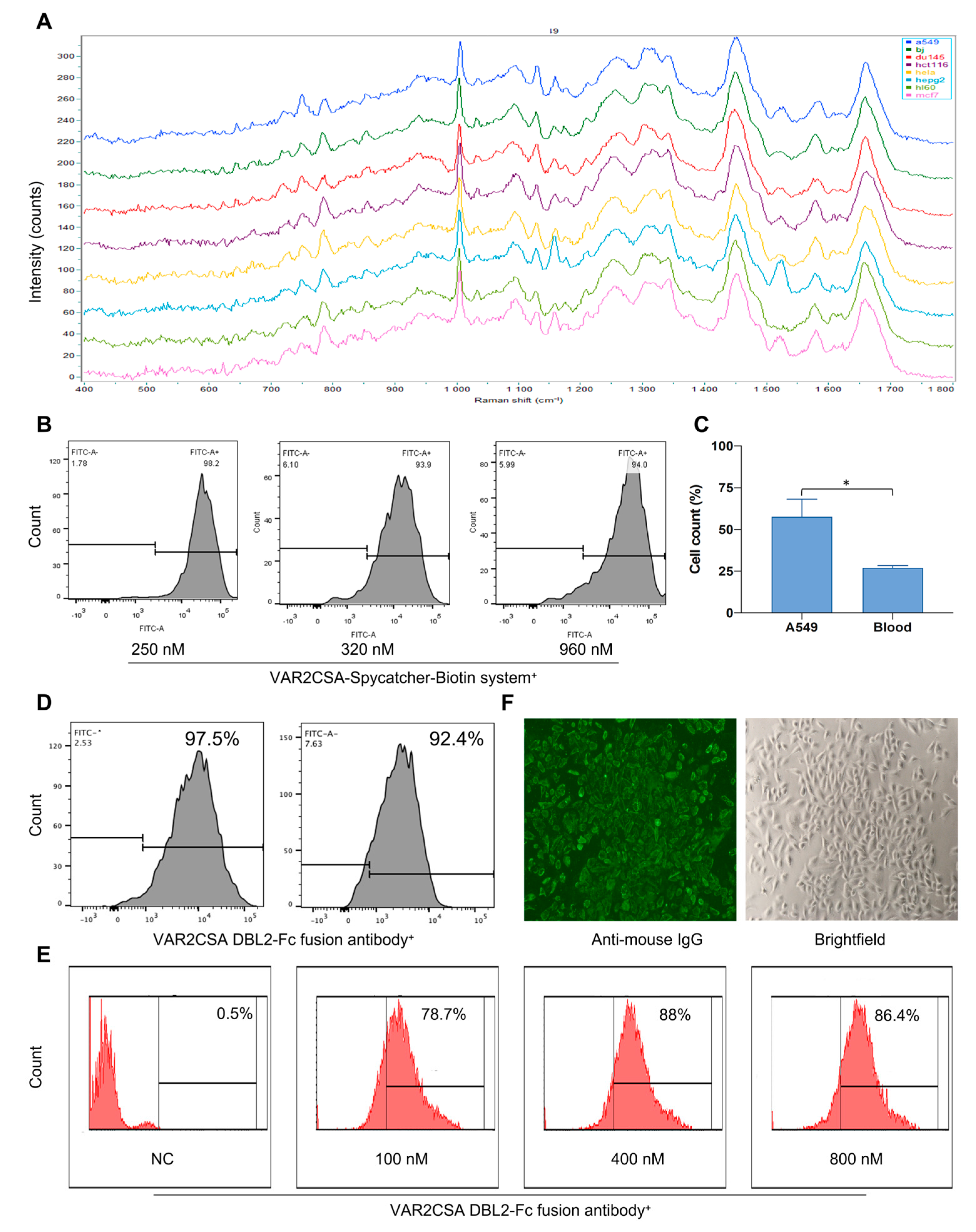
2.4. Raman Spectroscopy Can Predict Cell Type Successfully
2.5. Raman Spectroscopy Can Identify Tumor Cell Types Captured by Malaria Proteins
2.6. Raman Spectroscopy Can Identify Tumor Cells Captured by Malaria Proteins after Their Mixture with Peripheral Blood
2.7. VAR2CSA DBL2-Fc Fusion Antibody Has a Strong Binding Ability with Tumor Cells
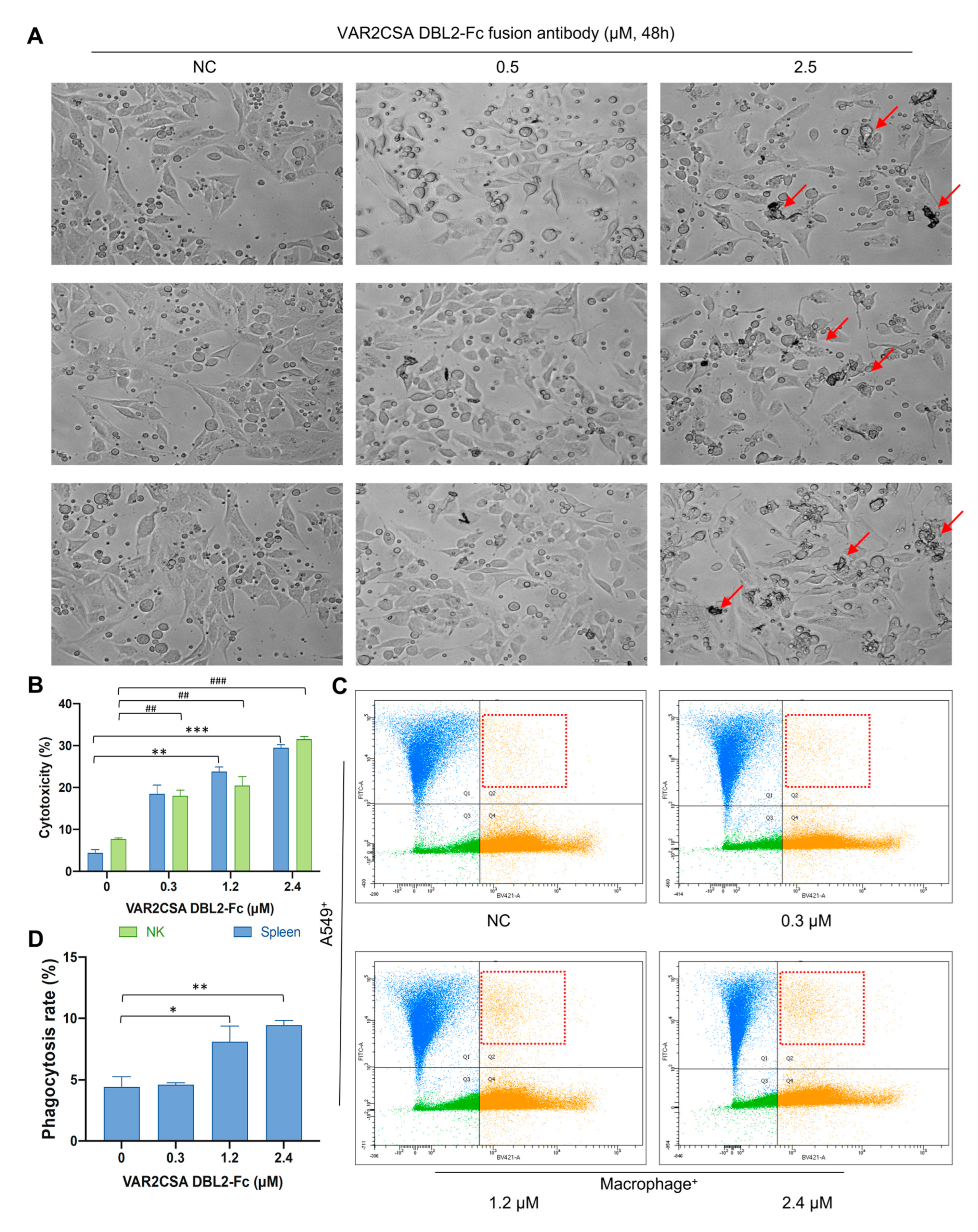
2.8. VAR2CSA DBL2-Fc Fusion Antibody Enhances Macrophage Antitumor Activity In Vitro
2.9. VAR2CSA DBL2-Fc Fusion Antibody Induces Macrophage-Dependent Phagocytosis in A549
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Antibodies
4.2. Cloning, Expression, Purification, and Molecular Weight Determination of VAR2CSA DBL1-2, VAR2CSA DBL2, and SpyCatcher
4.3. Gel Filtration Chromatography
4.4. Flow Cytometry
4.5. Capturing Cancer Cells by VAR2CSA-Coated Anti-Biotin Beads
4.6. Raman Microspectroscopy
4.7. Construction, Expression, and Purification of the VAR2CSA DBL2-Fc Fusion Antibody
4.8. ADCP
4.9. ADCC
4.10. Immunofluorescence Assays
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Rossi, E.; Fabbri, F. CTCs 2020: Great Expectations or Unreasonable Dreams. Cells 2019, 8, 989. [Google Scholar] [CrossRef]
- Nanduri, L.K.; Hissa, B.; Weitz, J.; Scholch, S.; Bork, U. The prognostic role of circulating tumor cells in colorectal cancer. Expert Rev. Anticancer Ther. 2019, 19, 1077–1088. [Google Scholar] [CrossRef]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Zhang, L.; Riethdorf, S.; Wu, G.; Wang, T.; Yang, K.; Peng, G.; Liu, J.; Pantel, K. Meta-analysis of the prognostic value of circulating tumor cells in breast cancer. Clin. Cancer Res. 2012, 18, 5701–5710. [Google Scholar] [CrossRef]
- Esmaeilsabzali, H.; Beischlag, T.V.; Cox, M.E.; Parameswaran, A.M.; Park, E.J. Detection and isolation of circulating tumor cells: Principles and methods. Biotechnol. Adv. 2013, 31, 1063–1084. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, H.; Yu, T.; Xie, J.; Chen, S.; Dong, H.; Sinko, P.J.; Lian, S.; Xu, J.; Wang, J.; et al. Isolation and characterization of living circulating tumor cells in patients by immunomagnetic negative enrichment coupled with flow cytometry. Cancer 2015, 121, 3036–3045. [Google Scholar] [CrossRef]
- Hardingham, J.E.; Grover, P.; Winter, M.; Hewett, P.J.; Price, T.J.; Thierry, B. Detection and Clinical Significance of Circulating Tumor Cells in Colorectal Cancer—20 Years of Progress. Mol. Med. 2015, 21 (Suppl. S1), S25–S31. [Google Scholar] [CrossRef]
- Poudineh, M.; Sargent, E.H.; Pantel, K.; Kelley, S.O. Profiling circulating tumour cells and other biomarkers of invasive cancers. Nat. Biomed. Eng. 2018, 2, 72–84. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H.; Brandt, B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer 2008, 8, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Mannavola, F.; Pelle, E.; Quaresmini, D.; Tucci, M.; Silvestris, F. Liquid biopsy of cancer: A multimodal diagnostic tool in clinical oncology. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef]
- Tayoun, T.; Faugeroux, V.; Oulhen, M.; Aberlenc, A.; Pawlikowska, P.; Farace, F. CTC-Derived Models: A Window into the Seeding Capacity of Circulating Tumor Cells (CTCs). Cells 2019, 8, 1145. [Google Scholar] [CrossRef]
- Dodo, K.; Fujita, K.; Sodeoka, M. Raman Spectroscopy for Chemical Biology Research. J. Am. Chem. Soc. 2022, 144, 19651–19667. [Google Scholar] [CrossRef]
- Oliva-Teles, L.; Pinto, R.; Vilarinho, R.; Carvalho, A.P.; Moreira, J.A.; Guimaraes, L. Environmental diagnosis with Raman Spectroscopy applied to diatoms. Biosens. Bioelectron. 2022, 198, 113800. [Google Scholar] [CrossRef] [PubMed]
- Haryanto, A.; Lee, C.W. Shell isolated nanoparticle enhanced Raman spectroscopy for mechanistic investigation of electrochemical reactions. Nano Converg. 2022, 9, 9. [Google Scholar] [CrossRef]
- Neng, J.; Zhang, Q.; Sun, P. Application of surface-enhanced Raman spectroscopy in fast detection of toxic and harmful substances in food. Biosens. Bioelectron. 2020, 167, 112480. [Google Scholar] [CrossRef]
- Yilmaz, H.; Yilmaz, D.; Taskin, I.C.; Culha, M. Pharmaceutical applications of a nanospectroscopic technique: Surface-enhanced Raman spectroscopy. Adv. Drug Deliv. Rev. 2022, 184, 114184. [Google Scholar] [CrossRef]
- Wang, W.T.; Zhang, H.; Yuan, Y.; Guo, Y.; He, S.X. Research Progress of Raman Spectroscopy in Drug Analysis. AAPS PharmSciTech 2018, 19, 2921–2928. [Google Scholar] [CrossRef]
- Shen, Y.; Hu, F.; Min, W. Raman Imaging of Small Biomolecules. Annu. Rev. Biophys. 2019, 48, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.R.; Hooper, D.C.; Zhang, L.; Wolverson, D.; Valev, V.K. Raman Techniques: Fundamentals and Frontiers. Nanoscale Res. Lett. 2019, 14, 231. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, S.; Manago, S.; De Luca, A.C. Raman Microscopy: Progress in Research on Cancer Cell Sensing. Sensors 2020, 20, 5525. [Google Scholar] [CrossRef]
- Czaplicka, M.; Nicinski, K.; Nowicka, A.; Szymborski, T.; Chmielewska, I.; Trzcinska-Danielewicz, J.; Girstun, A.; Kaminska, A. Effect of Varying Expression of EpCAM on the Efficiency of CTCs Detection by SERS-Based Immunomagnetic Optofluidic Device. Cancers 2020, 12, 3315. [Google Scholar] [CrossRef]
- Sha, M.Y.; Xu, H.; Natan, M.J.; Cromer, R. Surface-enhanced Raman scattering tags for rapid and homogeneous detection of circulating tumor cells in the presence of human whole blood. J. Am. Chem. Soc. 2008, 130, 17214–17215. [Google Scholar] [CrossRef]
- Ahmadal-Agroudi, M.; El-Mawla Megahed, L.A.; Abdallah, E.M.; Morsy, T.A. A Mini Overview of Malaria in Pregnancy. J. Egypt. Soc. Parasitol. 2017, 47, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; ter Kuile, F.O.; Nosten, F.; McGready, R.; Asamoa, K.; Brabin, B.; Newman, R.D. Epidemiology and burden of malaria in pregnancy. Lancet Infect. Dis. 2007, 7, 93–104. [Google Scholar] [CrossRef]
- Seiler, R.; Oo, H.Z.; Tortora, D.; Clausen, T.M.; Wang, C.K.; Kumar, G.; Pereira, M.A.; Orum-Madsen, M.S.; Agerbaek, M.O.; Gustavsson, T.; et al. An Oncofetal Glycosaminoglycan Modification Provides Therapeutic Access to Cisplatin-resistant Bladder Cancer. Eur. Urol. 2017, 72, 142–150. [Google Scholar] [CrossRef]
- Pudelko, A.; Wisowski, G.; Olczyk, K.; Kozma, E.M. The dual role of the glycosaminoglycan chondroitin-6-sulfate in the development, progression and metastasis of cancer. FEBS J. 2019, 286, 1815–1837. [Google Scholar] [CrossRef]
- Li, H.; Zhang, P.; Luo, J.; Hu, D.; Huang, Y.; Zhang, Z.R.; Fu, Y.; Gong, T. Chondroitin Sulfate-Linked Prodrug Nanoparticles Target the Golgi Apparatus for Cancer Metastasis Treatment. ACS Nano 2019, 13, 9386–9396. [Google Scholar] [CrossRef]
- Ma, R.; Lian, T.; Huang, R.; Renn, J.P.; Petersen, J.D.; Zimmerberg, J.; Duffy, P.E.; Tolia, N.H. Structural basis for placental malaria mediated by Plasmodium falciparum VAR2CSA. Nat. Microbiol. 2021, 6, 380–391. [Google Scholar] [CrossRef]
- Salanti, A.; Clausen, T.M.; Agerbaek, M.O.; Al Nakouzi, N.; Dahlback, M.; Oo, H.Z.; Lee, S.; Gustavsson, T.; Rich, J.R.; Hedberg, B.J.; et al. Targeting Human Cancer by a Glycosaminoglycan Binding Malaria Protein. Cancer Cell 2015, 28, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Pereira, M.A.; Al Nakouzi, N.; Oo, H.Z.; Agerbaek, M.O.; Lee, S.; Orum-Madsen, M.S.; Kristensen, A.R.; El-Naggar, A.; Grandgenett, P.M.; et al. Oncofetal Chondroitin Sulfate Glycosaminoglycans Are Key Players in Integrin Signaling and Tumor Cell Motility. Mol. Cancer Res. 2016, 14, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Bang-Christensen, S.R.; Pedersen, R.S.; Pereira, M.A.; Clausen, T.M.; Loppke, C.; Sand, N.T.; Ahrens, T.D.; Jorgensen, A.M.; Lim, Y.C.; Goksoyr, L.; et al. Capture and Detection of Circulating Glioma Cells Using the Recombinant VAR2CSA Malaria Protein. Cells 2019, 8, 998. [Google Scholar] [CrossRef]
- Agerbaek, M.O.; Bang-Christensen, S.R.; Yang, M.H.; Clausen, T.M.; Pereira, M.A.; Sharma, S.; Ditlev, S.B.; Nielsen, M.A.; Choudhary, S.; Gustavsson, T.; et al. The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat. Commun. 2018, 9, 3279. [Google Scholar] [CrossRef]
- Lepone, L.M.; Donahue, R.N.; Grenga, I.; Metenou, S.; Richards, J.; Heery, C.R.; Madan, R.A.; Gulley, J.L.; Schlom, J. Analyses of 123 Peripheral Human Immune Cell Subsets: Defining Differences with Age and between Healthy Donors and Cancer Patients Not Detected in Analysis of Standard Immune Cell Types. J. Circ. Biomark. 2016, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Scott Shaffer, J.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef]
- Paterlini-Brechot, P. Circulating Tumor Cells: Who is the Killer? Cancer Microenviron. 2014, 7, 161–176. [Google Scholar] [CrossRef]
- Qi, L.N.; Xiang, B.D.; Wu, F.X.; Ye, J.Z.; Zhong, J.H.; Wang, Y.Y.; Chen, Y.Y.; Chen, Z.S.; Ma, L.; Chen, J.; et al. Circulating Tumor Cells Undergoing EMT Provide a Metric for Diagnosis and Prognosis of Patients with Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4731–4744. [Google Scholar] [CrossRef]
- Kaminska, A.; Szymborski, T.; Witkowska, E.; Kijenska-Gawronska, E.; Swieszkowski, W.; Nicinski, K.; Trzcinska-Danielewicz, J.; Girstun, A. Detection of Circulating Tumor Cells Using Membrane-Based SERS Platform: A New Diagnostic Approach for ‘Liquid Biopsy’. Nanomaterials 2019, 9, 366. [Google Scholar] [CrossRef]
- Reza, K.K.; Dey, S.; Wuethrich, A.; Jing, W.; Behren, A.; Antaw, F.; Wang, Y.; Sina, A.A.; Trau, M. In Situ Single Cell Proteomics Reveals Circulating Tumor Cell Heterogeneity during Treatment. ACS Nano 2021, 15, 11231–11243. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, D.; Katopodis, P.; Stone, N.; Haskell, J.; Sheridan, H.; Gardner, B.; Urnovitz, H.; Schuetz, E.; Beck, J.; Hall, M.; et al. Liquid Biopsies in Lung Cancer: Four Emerging Technologies and Potential Clinical Applications. Cancers 2019, 11, 331. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef]
- Layoun, A.; Samba, M.; Santos, M.M. Isolation of murine peritoneal macrophages to carry out gene expression analysis upon Toll-like receptors stimulation. J. Vis. Exp. 2015, 98, e52749. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Microsphere | CD11b | CD45 | Cancer | |
|---|---|---|---|---|
| Plasma | 90,014 | 1352 | 1155 | 1911 |
| PBMC | 2186 | 3952 | 14,784 | 43,038 |
| Ficoll | 19,226 | 18,306 | 55,174 | 2764 |
| Red cell | 944 | 3476 | 7495 | 496 |
| a | CD11b/a | CD45/a | Cancer/a | |
|---|---|---|---|---|
| Plamsa | 1.072 | 1261.67 (0.23%) | 40,136.55 (0.07%) | 1783.32 (0.1%) |
| PBMC | 0.026 | 151,860.93 (28%) | 150,737.11 (38.46%) | 1,653,793.23 (96.61%) |
| Ficoll | 0.229 | 79,980.44 (14.75%) | 2,325,415.39 (16.32%) | 12,076.15 (0.71%) |
| Red cell | 0.011 | 309,305.09 (57.02%) | 2,777,519.64 (45.15%) | 44,135.59 (2.58%) |
| Tested Cell Type | Tested Cell Type | Predicted Cell Type | KNN Predicted Accuracy | SVM Predicted Accuracy | RF Predicted Accuracy | LDA Predicted Accuracy | PLS-DA Predicted Accuracy | XGB Predicted Accuracy |
|---|---|---|---|---|---|---|---|---|
| A549 | 63 | A549 | 98% | 100% | 100% | 100% | 100% | 100% |
| BJ | 79 | BJ | 96% | 100% | 100% | 100% | 100% | 100% |
| DU145 | 71 | DU145 | 100% | 100% | 100% | 100% | 92% | 100% |
| HCT116 | 72 | HCT116 | 97% | 100% | 100% | 100% | 99% | 100% |
| Hela | 64 | Hela | 98% | 100% | 100% | 100% | 97% | 100% |
| HepG2 | 78 | HepG2 | 99% | 100% | 100% | 100% | 97% | 100% |
| HL60 | 75 | HL60 | 99% | 100% | 100% | 100% | 100% | 100% |
| MCF7 | 79 | MCF7 | 97% | 100% | 100% | 100% | 99% | 100% |
| Test Cell Type | Tested Cell Number | Predicted Accuracy | Predicted Cell Type |
|---|---|---|---|
| A549 | 62 | 46.8% | A549 |
| Hela | 60 | 43.3% | Hela |
| MCF7 | 62 | 33.8% | HCT116 |
| HL60 | 50 | 60% | HL60 |
| DU145 | 58 | 32.7% | BJ |
| Algorithm | SVM | K-NN | PLS-DA | RF | LDA | XGB |
|---|---|---|---|---|---|---|
| HCT116 | 21.05% | 38.6% | 1.75% | 15.79% | 8.77% | 26.32% |
| MCF7 | 7.02% | 5.26% | 10.53% | 7.02% | 22.81% | 14.04% |
| A549 | 21.05% | 7.02% | 38.6% | 14.04% | 15.79% | 8.77% |
| Hela | 5.26% | 0.00% | 35.09% | 0.00% | 14.04% | 5.26% |
| HepG2 | 24.56% | 1.75% | 12.28% | 12.28% | 29.82% | 8.77% |
| DU145 | 17.54% | 38.6% | 1.75% | 26.32% | 7.02% | 26.32% |
| HL60 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| BJ | 3.51% | 8.77% | 0.00% | 24.56% | 1.75% | 10.53% |
| Predicted cell type | HepG2 | HCT116 | A549 | DU145 | HepG2 | HepG2/HCT116 |
| Algorithm | SVM | K-NN | PLS-DA | RF | LDA | XGB |
|---|---|---|---|---|---|---|
| HCT116 | 21.28% | 40.43% | 6.38% | 14.89% | 19.15% | 27.66% |
| MCF7 | 6.38% | 2.13% | 19.15% | 8.51% | 42.55% | 12.77% |
| A549 | 53.19% | 6.38% | 31.91% | 19.15% | 21.28% | 29.79% |
| Hela | 2.13% | 4.26% | 27.66% | 0.00% | 4.26% | 0.00% |
| HepG2 | 4.26% | 0.00% | 12.77% | 6.38% | 8.51% | 6.38% |
| DU145 | 2.13% | 19.15% | 2.13% | 4.26% | 0.00% | 2.13% |
| HL60 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| BJ | 10.64% | 27.66% | 0.00% | 46.81% | 4.26% | 21.28% |
| Predicted cell type | A549 | HCT116 | A549 | BJ | MCF7 | A549 |
| Algorithm | SVM | K-NN | PLS-DA | RF | LDA | XGB |
|---|---|---|---|---|---|---|
| HCT116 | 4.26% | 6.38% | 0.00% | 4.26% | 4.26% | 0.00% |
| MCF7 | 8.51% | 2.13% | 10.64% | 2.13% | 46.81% | 2.13% |
| A549 | 74.47% | 42.55% | 34.04% | 46.81% | 34.04% | 89.36% |
| Hela | 0.00% | 0.00% | 46.81% | 0.00% | 6.38% | 0.00% |
| HepG2 | 4.26% | 2.13% | 8.51% | 4.26% | 6.38% | 4.26% |
| DU145 | 0.00% | 2.13% | 0.00% | 0.00% | 2.13% | 0.00% |
| HL60 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| BJ | 8.51% | 44.68% | 0.00% | 42.55% | 0.00% | 4.26% |
| Predicted cell type | A549 | BJ | Hela | A549 | MCF7 | A549 |
| Algorithm | SVM | K-NN | PLS-DA | RF | LDA | XGB |
|---|---|---|---|---|---|---|
| HCT116 | 20.00% | 20.00% | 0.00% | 0.00% | 40.00% | 26.67% |
| MCF7 | 0.00% | 6.67% | 6.67% | 0.00% | 6.67% | 6.67% |
| A549 | 0.00% | 6.67% | 46.67% | 0.00% | 0.00% | 0.00% |
| Hela | 33.33% | 13.33% | 20.00% | 13.33% | 6.67% | 13.33% |
| HepG2 | 0.00% | 0.00% | 20.00% | 6.67% | 26.67% | 13.33% |
| DU145 | 20.00% | 46.67% | 6.67% | 13.33% | 6.67% | 6.67% |
| HL60 | 6.67% | 0.00% | 0.00% | 13.33% | 6.67% | 13.33% |
| BJ | 20.00% | 6.67% | 0.00% | 53.33% | 6.67% | 20.00% |
| Predicted cell type | Hela | DU145 | A549 | BJ | HCT116 | HCT116 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Zhang, Y.; Li, X.; Xu, J.; Zhao, C.; Yang, J. Raman Spectroscopy Combined with Malaria Protein for Early Capture and Recognition of Broad-Spectrum Circulating Tumor Cells. Int. J. Mol. Sci. 2023, 24, 12072. https://doi.org/10.3390/ijms241512072
Liu X, Zhang Y, Li X, Xu J, Zhao C, Yang J. Raman Spectroscopy Combined with Malaria Protein for Early Capture and Recognition of Broad-Spectrum Circulating Tumor Cells. International Journal of Molecular Sciences. 2023; 24(15):12072. https://doi.org/10.3390/ijms241512072
Chicago/Turabian StyleLiu, Xinning, Yidan Zhang, Xunrong Li, Jian Xu, Chenyang Zhao, and Jinbo Yang. 2023. "Raman Spectroscopy Combined with Malaria Protein for Early Capture and Recognition of Broad-Spectrum Circulating Tumor Cells" International Journal of Molecular Sciences 24, no. 15: 12072. https://doi.org/10.3390/ijms241512072
APA StyleLiu, X., Zhang, Y., Li, X., Xu, J., Zhao, C., & Yang, J. (2023). Raman Spectroscopy Combined with Malaria Protein for Early Capture and Recognition of Broad-Spectrum Circulating Tumor Cells. International Journal of Molecular Sciences, 24(15), 12072. https://doi.org/10.3390/ijms241512072