
Structural Characterization of Fluorescent Proteins Using Tunable Femtosecond Stimulated Raman Spectroscopy

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results and Discussion
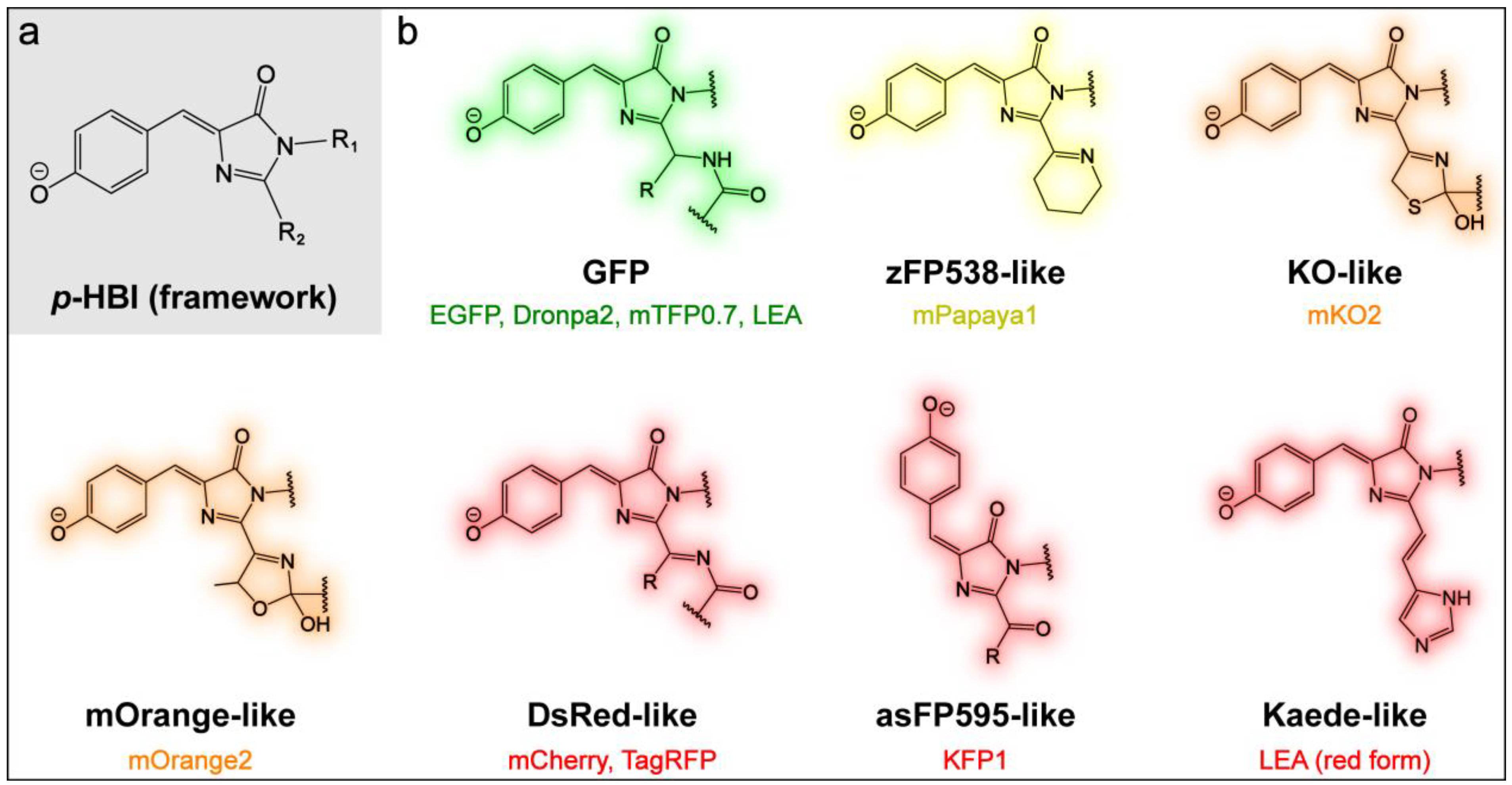
2.1. Effects of Sidechains in the GFP Model Chromophore
2.2. Effects of the Chromophore Environment
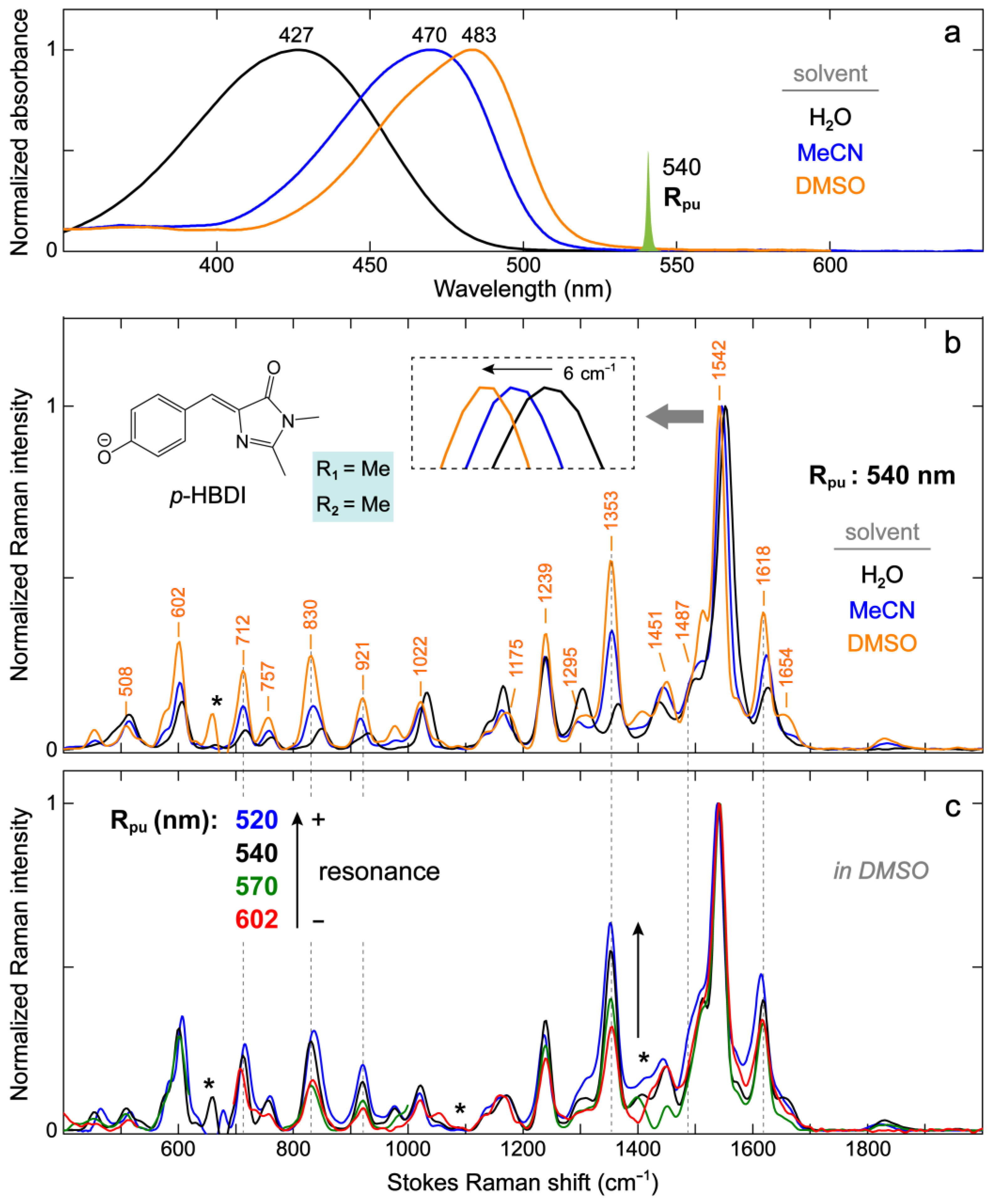
2.2.1. The Model GFP Chromophore in Various Solvents
2.2.2. GFP Chromophores in Various Protein Matrices
2.3. Effects of the Chromophore Structure
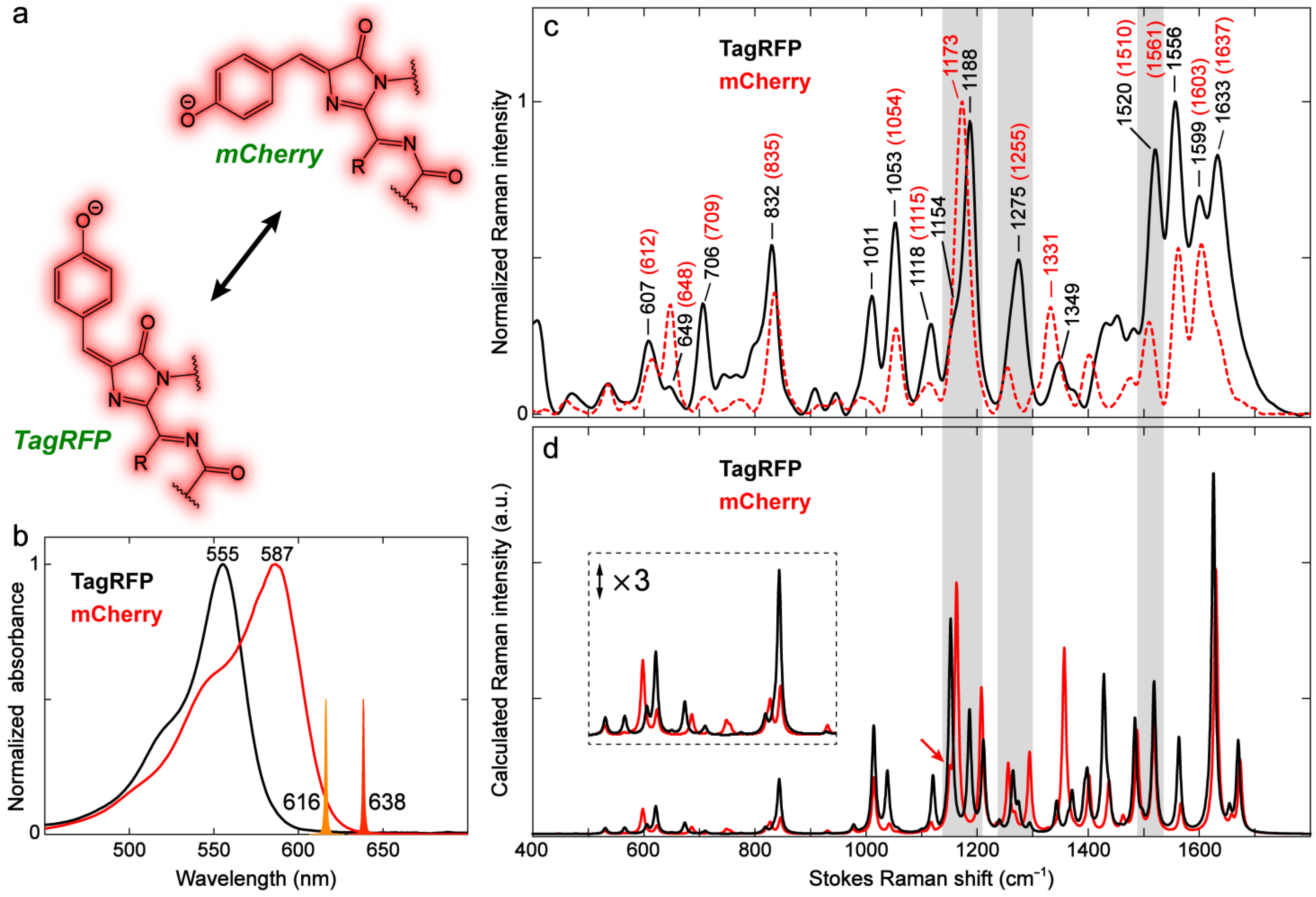
2.3.1. Yellow, Orange, and Red Fluorescent Protein (YFP, OFP, and RFP)
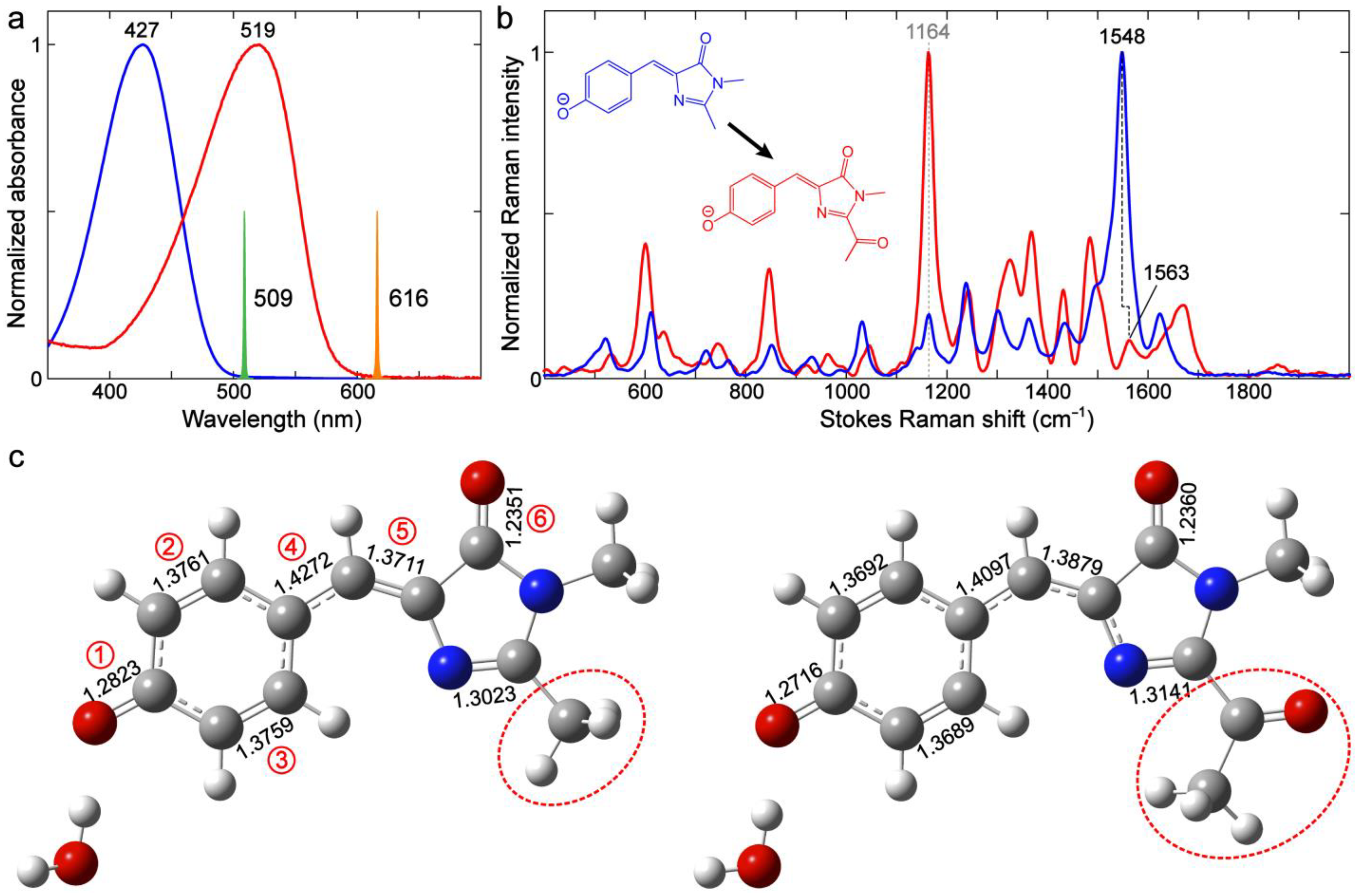
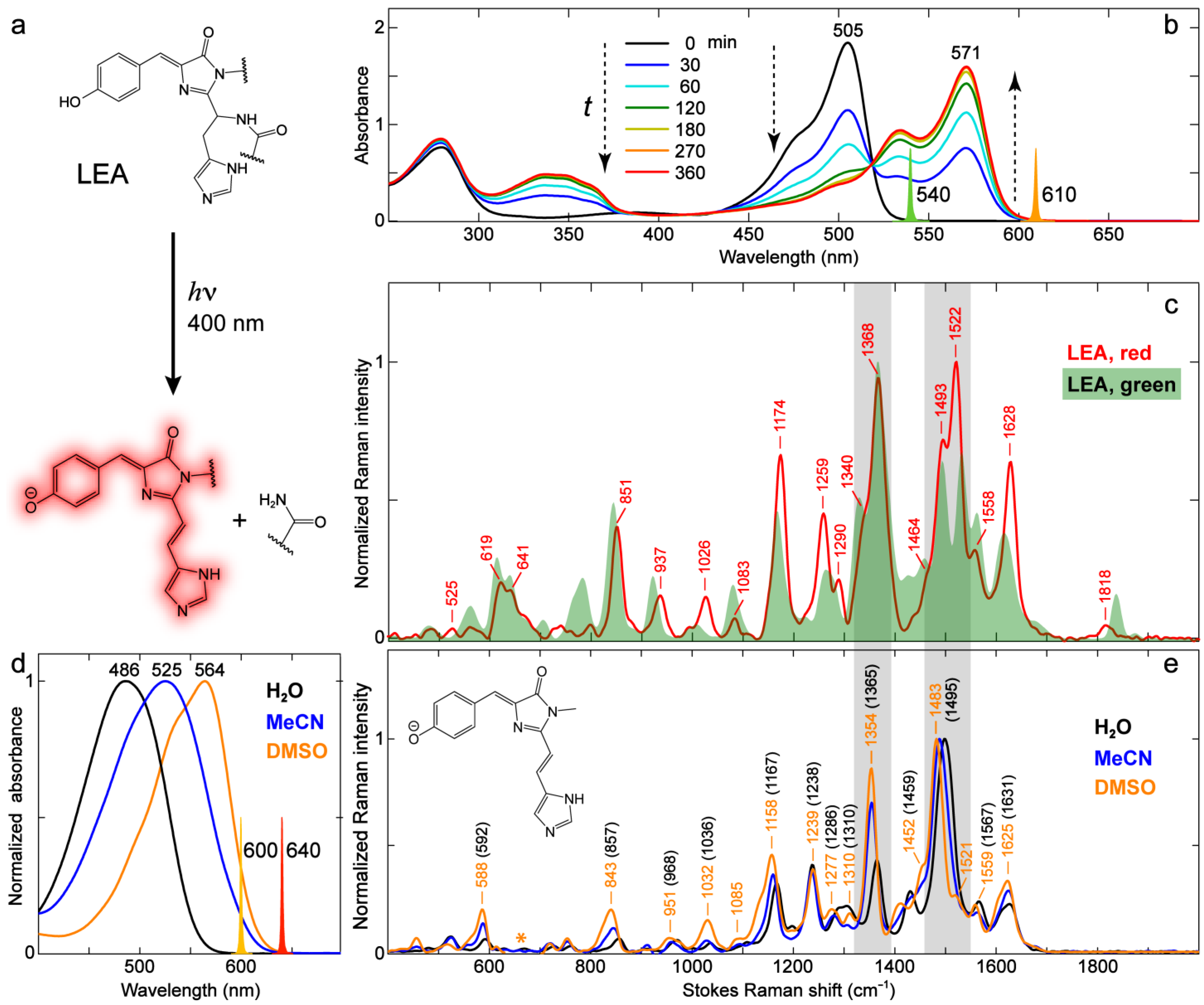
2.3.2. Kaede-Like RFP
2.4. Effects of the Chromophore Conformation
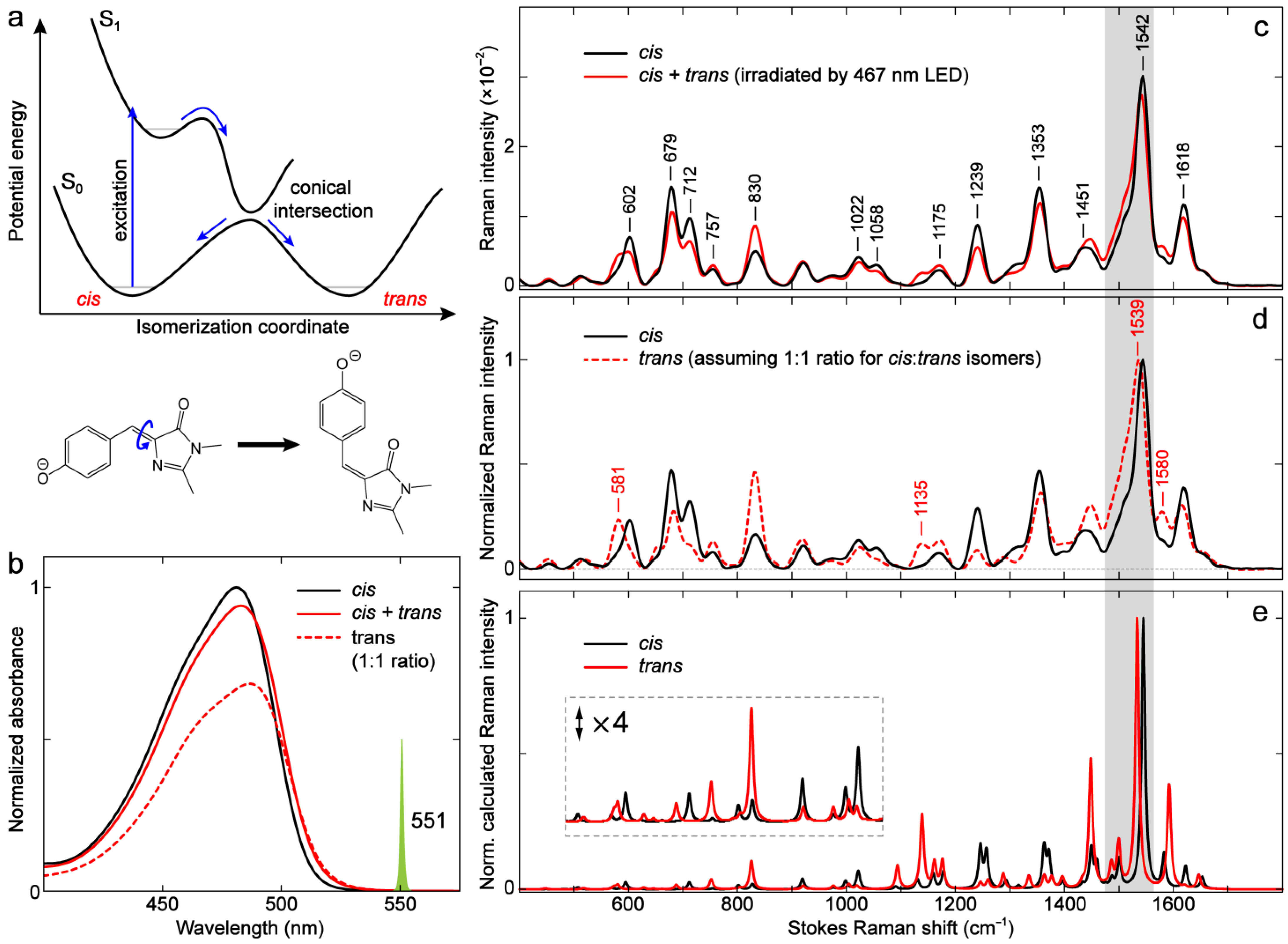
2.4.1. GFP Model Chromophores: cis vs. trans Isomers
2.4.2. DsRed-Like RFP: cis vs. trans Isomers
3. Materials and Methods
3.1. Protein Expression and Model Chromophore Preparation
3.1.1. EGFP, mKO2, and TagRFP
3.1.2. mPapaya1, mOrange2, LEA, and KFP1
3.1.3. Dronpa2 and mCherry
3.1.4. mTFP0.7
3.1.5. FP Model Chromophore Preparation
3.2. Steady-State Electronic Spectroscopy
3.3. Femtosecond Stimulated Raman Spectroscopy (FSRS)
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of Aequorin, a bioluminescent protein from the luminous Hydromedusan. Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Day, R.N.; Davidson, M.W. The fluorescent protein palette: Tools for cellular imaging. Chem. Soc. Rev. 2009, 38, 2887–2921. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Verkhusha, V.V. Chromophore transformations in red fluorescent proteins. Chem. Rev. 2012, 112, 4308–4327. [Google Scholar] [CrossRef] [PubMed]
- Chattoraj, M.; King, B.A.; Bublitz, G.U.; Boxer, S.G. Ultra-fast excited state dynamics in green fluorescent protein: Multiple states and proton transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 8362–8367. [Google Scholar] [CrossRef]
- Stoner-Ma, D.; Jaye, A.A.; Matousek, P.; Towrie, M.; Meech, S.R.; Tonge, P.J. Observation of excited-state proton transfer in green fluorescent proetin using ultrafast vibrational spectroscopy. J. Am. Chem. Soc. 2005, 127, 2864–2865. [Google Scholar] [CrossRef]
- Ando, R.; Hama, H.; Yamamoto-Hino, M.; Mizuno, H.; Miyawaki, A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 12651–12656. [Google Scholar] [CrossRef]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Röcker, C.; Salih, A.; Spindler, K.-D.; Nienhaus, G.U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef]
- Kim, H.; Grunkemeyer, T.J.; Modi, C.; Chen, L.; Fromme, R.; Matz, M.V.; Wachter, R.M. Acid–base catalysis and crystal structures of a least evolved ancestral GFP-like protein undergoing green-to-red photoconversion. Biochemistry 2013, 52, 8048–8059. [Google Scholar] [CrossRef]
- Brejc, K.; Sixma, T.K.; Kitts, P.A.; Kain, S.R.; Tsien, R.Y.; Ormö, M.; Remington, S.J. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA 1997, 94, 2306–2311. [Google Scholar] [CrossRef]
- Violot, S.; Carpentier, P.; Blanchoin, L.; Bourgeois, D. Reverse pH-dependence of chromophore protonation explains the large Stokes shift of the red fluorescent protein mKeima. J. Am. Chem. Soc. 2009, 131, 10356–10357. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zou, T.; Modi, C.; Dörner, K.; Grunkemeyer, T.J.; Chen, L.; Fromme, R.; Matz, M.V.; Ozkan, S.B.; Wachter, R.M. A hinge migration mechanism unlocks the evolution of green-to-red photoconversion in GFP-like proteins. Structure 2015, 23, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Konold, P.E.; Arik, E.; Weißenborn, J.; Arents, J.C.; Hellingwerf, K.J.; van Stokkum, I.H.M.; Kennis, J.T.M.; Groot, M.L. Confinement in crystal lattice alters entire photocycle pathway of the photoactive yellow protein. Nat. Commun. 2020, 11, 4248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Y.; Chen, C.; Zhu, R.; Jiang, J.; Weng, T.-C.; Ji, Q.; Huang, Y.; Fang, C.; Liu, W. Mapping the complete photocycle that powers a large Stokes shift red fluorescent protein. Angew. Chem. Int. Ed. 2023, 62, e202212209. [Google Scholar]
- Chang, J.; Romei, M.G.; Boxer, S.G. Structural evidence of photoisomerization pathways in fluorescent proteins. J. Am. Chem. Soc. 2019, 141, 15504–15508. [Google Scholar] [CrossRef]
- Fadini, A.; Hutchison, C.D.; Morozov, D.; Chang, J.; Maghlaoui, K.; Perrett, S.; Luo, F.; Kho, J.C.; Romei, M.G.; Morgan, R.M.L.; et al. Serial femtosecond crystallography reveals that photoactivation in a fluorescent protein proceeds via the hula twist mechanism. J. Am. Chem. Soc. 2023, in press. [CrossRef]
- Zimmer, M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 2002, 102, 759–782. [Google Scholar] [CrossRef]
- Jung, G. (Ed.) Fluorescent Proteins I: From Understanding to Design; Springer: Berlin/Heidelberg, Germany, 2012; Volume 11, 268p. [Google Scholar]
- Meech, S.R. Excited state reactions in fluorescent proteins. Chem. Soc. Rev. 2009, 38, 2922–2934. [Google Scholar] [CrossRef]
- Tang, L.; Liu, W.; Wang, Y.; Zhao, Y.; Oscar, B.G.; Campbell, R.E.; Fang, C. Unraveling ultrafast photoinduced proton transfer dynamics in a fluorescent protein biosensor for Ca2+ imaging. Chem. Eur. J. 2015, 21, 6481–6490. [Google Scholar] [CrossRef]
- Conyard, J.; Heisler, I.A.; Chan, Y.; Bulman Page, P.C.; Meech, S.R.; Blancafort, L. A new twist in the photophysics of the GFP chromophore: A volume-conserving molecular torsion couple. Chem. Sci. 2018, 9, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Fang, C. Photoswitchable fluorescent proteins: Mechanisms on ultrafast timescales. Int. J. Mol. Sci. 2022, 23, 6459. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Frontiera, R.R.; Tran, R.; Mathies, R.A. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200. [Google Scholar] [CrossRef] [PubMed]
- Laptenok, S.P.; Gil, A.A.; Hall, C.R.; Lukacs, A.; Iuliano, J.N.; Jones, G.A.; Greetham, G.M.; Donaldson, P.; Miyawaki, A.; Tonge, P.J.; et al. Infrared spectroscopy reveals multi-step multi-timescale photoactivation in the photoconvertible protein archetype Dronpa. Nat. Chem. 2018, 10, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Tang, L. Mapping structural dynamics of proteins with femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2020, 71, 239–265. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Browne, W.R.; Feringa, B.L.; Meech, S.R. Ultrafast motion in a third generation photomolecular motor. Nat. Commun. 2023, 14, 1253. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.F.; He, X.; Wachter, R.M.; Tonge, P.J. Probing the ground state structure of the green fluorescent protein chromophore using Raman spectroscopy. Biochemistry 2000, 39, 4423–4431. [Google Scholar] [CrossRef]
- Schellenberg, P.; Johnson, E.; Esposito, A.P.; Reid, P.J.; Parson, W.W. Resonance Raman scattering by the green fluorescent protein and an analogue of its chromophore. J. Phys. Chem. B 2001, 105, 5316–5322. [Google Scholar] [CrossRef]
- He, X.; Bell, A.F.; Tonge, P.J. Isotopic labeling and normal-mode analysis of a model green fluorescent protein chromophore. J. Phys. Chem. B 2002, 106, 6056–6066. [Google Scholar] [CrossRef]
- Fang, C.; Tang, L.; Chen, C. Unveiling coupled electronic and vibrational motions of chromophores in condensed phases. J. Chem. Phys. 2019, 151, 200901. [Google Scholar] [CrossRef]
- Huang, B.; Babcock, H.; Zhuang, X. Breaking the diffraction barrier: Super-resolution imaging of cells. Cell 2010, 143, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Nienhaus, K.; Nienhaus, G.U. Fluorescent proteins for live-cell imaging with super-resolution. Chem. Soc. Rev. 2014, 43, 1088–1106. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, M.; Li, D.; He, W.; Peng, J.; Betzig, E.; Xu, P. Highly photostable, reversibly photoswitchable fluorescent protein with high contrast ratio for live-cell superresolution microscopy. Proc. Natl. Acad. Sci. USA 2016, 113, 10364–10369. [Google Scholar] [CrossRef] [PubMed]
- Coquelle, N.; Sliwa, M.; Woodhouse, J.; Schirò, G.; Adam, V.; Aquila, A.; Barends, T.R.M.; Boutet, S.; Byrdin, M.; Carbajo, S.; et al. Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nat. Chem. 2018, 10, 31–37. [Google Scholar] [CrossRef]
- Woodhouse, J.; Nass Kovacs, G.; Coquelle, N.; Uriarte, L.M.; Adam, V.; Barends, T.R.M.; Byrdin, M.; de la Mora, E.; Bruce Doak, R.; Feliks, M.; et al. Photoswitching mechanism of a fluorescent protein revealed by time-resolved crystallography and transient absorption spectroscopy. Nat. Commun. 2020, 11, 741. [Google Scholar] [CrossRef]
- Bourgeois, D.; Adam, V. Reversible photoswitching in fluorescent proteins: A mechanistic view. IUBMB Life 2012, 64, 482–491. [Google Scholar] [CrossRef]
- Zhou, X.X.; Lin, M.Z. Photoswitchable fluorescent proteins: Ten years of colorful chemistry and exciting applications. Curr. Opin. Chem. Biol. 2013, 17, 682–690. [Google Scholar] [CrossRef]
- Buhrke, D.; Hildebrandt, P. Probing structure and reaction dynamics of proteins using time-resolved resonance Raman spectroscopy. Chem. Rev. 2020, 120, 3577–3630. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Fron, E.; Hosoi, H.; Kuramochi, H.; Takeuchi, S.; Mizuno, H.; Tahara, T. Excited-state proton transfer dynamics in LSSmOrange studied by time-resolved impulsive stimulated Raman spectroscopy. J. Phys. Chem. Lett. 2021, 12, 7466–7473. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Ai, H.-w.; Henderson, J.N.; Remington, S.J.; Campbell, R.E. Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: Structural characterization and applications in fluorescence imaging. Biochem. J. 2006, 400, 531–540. [Google Scholar] [CrossRef]
- Ando, R.; Flors, C.; Mizuno, H.; Hofkens, J.; Miyawaki, A. Highlighted generation of fluorescence signals using simultaneous two-color irradiation on Dronpa mutants. Biophys. J. 2007, 92, L97–L99. [Google Scholar] [CrossRef]
- Hoi, H.; Howe, E.S.; Ding, Y.; Zhang, W.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Davidson, M.W.; Campbell, R.E. An engineered monomeric Zoanthus sp. yellow fluorescent protein. Chem. Biol. 2013, 20, 1296–1304. [Google Scholar] [CrossRef]
- Karasawa, S.; Araki, T.; Nagai, T.; Mizuno, H.; Miyawaki, A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem. J. 2004, 381, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.G.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Lin, M.Z.; McKeown, M.R.; Steinbach, P.A.; Hazelwood, K.L.; Davidson, M.W.; Tsien, R.Y. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 2008, 5, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Belousov, V.V.; Zaraisky, A.G.; Novoselov, V.V.; Staroverov, D.B.; Zorov, D.B.; Lukyanov, S.; Lukyanov, K.A. Kindling fluorescent proteins for precise in vivo photolabeling. Nat. Biotechnol. 2003, 21, 191–194. [Google Scholar] [CrossRef]
- Merzlyak, E.M.; Goedhart, J.; Shcherbo, D.; Bulina, M.E.; Shcheglov, A.S.; Fradkov, A.F.; Gaintzeva, A.; Lukyanov, K.A.; Lukyanov, S.; Gadella, T.W.J.; et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 2007, 4, 555–557. [Google Scholar] [CrossRef]
- Krueger, T.D.; Tang, L.; Zhu, L.; Breen, I.L.; Wachter, R.M.; Fang, C. Dual illumination enhances transformation of an engineered green-to-red photoconvertible fluorescent protein. Angew. Chem. Int. Ed. 2020, 59, 1644–1652. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef] [PubMed]
- McCamant, D.W.; Kukura, P.; Yoon, S.; Mathies, R.A. Femtosecond broadband stimulated Raman spectroscopy: Apparatus and methods. Rev. Sci. Instrum. 2004, 75, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Dietze, D.R.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. ChemPhysChem 2016, 17, 1224–1251. [Google Scholar] [CrossRef] [PubMed]
- Webber, N.M.; Litvinenko, K.L.; Meech, S.R. Radiationless relaxation in a synthetic analogue of the green fluorescent protein chromophore. J. Phys. Chem. B 2001, 105, 8036–8039. [Google Scholar] [CrossRef]
- He, X.; Bell, A.F.; Tonge, P.J. Ground state isomerization of a model green fluorescent protein chromophore. FEBS Lett. 2003, 549, 35–38. [Google Scholar] [CrossRef]
- Martin, M.E.; Negri, F.; Olivucci, M. Origin, nature, and fate of the fluorescent state of the green fluorescent protein chromophore at the CASPT2//CASSCF resolution. J. Am. Chem. Soc. 2004, 126, 5452–5464. [Google Scholar] [CrossRef]
- Olsen, S.; Smith, S.C. Radiationless decay of red fluorescent protein chromophore models via twisted intramolecular charge-transfer states. J. Am. Chem. Soc. 2007, 129, 2054–2065. [Google Scholar] [CrossRef]
- Mandal, D.; Tahara, T.; Meech, S.R. Excited-state dynamics in the green fluorescent protein chromophore. J. Phys. Chem. B 2004, 108, 1102–1108. [Google Scholar] [CrossRef]
- Taylor, M.A.; Zhu, L.; Rozanov, N.D.; Stout, K.T.; Chen, C.; Fang, C. Delayed vibrational modulation of the solvated GFP chromophore into a conical intersection. Phys. Chem. Chem. Phys. 2019, 21, 9728–9739. [Google Scholar] [CrossRef]
- Boulanger, S.A.; Chen, C.; Tang, L.; Zhu, L.; Baleeva, N.S.; Myasnyanko, I.N.; Baranov, M.S.; Fang, C. Shedding light on ultrafast ring-twisting pathways of halogenated GFP chromophores from the excited to ground state. Phys. Chem. Chem. Phys. 2021, 23, 14636–14648. [Google Scholar] [CrossRef]
- Jones, C.M.; List, N.H.; Martínez, T.J. Resolving the ultrafast dynamics of the anionic green fluorescent protein chromophore in water. Chem. Sci. 2021, 12, 11347–11363. [Google Scholar] [CrossRef]
- Laptenok, S.P.; Conyard, J.; Page, P.C.B.; Chan, Y.; You, M.; Jaffrey, S.R.; Meech, S.R. Photoacid behaviour in a fluorinated green fluorescent protein chromophore: Ultrafast formation of anion and zwitterion states. Chem. Sci. 2016, 7, 5747–5752. [Google Scholar] [CrossRef]
- Song, W.; Strack, R.L.; Svensen, N.; Jaffrey, S.R. Plug-and-play fluorophores extend the spectral properties of Spinach. J. Am. Chem. Soc. 2014, 136, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Baranov, M.S.; Zhu, L.; Baleeva, N.S.; Smirnov, A.Y.; Zaitseva, S.O.; Yampolsky, I.V.; Solntsev, K.M.; Fang, C. Designing redder and brighter fluorophores by synergistic tuning of ground and excited states. Chem. Commun. 2019, 55, 2537–2540. [Google Scholar] [CrossRef] [PubMed]
- Povarova, N.V.; Zaitseva, S.O.; Baleeva, N.S.; Smirnov, A.Y.; Myasnyanko, I.N.; Zagudaylova, M.B.; Bozhanova, N.G.; Gorbachev, D.A.; Malyshevskaya, K.K.; Gavrikov, A.S.; et al. Red-shifted substrates for fast fluorogen-activating protein based on the GFP-like chromophores. Chem. Eur. J. 2019, 25, 9592–9596. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Romei, M.G.; Oltrogge, L.M.; Mathews, I.I.; Boxer, S.G. Unified model for photophysical and electro-optical properties of green fluorescent proteins. J. Am. Chem. Soc. 2019, 141, 15250–15265. [Google Scholar] [CrossRef]
- Fang, C.; Senes, A.; Cristian, L.; DeGrado, W.F.; Hochstrasser, R.M. Amide vibrations are delocalized across the hydrophobic interface of a transmembrane helix dimer. Proc. Natl. Acad. Sci. USA 2006, 103, 16740–16745. [Google Scholar] [CrossRef]
- Dong, J.; Solntsev, K.M.; Tolbert, L.M. Solvatochromism of the green fluorescence protein chromophore and its derivatives. J. Am. Chem. Soc. 2006, 128, 12038–12039. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Abboud, J.-L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, π*, α, and β, and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Altoe, P.; Bernardi, F.; Garavelli, M.; Orlandi, G.; Negri, F. Solvent effects on the vibrational activity and photodynamics of the green fluorescent protein chromophore: A quantum-chemical study. J. Am. Chem. Soc. 2005, 127, 3952–3963. [Google Scholar] [CrossRef] [PubMed]
- Gepshtein, R.; Huppert, D.; Agmon, N. Deactivation mechanism of the green fluorescent chromophore. J. Phys. Chem. B 2006, 110, 4434–4442. [Google Scholar] [CrossRef] [PubMed]
- Fried, S.D.; Boxer, S.G. Measuring electric fields and noncovalent interactions using the vibrational Stark effect. Acc. Chem. Res. 2015, 48, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Tao, Y.; Zou, W.; Chen, X.; Chen, X.; Freindorf, M.; Kraka, E. A critical evaluation of vibrational Stark effect (VSE) probes with the local vibrational mode theory. Sensors 2020, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Bauman, J.D.; Das, K.; Remorino, A.; Arnold, E.; Hochstrasser, R.M. Two-dimensional infrared spectra reveal relaxation of the nonnucleoside inhibitor TMC278 complexed with the HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 2008, 105, 1472–1477. [Google Scholar] [CrossRef]
- Weaver, J.B.; Kozuch, J.; Kirsh, J.M.; Boxer, S.G. Nitrile infrared intensities characterize electric fields and hydrogen bonding in protic, aprotic, and protein environments. J. Am. Chem. Soc. 2022, 144, 7562–7567. [Google Scholar] [CrossRef]
- Zheng, C.; Mao, Y.; Kozuch, J.; Atsango, A.O.; Ji, Z.; Markland, T.E.; Boxer, S.G. A two-directional vibrational probe reveals different electric field orientations in solution and an enzyme active site. Nat. Chem. 2022, 14, 891–897. [Google Scholar] [CrossRef]
- Mennucci, B. Hydrogen bond versus polar effects: An ab initio analysis on n→π* absorption spectra and N nuclear shieldings of diazines in solution. J. Am. Chem. Soc. 2002, 124, 1506–1515. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Rohrdanz, M.A.; Martins, K.M.; Herbert, J.M. A long-range-corrected density functional that performs well for both ground-state properties and time-dependent density functional theory excitation energies, including charge-transfer excited states. J. Chem. Phys. 2009, 130, 054112. [Google Scholar] [CrossRef]
- Park, J.W.; Shiozaki, T. On-the-fly CASPT2 surface-hopping dynamics. J. Chem. Theory Comput. 2017, 13, 3676–3683. [Google Scholar] [CrossRef] [PubMed]
- Raucci, U.; Chiariello, M.G.; Rega, N. Modeling excited-state proton transfer to solvent: A dynamics study of a super photoacid with a hybrid implicit/explicit solvent model. J. Chem. Theory Comput. 2020, 16, 7033–7043. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A.C. On the theory of Raman intensities. J. Chem. Phys. 1961, 34, 1476–1484. [Google Scholar] [CrossRef]
- Myers, A.B.; Mathies, R.A. Resonance Raman intensities: A probe of excited-state structure and dynamics. In Biological Applications of Raman Spectroscopy; Spiro, T.G., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1987; Volume 2, pp. 1–58. [Google Scholar]
- Stiel, A.C.; Trowitzsch, S.; Weber, G.; Andresen, M.; Eggeling, C.; Hell, S.W.; Jakobs, S.; Wahl, M.C. 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochem. J. 2007, 402, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.N.; Ai, H.-w.; Campbell, R.E.; Remington, S.J. Structural basis for reversible photobleaching of a green fluorescent protein homologue. Proc. Natl. Acad. Sci. USA 2007, 104, 6672–6677. [Google Scholar] [CrossRef]
- Romei, M.G.; Lin, C.-Y.; Mathews, I.I.; Boxer, S.G. Electrostatic control of photoisomerization pathways in proteins. Science 2020, 367, 76–79. [Google Scholar] [CrossRef]
- Krueger, T.D.; Tang, L.; Chen, C.; Zhu, L.; Breen, I.L.; Wachter, R.M.; Fang, C. To twist or not to twist: From chromophore structure to dynamics inside engineered photoconvertible and photoswitchable fluorescent proteins. Protein Sci. 2023, 32, e4517. [Google Scholar] [CrossRef]
- Arpino, J.A.J.; Rizkallah, P.J.; Jones, D.D. Crystal structure of enhanced green fluorescent protein to 1.35 Å resolution reveals alternative conformations for Glu222. PLoS ONE 2012, 7, e47132. [Google Scholar] [CrossRef]
- Fang, C.; Tang, L.; Oscar, B.G.; Chen, C. Capturing structural snapshots during photochemical reactions with ultrafast Raman spectroscopy: From materials transformation to biosensor responses. J. Phys. Chem. Lett. 2018, 9, 3253–3263. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Tang, L.; Oscar, B.G.; Zhu, L.; Fang, C. Panoramic portrait of primary molecular events preceding excited state proton transfer in water. Chem. Sci. 2016, 7, 5484–5494. [Google Scholar] [CrossRef]
- Donati, G.; Petrone, A.; Caruso, P.; Rega, N. The mechanism of a green fluorescent protein proton shuttle unveiled in the time-resolved frequency domain by excited state ab initio dynamics. Chem. Sci. 2018, 9, 1126–1135. [Google Scholar] [CrossRef]
- Andresen, M.; Wahl, M.C.; Stiel, A.C.; Gräter, F.; Schäfer, L.V.; Trowitzsch, S.; Weber, G.; Eggeling, C.; Grubmüller, H.; Hell, S.W.; et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13070–13074. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, H.; Zhang, J.; Ai, H.-w.; Fang, C. Structural origin and rational development of bright red noncanonical variants of green fluorescent protein. Phys. Chem. Chem. Phys. 2023, 25, 15624–15634. [Google Scholar] [CrossRef] [PubMed]
- Yampolsky, I.V.; Remington, S.J.; Martynov, V.I.; Potapov, V.K.; Lukyanov, S.; Lukyanov, K.A. Synthesis and properties of the chromophore of the asFP595 chromoprotein from Anemonia sulcata. Biochemistry 2005, 44, 5788–5793. [Google Scholar] [CrossRef]
- Henderson, J.N.; Remington, S.J. The kindling fluorescent protein: A transient photoswitchable marker. Physiology 2006, 21, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Tretyakova, Y.A.; Pakhomov, A.A.; Martynov, V.I. Chromophore structure of the kindling fluorescent protein asFP595 from Anemonia sulcata. J. Am. Chem. Soc. 2007, 129, 7748–7749. [Google Scholar] [CrossRef] [PubMed]
- Tozzini, V.; Nifosì, R. Ab initio molecular dynamics of the green fluorescent protein (GFP) chromophore: An insight into the photoinduced dynamics of green fluorescent proteins. J. Phys. Chem. B 2001, 105, 5797–5803. [Google Scholar] [CrossRef]
- Brakemann, T.; Weber, G.; Andresen, M.; Groenhof, G.; Stiel, A.C.; Trowitzsch, S.; Eggeling, C.; Grubmüller, H.; Hell, S.W.; Wahl, M.C.; et al. Molecular basis of the light-driven switching of the photochromic fluorescent protein Padron. J. Biol. Chem. 2010, 285, 14603–14609. [Google Scholar] [CrossRef]
- Kent, K.P.; Boxer, S.G. Light-activated reassembly of split green fluorescent protein. J. Am. Chem. Soc. 2011, 133, 4046–4052. [Google Scholar] [CrossRef]
- Kumpulainen, T.; Lang, B.; Rosspeintner, A.; Vauthey, E. Ultrafast elementary photochemical processes of organic molecules in liquid solution. Chem. Rev. 2017, 117, 10826–10939. [Google Scholar] [CrossRef]
- Yang, J.-S.; Huang, G.-J.; Liu, Y.-H.; Peng, S.-M. Photoisomerization of the green fluorescence protein chromophore and the meta- and para-amino analogues. Chem. Commun. 2008, 44, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Voliani, V.; Bizzarri, R.; Nifosì, R.; Abbruzzetti, S.; Grandi, E.; Viappiani, C.; Beltram, F. Cis−trans photoisomerization of fluorescent-protein chromophores. J. Phys. Chem. B 2008, 112, 10714–10722. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.; Helms, V.; McCammon, J.A.; Langhoff, P.W. Shedding light on the dark and weakly fluorescent states of green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 6177–6182. [Google Scholar] [CrossRef] [PubMed]
- Luin, S.; Voliani, V.; Lanza, G.; Bizzarri, R.; Nifosì, R.; Amat, P.; Tozzini, V.; Serresi, M.; Beltram, F. Raman study of chromophore states in photochromic fluorescent proteins. J. Am. Chem. Soc. 2009, 131, 96–103. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, L.; Fang, C. Femtosecond stimulated Raman line shapes: Dependence on resonance conditions of pump and probe pulses. Chin. J. Chem. Phys. 2018, 31, 492–502. [Google Scholar] [CrossRef]
- Zutterman, F.; Liégeois, V.; Champagne, B. TDDFT investigation of the Raman and resonant Raman spectra of fluorescent protein chromophore models. J. Phys. Chem. B 2022, 126, 3414–3424. [Google Scholar] [CrossRef]
- Petersen, J.; Wilmann, P.G.; Beddoe, T.; Oakley, A.J.; Devenish, R.J.; Prescott, M.; Rossjohn, J. The 2.0-Å crystal structure of eqFP611, a far red fluorescent protein from the sea anemone Entacmaea quadricolor. J. Biol. Chem. 2003, 278, 44626–44631. [Google Scholar] [CrossRef]
- Mukherjee, S.; Manna, P.; Hung, S.-T.; Vietmeyer, F.; Friis, P.; Palmer, A.E.; Jimenez, R. Directed evolution of a bright variant of mCherry: Suppression of nonradiative decay by fluorescence lifetime selections. J. Phys. Chem. B 2022, 126, 4659–4668. [Google Scholar] [CrossRef]
- Loos, D.C.; Habuchi, S.; Flors, C.; Hotta, J.-i.; Wiedenmann, J.; Nienhaus, G.U.; Hofkens, J. Photoconversion in the red fluorescent protein from the sea anemone Entacmaea quadricolor: Is cis−trans isomerization involved? J. Am. Chem. Soc. 2006, 128, 6270–6271. [Google Scholar] [CrossRef]
- Park, J.W.; Rhee, Y.M. Electric field keeps chromophore planar and produces high yield fluorescence in green fluorescent protein. J. Am. Chem. Soc. 2016, 138, 13619–13629. [Google Scholar] [CrossRef]
- Yarbrough, D.; Wachter, R.M.; Kallio, K.; Matz, M.V.; Remington, S.J. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-Å resolution. Proc. Natl. Acad. Sci. USA 2001, 98, 462–467. [Google Scholar] [CrossRef]
- Lambert, T.J. FPbase: A community-editable fluorescent protein database. Nat. Methods 2019, 16, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Hung, S.-T.; Douglas, N.; Manna, P.; Thomas, C.; Ekrem, A.; Palmer, A.E.; Jimenez, R. Engineering of a brighter variant of the fusionred fluorescent protein using lifetime flow cytometry and structure-guided mutations. Biochemistry 2020, 59, 3669–3682. [Google Scholar] [CrossRef]
- Ai, H.-W.; Campbell, R.E. Teal Fluorescent Proteins: Characterization of a Reversibly Photoswitchable Variant; SPIE BiOS; SPIE: San Jose, CA, USA, 2008; p. 68680D. [Google Scholar]
- Baranov, M.S.; Solntsev, K.M.; Lukyanov, K.A.; Yampolsky, I.V. A synthetic approach to GFP chromophore analogs from 3-azidocinnamates. Role of methyl rotors in chromophore photophysics. Chem. Commun. 2013, 49, 5778–5780. [Google Scholar] [CrossRef]
- Yampolsky, I.V.; Kislukhin, A.A.; Amatov, T.T.; Shcherbo, D.; Potapov, V.K.; Lukyanov, S.; Lukyanov, K.A. Synthesis and properties of the red chromophore of the green-to-red photoconvertible fluorescent protein Kaede and its analogs. Bioorg. Chem. 2008, 36, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Baleeva, N.S.; Myannik, K.A.; Yampolsky, I.V.; Baranov, M.S. Bioinspired fluorescent dyes based on a conformationally locked chromophore of the fluorescent protein Kaede. Eur. J. Org. Chem. 2015, 2015, 5716–5721. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, W.; Fang, C. A versatile femtosecond stimulated Raman spectroscopy setup with tunable pulses in the visible to near infrared. Appl. Phys. Lett. 2014, 105, 041106. [Google Scholar] [CrossRef]
- Liu, W.; Han, F.; Smith, C.; Fang, C. Ultrafast conformational dynamics of pyranine during excited state proton transfer in aqueous solution revealed by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 10535–10550. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Habuchi, S.; Cotlet, M.; Gensch, T.; Bednarz, T.; Haber-Pohlmeier, S.; Rozenski, J.; Dirix, G.; Michiels, J.; Vanderleyden, J.; Heberle, J.; et al. Evidence for the isomerization and decarboxylation in the photoconversion of the red fluorescent protein DsRed. J. Am. Chem. Soc. 2005, 127, 8977–8984. [Google Scholar] [CrossRef]
- Weigel, A.; Dobryakov, A.; Klaumünzer, B.; Sajadi, M.; Saalfrank, P.; Ernsting, N.P. Femtosecond stimulated Raman spectroscopy of flavin after optical excitation. J. Phys. Chem. B 2011, 115, 3656–3680. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, C.; Pontecorvo, E.; Cerullo, G.; Vos, M.H.; Scopigno, T. Direct observation of subpicosecond vibrational dynamics in photoexcited myoglobin. Nat. Chem. 2016, 8, 1137–1143. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R2 | Bond Length (Å) | ||||||
|---|---|---|---|---|---|---|---|---|
| CO, 1 | CC, 2 | CC, 3 | CC, 4 | CC, 5 | CO, 6 | CN, 7 | ||
| Me | Me | 1.2823 | 1.3761 | 1.3759 | 1.4272 | 1.3711 | 1.2351 | 1.3023 |
| Me | H | 1.2800 | 1.3747 | 1.3744 | 1.4234 | 1.3738 | 1.2363 | 1.2984 |
| H | Me | 1.2818 | 1.3757 | 1.3755 | 1.4263 | 1.3715 | 1.2340 | 1.3008 |
| Bond Length (Å) | |||||||
|---|---|---|---|---|---|---|---|
| CO, 1 | CC, 2 | CC, 3 | CC, 4 | CC, 5 | CO, 6 | CN, 7 | |
| water (implicit) | 1.2709 | 1.3741 | 1.3735 | 1.4229 | 1.3747 | 1.2372 | 1.3026 |
| water (1 explicit, –O−) | 1.2823 | 1.3761 | 1.3759 | 1.4272 | 1.3711 | 1.2351 | 1.3023 |
| water (1 explicit, C=O) | 1.2688 | 1.3725 | 1.3720 | 1.4191 | 1.3783 | 1.2462 | 1.3019 |
| water (2 explicit, –O− and C=O) | 1.2802 | 1.3747 | 1.3744 | 1.4237 | 1.3745 | 1.2438 | 1.3014 |
| MeCN (implicit) | 1.2703 | 1.3739 | 1.3733 | 1.4223 | 1.3750 | 1.2372 | 1.3024 |
| DMSO (implicit) | 1.2706 | 1.3740 | 1.3734 | 1.4226 | 1.3749 | 1.2372 | 1.3025 |
| p-HBDI (Water) | p-HBDI (DMSO) | EGFP | Dronpa2 | mTFP0.7 | LEA (Green Form) |
|---|---|---|---|---|---|
| 612 | 602 | 616 | 616 | 624 | 613 |
| 852 | 830 | 844 | 846 | 851 | 844 |
| 932 | 921 | 917 | 920 | 907 | 923 |
| – | – | 1079 | 1083 | 1071 | 1080 |
| 1164 | 1175 | 1163 | 1164 | 1161 | 1168 |
| 1238 | 1239 | 1256 | 1259 | 1248 | 1261 |
| 1302 | 1295 | 1324 | 1329 | 1318 | 1329 |
| 1363 | 1353 | 1362 | 1367 | 1365 | 1365 |
| 1496 | 1487 | 1492 | 1493 | 1492 | 1494 |
| 1548 | 1542 | 1532 | 1532 | 1541 | 1531 |
| 1578 | 1576 | 1570 | 1570 | 1574 | 1565 |
| 1623 | 1618 | 1614 | 1621 | 1622 | 1616 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Henderson, J.N.; Ruchkin, D.A.; Kirsh, J.M.; Baranov, M.S.; Bogdanov, A.M.; Mills, J.H.; Boxer, S.G.; Fang, C. Structural Characterization of Fluorescent Proteins Using Tunable Femtosecond Stimulated Raman Spectroscopy. Int. J. Mol. Sci. 2023, 24, 11991. https://doi.org/10.3390/ijms241511991
Chen C, Henderson JN, Ruchkin DA, Kirsh JM, Baranov MS, Bogdanov AM, Mills JH, Boxer SG, Fang C. Structural Characterization of Fluorescent Proteins Using Tunable Femtosecond Stimulated Raman Spectroscopy. International Journal of Molecular Sciences. 2023; 24(15):11991. https://doi.org/10.3390/ijms241511991
Chicago/Turabian StyleChen, Cheng, J. Nathan Henderson, Dmitry A. Ruchkin, Jacob M. Kirsh, Mikhail S. Baranov, Alexey M. Bogdanov, Jeremy H. Mills, Steven G. Boxer, and Chong Fang. 2023. "Structural Characterization of Fluorescent Proteins Using Tunable Femtosecond Stimulated Raman Spectroscopy" International Journal of Molecular Sciences 24, no. 15: 11991. https://doi.org/10.3390/ijms241511991
APA StyleChen, C., Henderson, J. N., Ruchkin, D. A., Kirsh, J. M., Baranov, M. S., Bogdanov, A. M., Mills, J. H., Boxer, S. G., & Fang, C. (2023). Structural Characterization of Fluorescent Proteins Using Tunable Femtosecond Stimulated Raman Spectroscopy. International Journal of Molecular Sciences, 24(15), 11991. https://doi.org/10.3390/ijms241511991