Abstract
Type 2 diabetes (T2D) is associated with a plethora of comorbidities, including osteoporosis, which occurs due to an imbalance between bone resorption and formation. Numerous mechanisms have been explored to understand this association, including the renin–angiotensin–aldosterone system (RAAS). An upregulated RAAS has been positively correlated with T2D and estrogen deficiency in comorbidities such as osteoporosis in humans and experimental studies. Therefore, research has focused on these associations in order to find ways to improve glucose handling, osteoporosis and the downstream effects of estrogen deficiency. Upregulation of RAAS may alter the bone microenvironment by altering the bone marrow inflammatory status by shifting the osteoprotegerin (OPG)/nuclear factor kappa-Β ligand (RANKL) ratio. The angiotensin-converting-enzyme/angiotensin II/Angiotensin II type 1 receptor (ACE/Ang II/AT1R) has been evidenced to promote osteoclastogenesis and decrease osteoblast formation and differentiation. ACE/Ang II/AT1R inhibits the wingless-related integration site (Wnt)/β-catenin pathway, which is integral in bone formation. While a lot of literature exists on the effects of RAAS and osteoporosis on T2D, the work is yet to be consolidated. Therefore, this review looks at RAAS activity in relation to osteoporosis and T2D. This review also highlights the relationship between RAAS activity, osteoporosis and estrogen deficiency in T2D.
1. Introduction
The renin–angiotensin–aldosterone system (RAAS) is a fluid and electrolyte regulatory system responsible for the maintenance of blood volume and pressure [1]. This system has been studied extensively and has been evidenced to be crucial in maintaining glucose homeostasis [2]. A plethora of studies have highlighted the relationship between hyperglycemia and alterations in RAAS, and the development of estrogen deficiency and osteoporosis [3,4]. RAAS components have been identified in bones, whereby the interaction between angiotensin II (Ang II) and the angiotensin II type 1 receptor (AT1R) inhibits osteoblast maturation [5]. Furthermore, upregulation of Ang II has been reported to elevate nuclear factor-κB ligand (RANKL) and decrease osteoprotegerin (OPG) expression, thus activating osteoclasts, which promote bone resorption [6]. Additionally, the upregulation of Ang II promotes the upregulation of aldosterone, which is another RAAS hormone that affects bone turnover by binding to mineralocorticoid receptors (MR) in osteoclasts, osteocytes and osteoblasts [7,8]. The Ang II/AT1R axis has been reported to stimulate proinflammatory cytokines that promote bone resorption and hinder osteoblast differentiation [9]. MRs are also expressed in the parathyroid tissue and several authors have demonstrated a positive correlation between the concentrations of aldosterone and serum parathyroid hormone (PTH), a calcium-regulating hormone [10]. Collectively, upregulation of the Ang II/AT1R interaction, the shift in the OPG/RANKL ratio and the increase in aldosterone contribute to decreased bone mineral density (BMD) [11], consequently altering the microarchitecture of the bone structure and leading to the development and progression of osteoporosis [11]. Other RAAS components such as angiotensin 1-7 (Ang 1-7), angiotensin 1-9 (Ang 1-9) and angiotensin-converting enzyme 2 (ACE2) are expressed in bone tissue and have been reported to negate the effects of Ang II/AT1R interaction and RANKL upregulation [12,13]. Furthermore, estrogen is a well-known bone anti-resorption hormone [14]. Estrogen exerts numerous physiological effects that maintain glucose homeostasis [15]. However, estrogen deficiency is associated with impaired glucose uptake, impaired insulin secretion, insulin resistance, increased gluconeogenesis and increased lipolysis, which are clinical markers of T2D [16,17]. Interestingly, RAAS is reportedly upregulated in T2D; therefore, this review seeks to highlight the relationship between RAAS and osteoporosis and their association with estrogen deficiency in T2D.
1.1. The Renin–Angiotensin–Aldosterone System
The renin–angiotensin–aldosterone system (RAAS) is a regulatory signaling pathway that maintains fluid and electrolyte homeostasis [18]. Renin is secreted by the kidneys in response to a decrease in blood pressure, blood volume, plasma sodium and potassium levels [19]. Renin has also been shown to induce the release of prorenin from the juxtaglomerular cells in the kidneys [20]. The release of prorenin results in a cascade of events where inactive angiotensin I (Ang I) is produced from angiotensinogen (AGT), which is subsequently cleaved by angiotensin-converting enzyme (ACE) into angiotensin II (Ang II). Ang II activates Ang II type 1 (AT1), which is responsible for inducing vasoconstriction in the systemic vasculature, subsequently raising blood pressure [21]. Furthermore, Ang II acts on the adrenal cortex to synthesize aldosterone, which promotes sodium and water reabsorption through activation of the mineralocorticoid receptor in the nephrons [2,21]. The renin–angiotensin–aldosterone system is pathologically activated in type II diabetes; this has been noted to result in various detrimental effects, including osteoporosis and estrogen deficiency [22,23,24]. Recent studies have evidenced the presence and activity of the renin–angiotensin system in T2D in various tissues, with observable changes in angiotensinogen, renin, ACE, aldosterone, angiotensin II, AT1R, AT2R, Ang 1-7 and Ang 1-9 in bones [22,25]. Furthermore, patients who suffer from T2D and hypertension usually have osteoporosis, with this phenomenon being highly prevalent in postmenopausal women [26].
1.2. Local RAAS and T2D
Several studies have highlighted the role of RAAS in the development and progression of insulin resistance, as RAAS is produced locally in numerous organs [27]. RAAS is upregulated in the skeletal muscle, which is a major site of glucose utilization [28]. Ang II suppresses phosphorylation of insulin receptor substrate (IRS)-1 in muscles, blocking increases in phosphatidylinositol 3 (PI3)-kinase and the consequent translocation of glucose transporter (GLUT) 4 to the cell membrane [29]. This results in hyperglycinemia due to impaired glucose uptake and insulin resistance [30]. Hyperglycemia induces p53 glycosylation, which has been implicated in angiotensinogen transcription and the subsequent generation of Ang II from the local RAAS [31], thus promoting the upregulation of local RAAS in various organs [25]. Furthermore, the upregulation of RAAS induces insulin resistance in adipose tissue [30]. In T2D, the renin-encoding gene is expressed and upregulated in adipose tissue, which also contains Cathepsin D and G enzymes that can create Ang I and Ang II via RAAS alternate pathways [32]. Nutrition was proposed as the link between RAAS upregulation and hyperglycemia. An animal study evidenced the upregulation of RASS mRNA, translocation of the RAAS genes and expression of RAAS proteins in rats fed on high-fat diets and high-fat-high-carbohydrate diets [33]. Angiotensin II induces the formation of prostacyclin in adipose tissue, which causes the conversion of preadipocytes to adipocytes and increases lipid synthesis and storage in adipocytes through Ang II/AT2R signaling and the ACE2/Ang 1-7/Mas receptor [34]. However, in overt hyperglycemia, which occurs in T2D, the expression of Ang II and AT1R is upregulated whilst the expression of AT2R and ACE2/Ang 1-7/Mas receptor is downregulated [28]. Ang II/AT1R signaling has been reported to induce lipogenesis and impair the differentiation of preadipocytes, thus affecting adipose storage capacity [28,33]. Consequently, the adipose tissue is overloaded with lipids, resulting in the ectopic redistribution of fats to other organs [33]. Additionally, a positive correlation has been noted between insulin resistance and the upregulation of Ang II/AT1R signaling [35]. Hence, the upregulation of adipose RAAS contributes to the development and progression of insulin resistance and T2D [35]. The increased lipogenesis contributes to the storage of fats in the liver [36]. Hypertriglyceridemia is closely associated with insulin resistance [37]. The liver and adipose tissue are the main sites of triglyceride synthesis [38]. Ang II/AT1R signaling, which is upregulated in T2D, is involved in the suppression of the protein expression and enzymatic activity of hepatic N-deacetylase (NDST) [39] This is a key regulatory enzyme involved in heparan sulphate (HS) biosynthesis in the diabetic state and suppression of hepatic NDST was suggested to lead to diabetic dyslipidemia [40,41,42]. Furthermore, Ang II-based activation of AT1R causes insulin resistance by increasing hepatic triglyceride levels, which is thought to contribute to the development of diabetes [28,43]. Diabetic dyslipidemia is associated with damage to vital organs such as the heart and kidneys [44,45]. Local RAAS in the kidneys has been evidenced, in various studies, to cause morphological changes that affect kidney function [44,46]. In T2D, systemic and kidney RAAS have been positively correlated with proteinuria and hypertension [46,47]. Increased blood pressure has been evidenced to cause stress on the heart, thus affecting its function [29,48]. Interestingly, local RAAS activity has also been reported to compromise the integrity of the heart [28,49]. In T2D, activation of the Ang II/AT1R pathway can promote cell growth and proliferation, apoptosis, oxidative stress generation, inflammation and fibrosis, which can contribute to cardiac remodeling and atherosclerosis [50,51]. Therefore, research has been focused on this system due to the detrimental effects of the upregulation of ACE/Ang II/AT1R signaling and downregulation of the AT2R and Ang 1-7/AT2R/Mas axis in the various organs (Figure 1) [28]. A study by Zheng and colleagues showed an improvement in insulin resistance and the mentioned downstream changes when the ACE/Ang II/AT1R axis was blocked [35]. Recently, local RAAS has been reported in bones and associated with bone fragility and the development of osteoporosis in T2D due to the shift in pro-resorptive and anti-resorptive factors in favor of pro-resorptive factors [52,53].
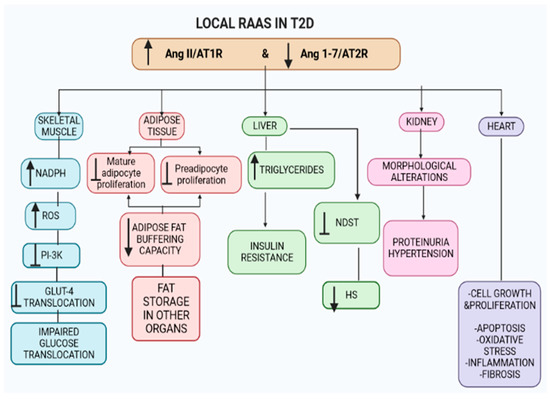
Figure 1.
Local RAAS in various organs. Angiotensin II (Ang II) and angiotensin II type 1 receptor (AT1R) signaling are upregulated, whilst angiotensin 1-7 (Ang 1-7) and angiotensin type 2 receptor (AT2R) axis are downregulated in the numerous organs, i.e., skeletal muscle (leading to insulin resistance) and adipose tissue (decreasing the adipose tissue buffering capacity). The shift in the axis in the liver results in increased hepatic triglycerides, insulin resistance and inhibition of N-deacetylase (NDST), reducing heparan sulphate (HS). Additionally, the shift in Ang II/AT1R and Ang 1-7/AT2R in the kidneys and heart causes morphological changes, leading to organ dysfunction. Key For all figures. ↑ = increase/upregulation; ↓ = decrease; ┴ = inhibition; → = convert.
2. Pro-Resorptive Factors
2.1. Parathyroid Hormone
Parathyroid hormone (PTH) is an 84-amino acid polypeptide released by the parathyroid glands in response to calcium deficiency [54]. PTH promotes renal tubular calcium reabsorption and stimulates renal 1,25 dihydroxy vitamin D synthesis, thus indirectly enhancing intestinal calcium absorption and bone remodeling [54]. PTH is critical in calcium homeostasis and the consequent formation of bone integrity by increasing the number of bone-forming cells as well as stimulating osteoblast development and reducing osteoblast cell death [54,55]. The parathyroid hormone further contributes to calcium homeostasis by promoting bone resorption when there is a calcium deficiency [55]. Studies have evidenced the expression of AT1R and mineralocorticoid receptors (MR) in the parathyroid glands [56,57]. Studies have shown that RAAS, along with the expression of AT1R and MR receptors, is upregulated during T2D, thus enhancing PTH release from the parathyroid glands [56,57]. Due to chronic hyperglycemia in T2D, the parathyroid glands are overstimulated, resulting in excessive release of PTH [58]. The increased PTH levels result in excessive bone resorption, which affects the structural integrity of the bone [58]. Hence, the upregulation of RAAS in T2D may stimulate the production of excessive amounts of PTH, leading to bone fracture and osteoporosis [3]. However, more studies are required to generate evidence for this theory. PTH is pivotal for calcium homeostasis as it promotes the production of 1,25-hydroxy vitamin D in the kidneys [59]. 1,25-hydroxy vitamin D restores calcium balance by promoting bone resorption, and the restored calcium homeostasis inhibits the release of PTH [59]. MR and AT1R are expressed in the parathyroid glands [56]. T2D leads to upregulation of aldosterone and the ACE/Ang II/AT1R axis, potentially resulting in the parathyroid glands being over-stimulated and leading to increased PTH and 1,25-hydroxy vitamin D concentrations that could further lead to osteoporosis [3,50,59,60]. Furthermore, PTH stimulates the release of aldosterone from the adrenal glands due to the expression of type 1 PTH receptors in the adrenal glands, indicating a cycle of continued activity [56].
2.2. Vitamin D3
Vitamin D3 plays a key role in calcium homeostasis and can be synthesized endogenously in the skin or absorbed via the intestines from our diet, converted to 25-hydroxyvitamin D (25(OH)2D) in the liver or to the bioactive form, 1,25-hydroxyvitamin D (1,25(OH)2D), in the kidneys [61]. In the postnatal stage, one of the principal functions of 1,25(OH)2D is to maintain calcium homeostasis by enhancing calcium absorption in the intestines [62]. When dietary calcium levels are low, 1,25(OH)2D increases transcellular intestinal calcium transport by upregulating the expression of the apical membrane calcium channel transient receptor potential vanilloid 6 (TRPV6) and the calcium-binding protein calbindin-D9k as these channels promote calcium reabsorption [61,63]. Additionally, 1,25(OH)2D promotes distal tubular calcium reabsorption in the kidney [64]. T2D causes renal morphological and functional alterations due to the upregulation of systemic and renal RAAS via the ACE/Ang II/AT1R axis, consequently affecting calcium homeostasis [58]. Calcium loss promotes an increase in 1,25(OH)2D, inducing bone breakdown—a process known as bone resorption—in an attempt to restore blood calcium levels [65]. A continued state of bone resorption contributes to bone fragility, increased risk of bone fracture and osteoporosis [66]. Interestingly, a recent study suggested an inverse relationship between 1,25(OH)2D and plasma renin, whereby 1,25(OH)2D has an inhibitory effect on renin [10]. However, PTH promotes aldosterone synthesis in the adrenal glands. Studies show that changes in PTH and 1,25(OH)2D are largely influenced by systemic and renal RAAS, affecting kidney function; however, growing evidence shows an interaction between local RAAS, inflammatory cytokines and bone remodeling [67,68]. Recent studies have demonstrated a positive correlation between PTH and 1,25(OH)2D and the production of Ang II and aldosterone [67,68]. Furthermore, MRs have not only been identified on the parathyroid glands but they have also been identified on osteoblasts, osteoclasts and bone cells, indicating that aldosterone has a direct effect on bone metabolism (Figure 2) [3]. Physiologically, 1,25(OH)2D interacts with the Wnt signaling cascade, which regulates osteoblast differentiation, to induce bone formation and mineralization in osteoblasts as well as osteogenic differentiation from human mesenchymal stem cells [69]. The promoter region of the gene encoding LRP5 is upregulated by interaction with 1,25(OH)2D–VDR, increasing LRP5 expression [70]. Interestingly, RAAS, through the ACE/Ang II/AT1R axis, has been evidenced to inhibit the Wnt signaling cascade [71]. RAAS affects 1,25(OH)2D, thus leading to increased osteoclast formation [12,72]. Furthermore, the 1,25(OH)2D–VDR interaction is impaired in T2D, further reducing bone formation and mineralization [73]. PTH, 1,25(OH)2D, Ang II, glucocorticoids and proinflammatory cytokines have been evidenced to cause osteoclastogenesis, leading to bone remodeling through the expression of nuclear factor-κB ligand (RANKL) [74,75,76,77]. The differentiation and bone-resorbing abilities of the osteoclast depend on RANKL and its receptor, nuclear factor-B (RANK) [77]. Osteoprotegerin (OPG) binds to RANK and inhibits RANKL–RANK binding via competitive inhibition, hence the OPG/RANKL ratio is a measure of osteoclast differentiation [77,78,79]. RANKL influences the immune system and regulates bone remodeling and regrowth [79].
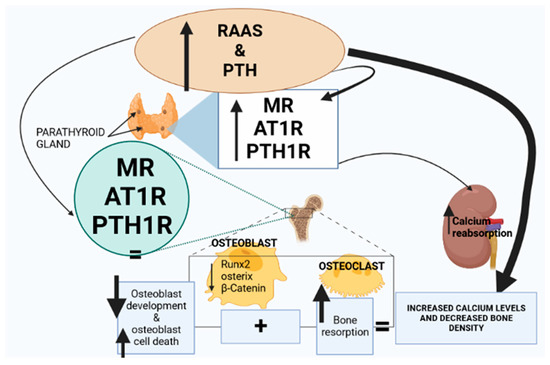
Figure 2.
Renin–angiotensin–aldosterone system (RAAS) activity and parathyroid hormone (PTH) expression are elevated in T2D. The expression of mineralocorticoid receptors (MRs), angiotensin type 1 receptor (AT1R) and parathyroid hormone 1 receptor (PTH1R) is upregulated in the parathyroid gland and the bone marrow. Consequently, Runx, osterix and β-catenin are downregulated, leading to a decrease in osteoblasts; however, osteoclast activity is elevated. Furthermore, the upregulation of MRs, AT1R and PTH1R in the parathyroid glands stimulates PTH hypersecretion, leading to increased calcium reabsorption in the kidneys and collectively resulting in increased plasma calcium and decreased bone density.
2.3. Proinflammatory Cytokines
Osteoblasts and osteoclasts, which are bone-forming and bone-resorbing cells, respectively, are necessary for maintaining bone homeostasis [80]. Osteoblasts synthesize proteins for the bone matrix and encourage bone deposition and mineralization [81]. Bone-related disorders such as postmenopausal osteoporosis, hyperparathyroidism and osteopetrosis are caused by an imbalance between osteoclast and osteoblast activity during bone remodeling as a result of proinflammatory cytokines, growth factors and hormones [82]. Proinflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) are involved in the breakdown of bone through enhancement of the development of osteoclast precursors into adult osteoclasts [83]. Furthermore, TNF-α promotes osteoclastogenesis by increasing RANKL expression in osteoblasts [83]. RANKL binding to RANK commits monocytic precursor cells to the osteoclastic lineage, thus promoting bone loss [83]. TNF-α is also involved in osteoclastogenesis through modulation of the wingless-related integration site (Wnt) signaling pathway, which is considered a key regulatory pathway for bone formation by osteoblasts [84,85]. TNF-α is a potent inducer of the production of Dickkopf-1 (Dkk-1), a Wnt antagonist that hinders the development of bone cells (Figure 3) [85,86]. Systemic and local RAAS is upregulated in T2D; hence, the ACE/Ang II/AT1R axis stimulates the activity of proinflammatory cytokines, resulting in increased osteoclastogenesis, reduced bone density and development of osteoporosis [87].
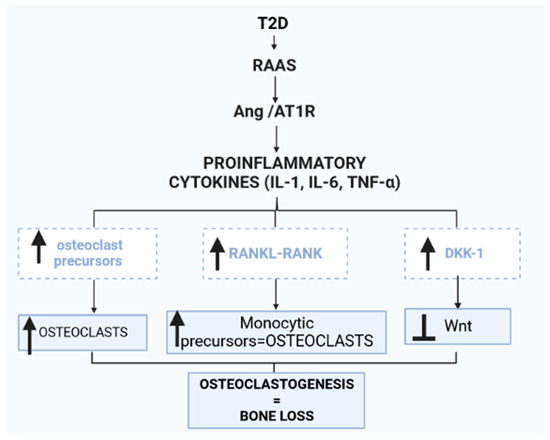
Figure 3.
Renin–angiotensin–aldosterone system (RAAS) activity in the bone marrow is increased in type 2 diabetes (T2D). The Ang II/AT1R axis is upregulated, stimulating proinflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α). The increased activity of proinflammatory cytokines in the bone marrow increases osteoclast precursors, thus increasing osteoclasts. Additionally, the nuclear factor-κB ligand (RANKL) and nuclear factor-κB (RANK) interaction is enhanced, thus promoting the conversion of monocytic precursors to osteoclasts. Osteoclast differentiation is stimulated by M-CSF and RANKL. M-CSF induces the proliferation and survival of osteoclast precursor cells through activation of ERK and Akt. RANKL recruits TRAF6 to activate MAPKs, Akt and NFATc1 to promote the differentiation of osteoclast precursors to osteoclasts. Furthermore, increased proinflammatory cytokine activity in the bone induces the production of dickkopf-1 (Dkk-1), a wingless-related integration site (Wnt) antagonist that hinders the development of bone cells.
2.4. ACE/Ang II/AT1R Axis and ACE2/Ang 1-7/Mas Receptor
Various components of RAAS are expressed in various bone cells and chondrocytes of epiphyseal plates under physiological environments, implicating local RAAS in epiphyseal elongation during bone development and repair [3,88]. Furthermore, a locally active RAAS influencing hematopoietic cell growth, generation, proliferation and differentiation has been identified in bone marrow cells (BMCs), hematopoietic-lineage BMCs and cultured marrow stromal cells (MSCs) [3,13,88]. Ang II/AT1R signaling in these cells affects the production of red blood cells and blood flow in capillaries in the bone marrow [89]. Ang II diminished osteoblastic differentiation and mineralization and reduced the percentage of mineralized nodules by activating AT1R in osteoblast UMR-106 cells [90]. The ACE/Ang II/AT1R axis stimulates bone marrow mononuclear cells to differentiate into multinuclear cells [91,92]. This axis also stimulates multinuclear cells to differentiate into osteoclasts and enhances tartrate-resistant acid phosphatase (TRAP)-positive multinuclear osteoclasts [91]. Osteoclasts are cells that mediate bone loss by increasing their resorptive activity and causing bone degradation, leading to the initiation of the normal bone remodeling process [93]. In pathological conditions such as T2D, systemic and local RAAS are upregulated, particularly the ACE/Ang II/AT1R axis, which may promote osteoclast over-production leading to bone degradation [25,28,94]. Chronic upregulation of RAAS, as observed in T2D, may result in continued bone breakdown, a decline in bone density, osteoporosis and an increased risk of bone fracture. Osteoporosis interventions have focused on RAAS blockers [95]. As previously established, osteoporosis occurs due to an imbalance in bone remodeling, which is marked by a decrease in osteoblastic activity and an increase in osteoclastic activity, resulting in a greater rate of bone resorption [66]. Ang II has been evidenced to play a key role in osteoclastogenesis, and thus osteoporosis, through various mechanisms such as those discussed in the subsequent sections [96].
2.4.1. Ang II and Expression of RANKL
A study demonstrated that Ang II resulted in a significant (eightfold) increase in RANKL mRNA expression through the ERK pathway in human osteoblasts and UMR-106 cells [97]. It also promoted the differentiation of mesenchymal stem cells into multinuclear cells, activated mature osteoclasts responsible for bone resorption and increased the number of TRAP-positive multinuclear osteoclasts, and hence, osteoclastogenesis [98,99]. Studies have suggested that an increase in RANKL upon activation of the AT1R–ERK signaling pathway may serve as a mediator of Ang II-induced osteoclast differentiation and activation [96,100]. In order to investigate this theory, Olmesartan (an AT1R blocker) or U0126 (an extracellular signaling kinase pathway MEK/ERK inhibitor) were added to osteoblasts, resulting in the amelioration of the osteoclastogenesis effects of Ang II [101]. Therefore, studies have alluded to ACE/Ang II/AT1R increasing RANKL expression, thus facilitating pathophysiological bone resorption [102].
2.4.2. Ang II Increases Cyclic Adenosine Monophosphate (cAMP)
RANKL and core-binding factor subunit alpha-1 (Cbfa1/Runx2), both of which are controlled by cAMP, are the main regulators of the differentiation of osteoblast and osteoclast cells [103]. Cbfa1/Runx2 is a transcription factor required for osteoblast development from mesenchymal progenitors and subsequent bone matrix mineralization [103,104]. Furthermore, Cbfa-1 is a potent transcription factor that regulates the expression of many of the osteoblast and chondrocyte functions and triggers the expression of major osteoblast-specific lineage genes [104,105]. Angiotensin II can promote an increase in intracellular cAMP and subsequently activate signaling pathways that are involved in the control of low-density-lipoproteins (LDLs), which in turn changes Cbfa1 expression [106,107]. LDL-mediated cholesterol administration improves osteoclast survival, and hence their lifespan, considerably [108,109]. Therefore, Ang II modifies Cbfa1 expression, consequently reducing the number of osteoblasts and resulting in poor bone formation by stimulating the cAMP signaling pathway and regulating other downstream targets such as LDLs [110,111]. Ang II triggers prostaglandins, which stimulate cAMP signaling, leading to upregulated expression of LDL receptors [112]. The upregulated expression of LDL receptors in the bone marrow leads to increased LDLs, which have been reported to take up space in the marrow, thus affecting osteoblast availability [113].
Furthermore, in vitro studies showed that free cholesterol reduced the proliferation and differentiation of osteoblasts and inhibited the expression of BMP2 and core binding factor alpha 1 (Cbfa1). As mentioned, local RAAS exists in other organs such as adipose tissue, where it alters adipose buffering capacity, thus increasing free cholesterol levels. Free cholesterol increases the level of malondialdehyde (MDA) and decreased the activity of superoxidase dismutase (SOD) in osteoblasts, indicating oxidative stress in the bone microenvironment. Oxidative stress inhibits and decreases osteoblast development and activity, which in turn reduces mineralization and osteogenesis. Furthermore, the increase in cAMP as a result of Ang II activates downstream signaling pathways that, in turn, downregulate the expression of Cbfa1/Runx2 while increasing the expression of RANKL [114]. The decrease in Cbfa1/Runx2 and the increase in RANKL decrease the quantity and function of osteoblasts, resulting in increased bone resorption and decreased bone formation [114].
2.4.3. Ang II Upregulates SOST Gene Expression
Ang II increases the expression of the SOST gene in osteocytes via activation of AT1R [5,85,90]. Sclerostin coding gene (SOST) encodes the secretory protein sclerostin that binds to LRP5/6 (low-density lipoprotein receptor-related protein) receptors on osteoblast cell membranes, inhibiting Wnt/b-catenin signaling and lowering osteoblastic bone production [85,115].
As already mentioned, Ang II/AT1R signaling is upregulated in T2D, resulting in the above cascade of events [90]. Therefore, local bone RAAS, specifically Ang II, has been positively associated with decreased bone density, increased bone turnover and impaired bone microarchitecture, leading to the development and progression of osteoporosis [116]. However, the ACE2/Ang 1-7/MasR receptor arm of bone RAAS has been demonstrated to increase the expression of OPG, a glycoprotein that regulates bone remodeling [13]. OPG regulates bone remodeling by controlling osteoclast activity, hence interfering with the interaction between RANK and its ligand RANKL [76]. In order to regulate cell proliferation, the apoptosis regulator gene RANKL interacts with OPG, a ligand for the RANK receptor, under physiological conditions (Figure 4) [76]. However, the ACE/Ang II/AT1R axis is upregulated and the ACE2/Ang 1-7/MasR arm is downregulated in T2D, consequently decreasing the expression of OPG protein [13,117]. Thus, the shift in the RANKL/OPG ratio is regarded as one of the markers of bone fragility and the development of bone disorders such as osteoporosis [13].
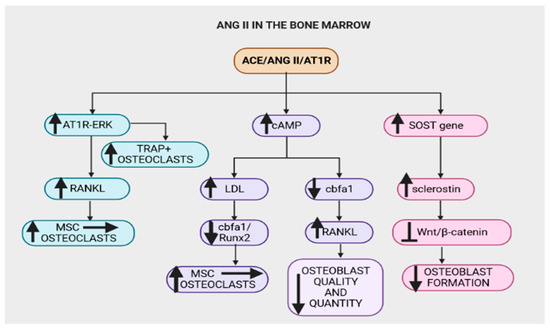
Figure 4.
The Angiotensin-converting-enzyme (ACE)/angiotensin II (Ang II)/angiotensin type 1 receptor (AT1R) axis (ACE/Ang II/AT1R) in the bone marrow contributes to bone degradation by altering the microarchitecture of the bone structure. The ACE/Ang II/AT1R axis upregulates the AT1R–extracellular signal-regulated kinases (AT1R–ERK) pathway, which increases the formation of tartrate-resistant acid phosphatase positive (TRAP+) osteoclasts and nuclear factor-κB ligand (RANKL). RANKL promotes the conversion of mesenchymal stem cells (MSCs) to osteoclasts. Furthermore, this axis increases cAMP, downregulates core-binding factor subunit alpha-1/Runt-related transcription factor 2 (cbfα1/Runx2) and increases RANKL, subsequently decreasing osteoblast quantity and quality. Runx2 enhances the proliferation of suture mesenchymal cells and induces their commitment to osteoblast lineage cells. A decrease in Runx2 reduces the conversion of MSCs to osteoblasts, thus monocytic precursors are converted to osteoclasts. The ACE/Ang II/AT1R axis increases the expression of sclerostin coding gene (SOST) and hence the sclerostin protein that inhibits the wingless-related integration site (Wnt)/β-catenin pathway, thus significantly reducing osteoblast formation.
2.5. ANG 1-7
The ACE2/Ang1-7/Mas receptor cascade is thought to be the beneficial arm in the biological consequences of systemic and local RAAS. Ang (1-7) improves abnormal bone metabolism and micro-architecture [12,13,118]. Furthermore, Ang (1-7) significantly increases mineralization while inhibiting osteoclastogenesis [12,118]. To test this theory, A-779 was used to block the Mas receptor, resulting in significantly reduced beneficial effects of Ang (1-7) on bone health, indicating that the Mas receptor plays a critical role in regulating Ang (1-7)’s osteoprotective properties [13,119,120]. The role of ACE/Ang II/AT1R in osteoclastogenesis due to the activated proinflammatory cytokines has been discussed [96]. Interestingly, evidence suggests that the ACE2/Ang1-7/Mas receptor axis counteracts the proinflammatory cytokine effects, thus improving bone health. Ang-(1-7) reduces the expression of proinflammatory cytokines associated with bone resorption, namely IL-6 and TNF-α [121,122,123,124]. In osteoporosis-related alveolar bone resorption, IL-6—an osteoclastogenesis-promoting cytokine—promotes the conversion of forkhead box P3 (FoxP3) T cells to T helper 17 (Th17) cells, which guard against germs but induce osteoclast formation and thus, bone injury [125,126,127]. Additionally, the ACE2/Ang1-7/MasR axis has been observed to reduce the expression of RANKL and IL-1β mRNA levels, hence reducing osteoclastogenesis (Figure 5) [12,124]. However, in T2D, the conventional RAAS axis is upregulated and the ACE2/Ang1-7/MasR axis is downregulated and may thus compromise bone health [124,128,129]. Anti-resorptive factors such as calcium, estrogen and TGFb promote bone health via the ACE2/Ang1-7/MasR axis and the Ang (1-7)/AT2R pathway [13,130].
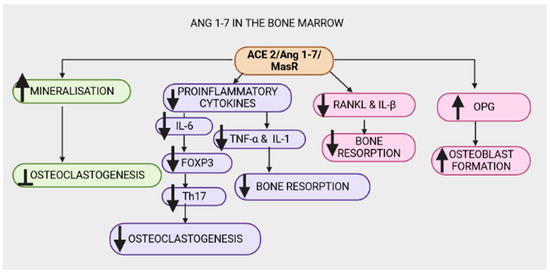
Figure 5.
The Angiotensin-converting-enzyme 2 (ACE2)/angiotensin 1-7(Ang 1-7)/MasR (ACE2/Ang 1-7/MasR) axis exhibits osteoprotective properties in the bone marrow by increasing mineralization and inhibiting osteoclastogenesis. The ACE2/Ang 1-7/MasR axis counteracts the ACE/Ang II/AT1R signal by decreasing proinflammatory cytokines (interleukin-1 (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α)). A decrease in IL-6 reduces the conversion of forkhead box P3 (FoxP3) T cells to T helper 17 (Th17) cells, consequently reducing osteoclastogenesis. Additionally, a reduction in the proinflammatory cytokines TNF-α, interleukin-1β (IL-1β) and nuclear factor-κB ligand (RANKL) decreases bone resorption. The ACE2/Ang1-7/MasR axis increases osteoprotegerin (OPG), resulting in increased osteoblast formation.
3. Anti-Resorptive Factors
3.1. Transforming Growth Factor β (TGF-β)
Osteoblasts develop from mesenchymal stem cells (MSCs) in the bone marrow, while osteoclasts develop from hematopoietic stem cells (HSCs) [131,132]. TGF-β1 regulates both osteoblast and osteoclast differentiation, thus balancing bone production and resorption [133]. TGF-β1 maintains bone density by promoting osteoblast proliferation, inhibiting osteoblast apoptosis and attracting osteoblastic precursors or matrix-producing osteoblasts to the location via chemotactic attraction [133,134]. Additionally, in the first phases of osteoblast development, TGF-1β increases the synthesis of extracellular bone matrix protein by osteoblasts, thus maintaining bone health [134]. Estrogen increases osteoblast proliferation and differentiation by increasing the synthesis of TGF- β1 in osteoblasts [135]. In addition, estrogen inhibits bone loss by encouraging osteoclast apoptosis through a TGF-dependent process [136]. However, in T2D, there is an estrogen deficiency associated with impaired estrogen receptor expression that results in reduced TGF-β1 [14,137,138]. Hence, estrogen deficiency due to the upregulation of RAAS is positively correlated with decreased TGF-β1 expression, leading to bone loss [139,140]. Furthermore, studies have highlighted a positive correlation between increased glucocorticoids and T2D [141,142]. Glucocorticoids are known to promote the apoptosis of osteoblasts and inhibit their proliferation and differentiation while promoting the differentiation of osteoclasts [143,144]. Glucocorticoids upregulate TGF-β1 expression in osteoblasts; however, unlike estrogen, glucocorticoids synergize with TGF-β1 to enhance osteoclast formation by stimulating the priming of osteoclast progenitors for differentiation into osteoclasts [145]. Therefore, the upregulated glucocorticoids contribute to the progression of insulin resistance to the development of T2D due to increasing glucose levels [141,146]. Furthermore, local bone RAAS activity has been reported in glucocorticoid-induced osteoporosis, hence therapies that target the RAAS system have been reported to improve glucose homeostasis and hence the declining bone health [5,52,76,147]. Numerous cell types, including osteoblasts, B lymphocytes and osteogenic stromal stem cells produce OPG in response to anti-resorptive agents such as estrogen and TGFβ-related bone morphogenic proteins [148,149].
3.2. Estrogen
3.2.1. Estrogen Physiology
Estrogen is a sex hormone—predominantly synthesized in the ovaries in females and testes in males—that governs the growth, development and physiology of the reproductive system [150]. The neuroendocrine, skeletal, adipogenic and cardiovascular systems are similarly impacted by estrogen due to the presence of estrogen receptors in these organs [151]. Estrogen is associated with glucose handling as it has been shown to stimulate insulin sensitization in the skeletal muscle and adipose tissue through the insulin signaling pathway [152]. Therefore, estrogen, due to the ERs in the skeletal muscle and adipose tissue, is associated with impaired insulin signaling and the development and progression of hyperglycemia reported in T2D [152,153,154,155].
Studies have determined the presence of estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) in the bone marrow [156]. Osteoblasts, osteoclasts and osteocytes express ERs, which have beneficial effects on bone integrity [156]. The binding of estrogen to ERs modulates the expression of genes that encode proteins that are estrogen targets, including IL-1β, insulin-like growth factor 1 (IGF1) and TGF-β1 [157,158,159]. As discussed, TGF-β1 promotes osteoblast proliferation, inhibits osteoblast apoptosis and recruits osteoblastic precursors or matrix-producing osteoblasts to maintain bone density [133,149,160]. Furthermore, the binding of estrogen to ER promotes the upregulation of OPG and the downregulation of RANKL, thus impairing osteoclastogenesis and bone resorption [161,162]. Additionally, the estrogen–ER interaction activates Wnt/β-catenin signaling to increase osteogenesis by curbing the differentiation of MSC to adipocytes and facilitating the differentiation of mesenchymal stem cells to osteoblasts, thus promoting bone formation [163]. Sclerostin is a Wnt antagonist that competitively binds to LRP5/6 to inhibit the Wnt signaling pathway [164]. Reports show that sclerostin is significantly increased in postmenopausal women in comparison to premenopausal women [165]. Therefore, due to reduced estrogen levels in postmenopausal women, sclerostin is significantly increased, leading to morphological changes in the bone marrow microarchitecture due to the inhibition of osteoblastogenesis and the increase in bone resorption [165,166,167]. Moreover, reports have suggested that the ACE/Ang II/AT1R axis upregulates the SOST gene that encodes sclerostin, thus promoting osteoclastogenesis [168]. The predominant form of estrogen, estradiol (E2), has been shown to increase the mRNA levels of the AGT gene [24]. Interestingly, E2 has been evidenced to downregulate AT1R expression and reduce renin and ACE activity [169]. Additionally, AT1R mRNA is regulated post-transcriptionally by estrogen-sensitive binding proteins [170,171]. Although estrogen’s effect on Ang II has not been fully elucidated, it can attenuate Ang II responses to AT1R [171]. Estrogen treatment elevates Ang (1-7) levels in transgenic mice [172,173]. Structural and biochemical changes in rats with ovariectomy-induced osteoporosis are improved by the ANG (1-7) axis; therefore, estrogen might use this axis to exert its osteoprotective effects [13,174]. Moreover, estrogen has been evidenced to mediate the transcription of ACE2; thus ACE2 is downregulated and AT1R is upregulated in estrogen deficiency, leading to estrogen deficiency-induced osteoporosis [174]. Ang II also downregulates the mRNA expression of osteocalcin, which is uniquely produced during the maturation of osteoblastic cells [175]. Additionally, Ang II reduces the activity of alkaline phosphatase (ALP), which is a hallmark of osteoblastic differentiation [175,176]. Thus, estrogen is critical for balancing bone resorption and formation [26,177].
3.2.2. Estrogen Deficiency in T2D
A bi-directional relationship between estrogen deficiency and T2D has been studied, where hyperglycemia is linked to reduced estrogen bioavailability and a lack of estrogen is shown to contribute to the progression of insulin resistance [153,178]. Postmenopausal women have been shown to suffer from progressive impaired glucose tolerance, declining bone mass density and increased bone turnover due to estrogen deficiency [179,180]. Furthermore, estrogen regulates the expression of its own receptors, hence estrogen deficiency is associated with reduced ER expression in the bone marrow [181,182]. As previously stated, osteoblasts express ERs. As T2D is associated with estrogen deficiency, ERs are also reduced because estrogen regulates the expression of its own receptors, thus potentially leading to impaired bone metabolism [16,161]. Rats lacking estrogen exhibit an imbalance between the traditional ACE/Ang II/AT1R receptor pathway and the ACE2/Ang1-7/Mas-receptor pathway in their femurs [183,184]. Hence, focused interventions have proven that blocking Ang II action with an AT1R antagonist prevents bone loss associated with estrogen deficiency [185,186]. The hyperglycinemia noted in T2D is positively correlated with estrogen deficiency and the upregulation of the ACE/Ang II/AT1R axis and downregulation of the ACE2/Ang 1-7/Mas receptor axis and AT2R, which may promote osteoporosis [185,186]. Furthermore, due to the effect of estrogen on glucose handling, estrogen deficiency and the upregulation of local RAAS are linked to increased low-density lipoproteins (LDLs) [184,187]. Increased LDLs were shown to correlate with low bone mass in postmenopausal women, alluding to a possible relationship between triglycerides and the maintenance of bone mass (Figure 6) [188,189]. The relationship between estrogen deficiency, increased systemic and local RAAS activity and hypertension has recently received attention [190]. A plethora of studies have evidenced the correlation between systemic RAAS and hypertension [19,190]. Over the past decade, the focus has shifted to local RAAS, including bone RAAS, osteoporosis and hypertension [191]. Data demonstrates that these are enhanced in postmenopausal women who have significantly reduced estrogen levels [24,186]. Hence, a relationship between estrogen deficiency, RAAS activity and osteoporosis has gained traction [186]. Interestingly, hypertension and systemic and local RAAS upregulation has been demonstrated in T2D [51,192]. Hence, the correlation between RAAS, hypertension, osteoporosis and estrogen deficiency in T2D is of interest for formulating effective treatment strategies. Henceforth, this review focuses on the plethora of mechanisms involving RAAS and their association with osteoporosis and estrogen deficiency in association with T2D.
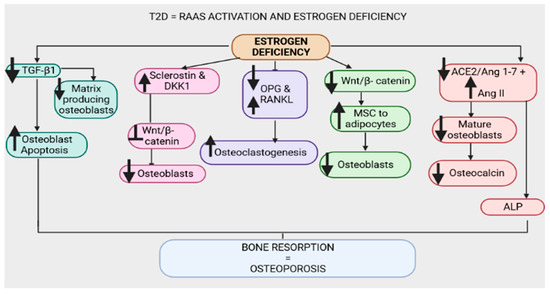
Figure 6.
Upregulation of the renin–angiotensin–aldosterone system and estrogen deficiency have been evidenced in type 2 diabetes (T2D). Estrogen deficiency downregulates transforming growth factor β1 (TGF-β1), leading to enhanced osteoblast apoptosis and reduced matrix-producing osteoblasts. Furthermore, estrogen deficiency is associated with increased sclerostin and dickkopf-1 (Dkk-1), which inhibit the wingless-related integration site (Wnt)/β-catenin pathway, leading to a reduction in osteoblasts. Estrogen deficiency promotes the upregulation of nuclear factor-κB ligand (RANKL) and the downregulation of osteoprotegerin (OPG), leading to osteoclastogenesis and bone resorption. The wingless-related integration site (Wnt)/β-catenin pathway is downregulated in estrogen deficiency, thus enhancing the conversion of mesenchymal stem cells (MSCs) to adipocytes and reducing osteoblasts. Estrogen deficiency downregulates the angiotensin-converting-enzyme 2 (ACE2)/angiotensin 1-7 (Ang 1-7)/MasR (ACE2/Ang 1-7/MasR) axis and upregulates the angiotensin-converting-enzyme (ACE)/angiotensin II (Ang II)/angiotensin type 1 receptor (AT1R) axis (ACE/Ang II/AT1R), hence decreasing osteocalcin, alkaline phosphatase (ALP) activity and the number of mature osteoblasts.
4. Conclusions
Hypertension, osteoporosis, adiposity and estrogen deficiency have been identified as risk factors for T2D [193,194,195]. The renin–angiotensin–aldosterone system is positively correlated with T2D and hypertension [196,197,198,199]. Anti-hypertensive medications that block the RAAS have been evidenced to have osteoprotective effects [3,200,201,202,203,204,205]. Furthermore, RAAS blockers are directly correlated with improved glucose handling and inhibition of progressive insulin resistance [199,206]. In this review, the mechanistic action of this system in association with the shift in bone formation and resorption has been highlighted. The effects of the classical arm and the physiologically beneficial arm were analyzed and this system was shown to affect the inflammatory status, subsequently affecting the microarchitecture of the bone [3,207,208]. Additionally, the presence of RAAS receptors, including mineralocorticoid receptors in the parathyroid and adrenal glands and osteoblasts has been evidenced, further emphasizing the role of RAAS in calcium handling and its effects in the bone microenvironment [209,210,211,212].
In addition to the relationship between RAAS, hypertension and osteoporosis, these findings may suggest that the upregulation of RAAS in T2D acts on its receptors and contributes to the development of osteoporosis. This review further focused on the relationship between RAAS, osteoporosis and estrogen deficiency in T2D. Estrogen deficiency has been proposed as a biomarker for T2D due to the critical role of estrogen in glucose handling [16,213]. Additionally, several studies have investigated the role of local and systemic RAAS in relation to hypertension and glucose homeostasis [214,215]. Interestingly, estrogen deficiency has been associated with hypertension and osteoporosis in postmenopausal women. This review focused on the relationship between estrogen deficiency, osteoporosis and hypertension in T2D by exploring mechanistic pathways that may be involved, for example, RAAS.
The association between RAAS and osteoporosis has been identified independently of T2D. However, a study investigating RAAS activity in T2D and its possible role in the development of osteoporosis in T2D is required.
Author Contributions
Conceptualization, B.C.M., P.M. and A.K.; Writing-original draft preparation, B.C.M.; Writing review and editing, B.C.M., P.M. and A.K.; Figures and table preparation, B.C.M. and P.M.; Supervision, P.S.N., N.H.S. and A.K. All authors have read and agreed to the published version of the manuscript.
Funding
The work is funded by the National Research Foundation (South Africa), grant number 121739.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to express their gratitude to the National Research Foundation (NRF) and the University of KwaZulu-Natal College of Health Science (CHS).
Conflicts of Interest
The authors declared no conflict of interest.
References
- Harrison-Bernard, L.M. The renal renin-angiotensin system. Adv. Physiol. Educ. 2009, 33, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.M.; Brown, N.J. The renin–angiotensin–aldosterone system and glucose homeostasis. Trends Pharmacol. Sci. 2011, 32, 734–739. [Google Scholar] [CrossRef]
- Mo, C.; Ke, J.; Zhao, D.; Zhang, B. Role of the renin–angiotensin–aldosterone system in bone metabolism. J. Bone Miner. Metab. 2020, 38, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Sumino, H.; Ichikawa, S.; Kasama, S.; Takahashi, T.; Kumakura, H.; Takayama, Y.; Kanda, T.; Murakami, M.; Kurabayashi, M. Effects of raloxifene on the renin–angiotensin–aldosterone system and blood pressure in hypertensive and normotensive osteoporotic postmenopausal women. Geriatr. Gerontol. Int. 2010, 10, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Shuai, B.; Yang, Y.; Shen, L.; Zhu, R.; Xu, X.; Ma, C.; Lv, L.; Zhao, J.; Rong, J. Local renin-angiotensin system is associated with bone mineral density of glucocorticoid-induced osteoporosis patients. Osteoporos. Int. 2015, 26, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef]
- Bislev, L.S.; Sikjær, T.; Rolighed, L.; Rejnmark, L. Relationship between aldosterone and parathyroid hormone, and the effect of angiotensin and aldosterone inhibition on bone health. Clin. Rev. Bone Miner. Metab. 2015, 13, 194–205. [Google Scholar] [CrossRef]
- Gao, X.; Yamazaki, Y.; Tezuka, Y.; Omata, K.; Ono, Y.; Morimoto, R.; Nakamura, Y.; Satoh, F.; Sasano, H. The effect of extracellular calcium metabolism on aldosterone biosynthesis in physiological and pathological status. Horm. Metab. Res. 2020, 52, 448–453. [Google Scholar] [CrossRef]
- Hou, X.; Tian, F. STAT3-mediated osteogenesis and osteoclastogenesis in osteoporosis. Cell Commun. Signal. 2022, 20, 112. [Google Scholar] [CrossRef]
- Vaidya, A.; Brown, J.M.; Williams, J.S. The renin–angiotensin–aldosterone system and calcium-regulatory hormones. J. Hum. Hypertens. 2015, 29, 515–521. [Google Scholar] [CrossRef]
- Wright, H.; McCarthy, H.S.; Middleton, J.; Marshall, M.J. RANK, RANKL and osteoprotegerin in bone biology and disease. Curr. Rev. Musculoskelet. Med. 2009, 2, 56–64. [Google Scholar] [CrossRef]
- Queiroz-Junior, C.M.; Santos, A.C.P.M.; Galvão, I.; Souto, G.R.; Mesquita, R.A.; Sá, M.A.; Ferreira, A.J. The angiotensin converting enzyme 2/angiotensin-(1-7)/Mas Receptor axis as a key player in alveolar bone remodeling. Bone 2019, 128, 115041. [Google Scholar] [CrossRef]
- Abuohashish, H.M.; Ahmed, M.M.; Sabry, D.; Khattab, M.M.; Al-Rejaie, S.S. Angiotensin (1-7) ameliorates the structural and biochemical alterations of ovariectomy-induced osteoporosis in rats via activation of ACE-2/Mas receptor axis. Sci. Rep. 2017, 7, 2293. [Google Scholar] [CrossRef]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Ropero, A.B.; Alonso-Magdalena, P.; Quesada, I.; Nadal, A. The role of estrogen receptors in the control of energy and glucose homeostasis. Steroids 2008, 73, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F. Is estradiol a biomarker of type 2 diabetes risk in postmenopausal women? Diabetes 2017, 66, 568–570. [Google Scholar] [CrossRef]
- Paschou, S.A.; Papanas, N. Type 2 diabetes mellitus and menopausal hormone therapy: An update. Diabetes Ther. 2019, 10, 2313–2320. [Google Scholar] [CrossRef]
- Ivy, J.R.; Bailey, M.A. Pressure natriuresis and the renal control of arterial blood pressure. J. Physiol. 2014, 592, 3955–3967. [Google Scholar] [CrossRef] [PubMed]
- Fountain, J.H.; Kaur, J.; Lappin, S.L. Physiology, renin angiotensin system. In StatPearls [Internet]; StatPearls Publishing: London, UK, 2023. [Google Scholar]
- Muñoz-Durango, N.; Fuentes, C.A.; Castillo, A.E.; González-Gómez, L.M.; Vecchiola, A.; Fardella, C.E.; Kalergis, A.M. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: Molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int. J. Mol. Sci. 2016, 17, 797. [Google Scholar] [CrossRef]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System; BioMed Central: London, UK, 2017. [Google Scholar]
- Sheu, A.; Greenfield, J.R.; White, C.P.; Center, J.R. Assessment and treatment of osteoporosis and fractures in type 2 diabetes. Trends Endocrinol. Metab. 2022, 33, 333–344. [Google Scholar] [CrossRef]
- Compston, J. Type 2 diabetes mellitus and bone. J. Intern. Med. 2018, 283, 140–153. [Google Scholar] [CrossRef]
- O’Donnell, E.; Floras, J.S.; Harvey, P.J. Estrogen status and the renin angiotensin aldosterone system. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2014, 307, R498–R500. [Google Scholar] [CrossRef]
- Nabi, A.N.; Ebihara, A. Diabetes and Renin-Angiotensin-Aldosterone System: Pathophysiology and Genetics. In Renin-Angiotensin Aldosterone System; Intechopen: London, UK, 2021; Volume 21. [Google Scholar]
- Cheng, C.-H.; Chen, L.-R.; Chen, K.-H. Osteoporosis due to hormone imbalance: An overview of the effects of estrogen deficiency and glucocorticoid overuse on bone turnover. Int. J. Mol. Sci. 2022, 23, 1376. [Google Scholar] [CrossRef] [PubMed]
- Underwood, P.C.; Adler, G.K. The renin angiotensin aldosterone system and insulin resistance in humans. Curr. Hypertens. Rep. 2013, 15, 59–70. [Google Scholar] [CrossRef]
- Ramalingam, L.; Menikdiwela, K.; LeMieux, M.; Dufour, J.M.; Kaur, G.; Kalupahana, N.; Moustaid-Moussa, N. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.-S.; Liu, C.; Tian, R.; Nishiyama, A.; Raij, L. Skeletal muscle insulin resistance in salt-sensitive hypertension: Role of angiotensin II activation of NF κ B. Cardiovasc. Diabetol. 2015, 14, 45. [Google Scholar] [CrossRef]
- Gutierrez-Rodelo, C.; Arellano-Plancarte, A.; Hernandez-Aranda, J.; Landa-Galvan, H.V.; Parra-Mercado, G.K.; Moreno-Licona, N.J.; Hernandez-Gonzalez, K.D.; Catt, K.J.; Villalobos-Molina, R.; Olivares-Reyes, J.A. Angiotensin II Inhibits Insulin Receptor Signaling in Adipose Cells. Int. J. Mol. Sci. 2022, 23, 6048. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61. [Google Scholar] [CrossRef]
- Ting, R.; Dutton, H.; Sorisky, A. In vitro studies of the renin-angiotensin system in human adipose tissue/adipocytes and possible relationship to SARS-CoV-2: A scoping review. Adipocyte 2023, 12, 2194034. [Google Scholar] [CrossRef] [PubMed]
- Mkhize, B.C.; Mosili, P.; Ngubane, P.S.; Sibiya, N.H.; Khathi, A. Diet-induced prediabetes: Effects on the activity of the renin–angiotensin–aldosterone system in selected organs. J. Diabetes Investig. 2022, 13, 768–780. [Google Scholar] [CrossRef]
- Karlsson, C.; Lindell, K.; Ottosson, M.; Sjöström, L.; Carlsson, B.R.; Carlsson, L.M. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II. J. Clin. Endocrinol. Metab. 1998, 83, 3925–3929. [Google Scholar] [CrossRef]
- Zheng, J.; Ding, J.; Liao, M.; Qiu, Z.; Yuan, Q.; Mai, W.; Dai, Y.; Zhang, H.; Wu, H.; Wang, Y. Immunotherapy against angiotensin II receptor ameliorated insulin resistance in a leptin receptor-dependent manner. FASEB J. 2021, 35, e21157. [Google Scholar] [CrossRef] [PubMed]
- Trouwborst, I.; Bowser, S.M.; Goossens, G.H.; Blaak, E.E. Ectopic fat accumulation in distinct insulin resistant phenotypes; targets for personalized nutritional interventions. Front. Nutr. 2018, 5, 77. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liu, H.; Yu, J.; He, S.; Li, P.; Ma, C.; Zhang, H.; Xu, L.; Ping, F.; Li, W. Triglyceride is independently correlated with insulin resistance and islet beta cell function: A study in population with different glucose and lipid metabolism states. Lipids Health Dis. 2020, 19, 121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Zhou, S.-F.; Yu, Z.-L.; Wang, X.-Y.; Zhu, P.-L.; Chu, Z.-S.; Pan, S.-Y.; Xie, M.; Ko, K.-M. Biochemical mechanism underlying hypertriglyceridemia and hepatic steatosis/hepatomegaly induced by acute schisandrin B treatment in mice. Lipids Health Dis. 2017, 16, 8. [Google Scholar] [CrossRef]
- Song, R.; Van Buren, T.; Yosypiv, I.V. Histone deacetylases are critical regulators of the renin-angiotensin system during ureteric bud branching morphogenesis. Pediatr. Res. 2010, 67, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Dou, W.; Xu, Y.; Pagadala, V.; Pedersen, L.C.; Liu, J. Role of deacetylase activity of N-deacetylase/N-sulfotransferase 1 in forming N-sulfated domain in heparan sulfate. J. Biol. Chem. 2015, 290, 20427–20437. [Google Scholar] [CrossRef]
- Chen, K.; Liu, M.L.; Schaffer, L.; Li, M.; Boden, G.; Wu, X.; Williams, K.J. Type 2 diabetes in mice induces hepatic overexpression of sulfatase 2, a novel factor that suppresses uptake of remnant lipoproteins. Hepatology 2010, 52, 1957–1967. [Google Scholar] [CrossRef]
- Williams, K.J.; Liu, M.-L.; Zhu, Y.; Xu, X.; Davidson, W.R.; McCue, P.; Sharma, K. Loss of heparan N-sulfotransferase in diabetic liver: Role of angiotensin II. Diabetes 2005, 54, 1116–1122. [Google Scholar] [CrossRef][Green Version]
- Borém, L.M.; Neto, J.F.; Brandi, I.V.; Lelis, D.F.; Santos, S.H. The role of the angiotensin II type I receptor blocker telmisartan in the treatment of non-alcoholic fatty liver disease: A brief review. Hypertens. Res. 2018, 41, 394–405. [Google Scholar] [CrossRef]
- Watson, A.M.; Gould, E.A.; Moody, S.C.; Sivakumaran, P.; Sourris, K.C.; Chow, B.S.; Koïtka-Weber, A.; Allen, T.J.; Jandeleit-Dahm, K.A.; Cooper, M.E. Disparate effects of diabetes and hyperlipidemia on experimental kidney disease. Front. Physiol. 2020, 11, 518. [Google Scholar] [CrossRef]
- Chen, S.-C.; Tseng, C.-H. Dyslipidemia, kidney disease, and cardiovascular disease in diabetic patients. Rev. Diabet. Stud. RDS 2013, 10, 88. [Google Scholar] [CrossRef]
- Park, C.H.; Kim, H.W.; Park, J.T.; Chang, T.I.; Yoo, T.-H.; Lee, J.; Sung, S.; Jung, J.Y.; Hyun, Y.Y.; Oh, K.-H. Intrarenal Renin-Angiotensin System Activation Alters Relationship Between Systolic Blood Pressure and Progression of Chronic Kidney Disease. Hypertension 2023, 80, 1024–1034. [Google Scholar] [CrossRef]
- Leoncini, G.; Viazzi, F.; De Cosmo, S.; Russo, G.; Fioretto, P.; Pontremoli, R. Blood pressure reduction and RAAS inhibition in diabetic kidney disease: Therapeutic potentials and limitations. J. Nephrol. 2020, 33, 949–963. [Google Scholar] [CrossRef]
- Tackling, G.; Borhade, M.B. Hypertensive heart disease. In StatPearls [Internet]; StatPearls Publishing: Bethesda, United States, 2021. [Google Scholar]
- De Mello, W.C. Local renin angiotensin aldosterone systems and cardiovascular diseases. Med. Clin. 2017, 101, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Michelli, A.; Zuolo, G.; Candido, R.; Fabris, B. Update on RAAS modulation for the treatment of diabetic cardiovascular disease. J. Diabetes Res. 2016, 2016, 8917578. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension 2018, 72, 537–548. [Google Scholar] [CrossRef]
- Yongtao, Z.; Kunzheng, W.; Jingjing, Z.; Hu, S.; Jianqiang, K.; Ruiyu, L.; Chunsheng, W. Glucocorticoids activate the local renin–angiotensin system in bone: Possible mechanism for glucocorticoid-induced osteoporosis. Endocrine 2014, 47, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Sassi, F.; Buondonno, I.; Luppi, C.; Spertino, E.; Stratta, E.; Di Stefano, M.; Ravazzoli, M.; Isaia, G.; Trento, M.; Passera, P. Type 2 diabetes affects bone cells precursors and bone turnover. BMC Endocr. Disord. 2018, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, N.; Yonaha, T.; Yamanouchi, M.; Sumi, H.; Taki, Y.; Shibagaki, Y.; Shiizaki, K.; Yano, S. Bone responsiveness to parathyroid hormone is negatively associated with parathyroid hormone-lowering drug use in patients undergoing hemodialysis: A cross-sectional study. BMC Nephrol. 2021, 22, 275. [Google Scholar] [CrossRef]
- Khan, M.; Jose, A.; Sharma, S. Physiology, parathyroid hormone. In StatPearls [Internet]; StatPearls Publishing: Bethesda, United States, 2021. [Google Scholar]
- Zheng, M.-H.; Li, F.-X.-Z.; Xu, F.; Lin, X.; Wang, Y.; Xu, Q.-S.; Guo, B.; Yuan, L.-Q. The interplay between the renin-angiotensin-aldosterone system and parathyroid hormone. Front. Endocrinol. 2020, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, S.; Brown, J.M.; Connors, M.; Williams, J.S.; Adler, G.K.; Vaidya, A. Angiotensin-converting enzyme inhibition and parathyroid hormone secretion. Int. J. Endocrinol. 2017, 2017, 4138783. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Latiwesh, O.B.; Ali, A.; Tabrez, E.; Mehra, L.; Nwachukwu, F. Parathyroid gland response to vitamin D deficiency in type 2 diabetes mellitus: An observational study. Cureus 2018, 10, e3656. [Google Scholar] [CrossRef] [PubMed]
- Raposo, L.; Martins, S.; Ferreira, D.; Guimarães, J.T.; Santos, A.C. Vitamin D, parathyroid hormone and metabolic syndrome–the PORMETS study. BMC Endocr. Disord. 2017, 17, 71. [Google Scholar] [CrossRef]
- Williams, A.; Zhao, S.; Brock, G.; Kline, D.; Echouffo-Tcheugui, J.B.; Effoe, V.S.; Bertoni, A.G.; Michos, E.D.; de Boer, I.H.; Kestenbaum, B. Vitamin D, parathyroid hormone, glucose metabolism and incident diabetes in the multiethnic study of atherosclerosis. BMJ Open Diabetes Res. Care 2022, 10, e002931. [Google Scholar] [CrossRef]
- Keane, J.T.; Elangovan, H.; Stokes, R.A.; Gunton, J.E. Vitamin D and the liver—Correlation or cause? Nutrients 2018, 10, 496. [Google Scholar] [CrossRef]
- Ashley, B.; Simner, C.; Manousopoulou, A.; Jenkinson, C.; Hey, F.; Frost, J.M.; Rezwan, F.I.; White, C.H.; Lofthouse, E.M.; Hyde, E. Placental uptake and metabolism of 25 (OH) vitamin D determine its activity within the fetoplacental unit. Elife 2022, 11, e71094. [Google Scholar] [CrossRef]
- Elangovan, H.; Chahal, S.; Gunton, J.E. Vitamin D in liver disease: Current evidence and potential directions. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 907–916. [Google Scholar] [CrossRef]
- Gunnarsson, Ö.; Indridason, O.; Franzson, L.; Sigurdsson, G. Factors associated with elevated or blunted PTH response in vitamin D insufficient adults. J. Intern. Med. 2009, 265, 488–495. [Google Scholar] [CrossRef]
- Harrington, J.; Perumal, N.; Al Mahmud, A.; Baqui, A.; Roth, D.E. Vitamin D and fetal–neonatal calcium homeostasis: Findings from a randomized controlled trial of high-dose antenatal vitamin D supplementation. Pediatr. Res. 2014, 76, 302–309. [Google Scholar] [CrossRef]
- Föger-Samwald, U.; Dovjak, P.; Azizi-Semrad, U.; Kerschan-Schindl, K.; Pietschmann, P. Osteoporosis: Pathophysiology and therapeutic options. EXCLI J. 2020, 19, 1017. [Google Scholar] [PubMed]
- Lenzini, L.; Prisco, S.; Vanderriele, P.E.; Lerco, S.; Torresan, F.; Maiolino, G.; Seccia, T.M.; Iacobone, M.; Rossi, G.P. PTH modulation by aldosterone and angiotensin II is blunted in hyperaldosteronism and rescued by adrenalectomy. J. Clin. Endocrinol. Metab. 2019, 104, 3726–3734. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Williams, J.S.; Luther, J.M.; Garg, R.; Garza, A.E.; Pojoga, L.H.; Ruan, D.T.; Williams, G.H.; Adler, G.K.; Vaidya, A. Human interventions to characterize novel relationships between the renin–angiotensin–aldosterone system and parathyroid hormone. Hypertension 2014, 63, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhu, N.; Wang, Y.; Cheng, G. 1,25(OH)2D3 inhibits osteogenic differentiation through activating β-catenin signaling via downregulating bone morphogenetic protein 2. Mol. Med. Rep. 2020, 22, 5023–5032. [Google Scholar] [CrossRef]
- Haussler, M.R.; Livingston, S.; Sabir, Z.L.; Haussler, C.A.; Jurutka, P.W. Vitamin D receptor mediates a myriad of biological actions dependent on its 1, 25-dihydroxyvitamin D ligand: Distinct regulatory themes revealed by induction of Klotho and fibroblast growth Factor-23. JBMR Plus 2021, 5, e10432. [Google Scholar] [CrossRef]
- Ali, R.M.; Al-Shorbagy, M.Y.; Helmy, M.W.; El-Abhar, H.S. Role of Wnt4/β-catenin, Ang II/TGFβ, ACE2, NF-κB, and IL-18 in attenuating renal ischemia/reperfusion-induced injury in rats treated with Vit D and pioglitazone. Eur. J. Pharmacol. 2018, 831, 68–76. [Google Scholar] [CrossRef]
- McMullan, C.J.; Borgi, L.; Curhan, G.C.; Fisher, N.; Forman, J.P. The effect of vitamin D on renin-angiotensin-system activation and blood pressure-a randomized control trial. J. Hypertens. 2017, 35, 822. [Google Scholar] [CrossRef]
- Contreras-Bolívar, V.; García-Fontana, B.; García-Fontana, C.; Muñoz-Torres, M. Mechanisms involved in the relationship between vitamin D and insulin resistance: Impact on clinical practice. Nutrients 2021, 13, 3491. [Google Scholar] [CrossRef]
- Goltzman, D. Approach to hypercalcemia. In Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2019. [Google Scholar]
- Zupan, J.; Jeras, M.; Marc, J. Osteoimmunology and the influence of pro-inflammatory cytokines on osteoclasts. Biochem. Medica. 2013, 23, 43–63. [Google Scholar] [CrossRef]
- Liu, X.; Sun, Y.; Wei, Q.; Jiang, W.; Jiao, M.; Yan, J.; Tian, R.; Yang, P.; Wang, K.; Wang, C. Captopril alleviates glucocorticoid-induced osteonecrosis of the femoral head by mediating the ACE2/Ang-(1-7)/Mas receptor cascade. Eur. J. Pharmacol. 2022, 921, 174871. [Google Scholar] [CrossRef]
- Guan, X.-X.; Zhou, Y.; Li, J.-Y. Reciprocal roles of angiotensin II and angiotensin II receptors blockade (ARB) in regulating Cbfa1/RANKL via cAMP signaling pathway: Possible mechanism for hypertension-related osteoporosis and antagonistic effect of ARB on hypertension-related osteoporosis. Int. J. Mol. Sci. 2011, 12, 4206–4213. [Google Scholar]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Koh, J.-T.; Kim, N. The ATF3–OPG Axis Contributes to Bone Formation by Regulating the Differentiation of Osteoclasts, Osteoblasts, and Adipocytes. Int. J. Mol. Sci. 2022, 23, 3500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, J.; Liu, P.; Wang, Q.; Liu, L.; Zhao, H. The RANK/RANKL/OPG system and tumor bone metastasis: Potential mechanisms and therapeutic strategies. Front. Endocrinol. 2022, 13, 3320. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.-H. Osteoblast-osteoclast communication and bone homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef]
- Dirckx, N.; Moorer, M.C.; Clemens, T.L.; Riddle, R.C. The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol. 2019, 15, 651–665. [Google Scholar] [CrossRef]
- Al-Daghri, N.M.; Aziz, I.; Yakout, S.; Aljohani, N.J.; Al-Saleh, Y.; Amer, O.E.; Sheshah, E.; Younis, G.Z.; Al-Badr, F.B.M. Inflammation as a contributing factor among postmenopausal Saudi women with osteoporosis. Medicine 2017, 96, e5780. [Google Scholar] [CrossRef]
- Yang, D.-H.; Yang, M.-Y. The role of macrophage in the pathogenesis of osteoporosis. Int. J. Mol. Sci. 2019, 20, 2093. [Google Scholar] [CrossRef] [PubMed]
- Cici, D.; Corrado, A.; Rotondo, C.; Cantatore, F.P. Wnt signaling and biological therapy in rheumatoid arthritis and spondyloarthritis. Int. J. Mol. Sci. 2019, 20, 5552. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Oursler, M.J. Developments in sclerostin biology: Regulation of gene expression, mechanisms of action, and physiological functions. Curr. Osteoporos. Rep. 2014, 12, 107–114. [Google Scholar] [CrossRef]
- Li, S.; Yin, Y.; Yao, L.; Lin, Z.; Sun, S.; Zhang, J.; Li, X. TNF-α treatment increases DKK1 protein levels in primary osteoblasts via upregulation of DKK1 mRNA levels and downregulation of miR-335-5p. Mol. Med. Rep. 2020, 22, 1017–1025. [Google Scholar] [CrossRef]
- Do Carmo, L.; Harrison, D.G. Hypertension and osteoporosis: Common pathophysiological mechanisms. Med. Nov. Technol. Devices 2020, 8, 100047. [Google Scholar] [CrossRef]
- Ciftciler, R.; Haznedaroglu, I.C. Pathobiological interactions of local bone marrow renin-angiotensin system and central nervous system in systemic arterial hypertension. Front. Endocrinol. 2020, 11, 425. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.E.; Dizerega, G.S. Contribution of the local RAS to hematopoietic function: A novel therapeutic target. Front. Endocrinol. 2013, 4, 157. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, J.; Caballero, R.; Delpón, E. The renin–angiotensin system and bone. Clin. Rev. Bone Miner. Metab. 2015, 13, 125–148. [Google Scholar] [CrossRef]
- Zhang, F.; Dong, Z.; Gao, S.; Chen, G.; Liu, D. AT1R-mediated apoptosis of bone marrow mesenchymal stem cells is associated with mtROS production and mtDNA reduction. Oxidative Med. Cell. Longev. 2019, 2019, 4608165. [Google Scholar] [CrossRef] [PubMed]
- Jarajapu, Y.P. Targeting angiotensin-converting enzyme-2/angiotensin-(1-7)/Mas receptor axis in the vascular progenitor cells for cardiovascular diseases. Mol. Pharmacol. 2021, 99, 29–38. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-related cytokines regulate osteoclast formation and bone resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef]
- Rahimi, Z.; Moradi, M.; Nasri, H. A systematic review of the role of renin angiotensin aldosterone system genes in diabetes mellitus, diabetic retinopathy and diabetic neuropathy. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2014, 19, 1090. [Google Scholar]
- Romero-Díaz, C.; Duarte-Montero, D.; Gutiérrez-Romero, S.A.; Mendivil, C.O. Diabetes and bone fragility. Diabetes Ther. 2021, 12, 71–86. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, Y.; Wang, C.; Wang, X.; Wang, Y.; Zhang, H. Angiotensin II upregulates RANKL/NFATC1 expression in synovial cells from patients with rheumatoid arthritis through the ERK1/2 and JNK pathways. J. Orthop. Surg. Res. 2021, 16, 297. [Google Scholar] [CrossRef]
- Tiyasatkulkovit, W.; Charoenphandhu, N.; Wongdee, K.; Thongbunchoo, J.; Krishnamra, N.; Malaivijitnond, S. Upregulation of osteoblastic differentiation marker mRNA expression in osteoblast-like UMR106 cells by puerarin and phytoestrogens from Pueraria mirifica. Phytomedicine 2012, 19, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhi, X.; Wang, J.; Su, J. RANKL signaling in bone marrow mesenchymal stem cells negatively regulates osteoblastic bone formation. Bone Res. 2018, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Kodama, J.; Kaito, T. Osteoclast multinucleation: Review of current literature. Int. J. Mol. Sci. 2020, 21, 5685. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-B.; Agidigbi, T.S.; Kang, I.-S.; Kim, C. ERK Inhibition Increases RANKL-Induced Osteoclast Differentiation in RAW 264.7 Cells by Stimulating AMPK Activation and RANK Expression and Inhibiting Anti-Osteoclastogenic Factor Expression. Int. J. Mol. Sci. 2022, 23, 13512. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Yang, G.; Zhou, L. U0126 prevents ERK pathway phosphorylation and interleukin-1beta mRNA production after cerebral ischemia. Chin. Med. Sci. J. Chung-Kuo I Hsueh K’o Hsueh Tsa Chih 2004, 19, 270–275. [Google Scholar]
- Akagi, T.; Mukai, T.; Mito, T.; Kawahara, K.; Tsuji, S.; Fujita, S.; Uchida, H.A.; Morita, Y. Effect of angiotensin II on bone erosion and systemic bone loss in mice with tumor necrosis factor-mediated arthritis. Int. J. Mol. Sci. 2020, 21, 4145. [Google Scholar] [CrossRef]
- Lv, W.-T.; Du, D.-H.; Gao, R.-J.; Yu, C.-W.; Jia, Y.; Jia, Z.-F.; Wang, C.-J. Regulation of hedgehog signaling offers a novel perspective for bone homeostasis disorder treatment. Int. J. Mol. Sci. 2019, 20, 3981. [Google Scholar] [CrossRef]
- Bruderer, M.; Richards, R.; Alini, M.; Stoddart, M.J. Role and regulation of RUNX2 in osteogenesis. Eur. Cell Mater. 2014, 28, 269–286. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Transcriptional programming in arteriosclerotic disease: A multifaceted function of the Runx2 (runt-related transcription factor 2). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Shang, Z.; Wei, T. Role of Cx43-Based Gap Junction in Murine Osteoblast-Like Mc3t3-E1Cells Exposed to 17-Βestradiol. Mol. Biol. 2017, 6, 1000197. [Google Scholar] [CrossRef]
- Passos, L.S.; Lupieri, A.; Becker-Greene, D.; Aikawa, E. Innate and adaptive immunity in cardiovascular calcification. Atherosclerosis 2020, 306, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, N.; Shahbazi, M.-A.; Maleki, A.; Hamidi, M.; Ramazani, A.; Santos, H.A. Emerging insights on drug delivery by fatty acid mediated synthesis of lipophilic prodrugs as novel nanomedicines. J. Control. Release 2020, 326, 556–598. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.-Y.; Zhan, J.-K.; Liu, Y.-S. Roles and functions of antisense lncRNA in vascular aging. Ageing Res. Rev. 2021, 72, 101480. [Google Scholar] [CrossRef]
- Zhou, Y.; Guan, X.; Chen, X.; Yu, M.; Wang, C.; Chen, X.; Shi, J.; Liu, T.; Wang, H. Angiotensin II/angiotensin II receptor blockade affects osteoporosis via the AT1/AT2-mediated cAMP-dependent PKA pathway. Cells Tissues Organs 2017, 204, 25–37. [Google Scholar] [CrossRef]
- Momenzadeh, M.; Khosravian, M.; Lakkakula, B.V. Potential of renin-angiotensin system inhibition to improve metabolic bone disorders. J. Nephropharmacol. 2020, 10, e16. [Google Scholar] [CrossRef]
- Yu, Y.; Lucitt, M.B.; Stubbe, J.; Cheng, Y.; Friis, U.G.; Hansen, P.B.; Jensen, B.L.; Smyth, E.M.; FitzGerald, G.A. Prostaglandin F2α elevates blood pressure and promotes atherosclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7985–7990. [Google Scholar] [CrossRef]
- Mineo, C. Lipoprotein receptor signalling in atherosclerosis. Cardiovasc. Res. 2020, 116, 1254–1274. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Vlashi, R.; Zhang, X.; Wu, M.; Chen, G. Wnt signaling: Essential roles in osteoblast differentiation, bone metabolism and therapeutic implications for bone and skeletal disorders. Genes Dis. 2022, 10, 1291–1317. [Google Scholar] [CrossRef]
- Roy, B.; Curtis, M.E.; Fears, L.S.; Nahashon, S.N.; Fentress, H.M. Molecular mechanisms of obesity-induced osteoporosis and muscle atrophy. Front. Physiol. 2016, 7, 439. [Google Scholar] [CrossRef]
- Hao, P.-P.; Chen, Y.-G.; Liu, Y.-P.; Zhang, M.-X.; Yang, J.-M.; Gao, F.; Zhang, Y.; Zhang, C. Association of plasma angiotensin-(1–7) level and left ventricular function in patients with type 2 diabetes mellitus. PLoS ONE 2013, 8, e62788. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Song, Y.; Zhao, X.; Wong, M.S.; Zhang, W. Renin inhibitor aliskiren exerts beneficial effect on trabecular bone by regulating skeletal renin-angiotensin system and kallikrein-kinin system in ovariectomized mice. Osteoporos. Int. 2016, 27, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Mińczuk, K.; Schlicker, E.; Malinowska, B. Cross-Talk between CB1, AT1, AT2 and Mas Receptors Responsible for Blood Pressure Control in the Paraventricular Nucleus of Hypothalamus in Conscious Spontaneously Hypertensive Rats and Their Normotensive Controls. Cells 2022, 11, 1542. [Google Scholar] [CrossRef]
- Sampaio, W.O.; Souza dos Santos, R.A.; Faria-Silva, R.; da Mata Machado, L.T.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007, 49, 185–192. [Google Scholar] [CrossRef]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-e-Silva, A.C. The anti-inflammatory potential of ACE2/angiotensin-(1-7)/Mas receptor axis: Evidence from basic and clinical research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef]
- Villalobos, L.A.; San Hipólito-Luengo, Á.; Ramos-González, M.; Cercas, E.; Vallejo, S.; Romero, A.; Romacho, T.; Carraro, R.; Sánchez-Ferrer, C.F.; Peiró, C. The angiotensin-(1-7)/mas axis counteracts angiotensin II-dependent and-independent pro-inflammatory signaling in human vascular smooth muscle cells. Front. Pharmacol. 2016, 7, 482. [Google Scholar] [CrossRef]
- Saponaro, F.; Rutigliano, G.; Sestito, S.; Bandini, L.; Storti, B.; Bizzarri, R.; Zucchi, R. ACE2 in the era of SARS-CoV-2: Controversies and novel perspectives. Front. Mol. Biosci. 2020, 7, 588618. [Google Scholar] [CrossRef]
- Al-Azzawi, I.S.; Mohammed, N.S.; Saad, I. The impact of angiotensin converting enzyme-2 (ACE-2) on bone remodeling marker osteoprotegerin (OPG) in post-COVID-19 Iraqi patients. Cureus 2022, 14, e29926. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Dar, H.Y.; Mishra, P.K. Immunoporosis: Immunology of osteoporosis—Role of T cells. Front. Immunol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Hua, F.; Ding, W.; Ding, K.; Zhang, Y.; Xu, C. The correlation between the Th17/Treg cell balance and bone health. Immun. Ageing 2020, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.M.; Srivastava, K.; Mansoori, M.N.; Trivedi, R.; Chattopadhyay, N.; Singh, D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: A new candidate in the pathogenesis of osteoporosis. PLoS ONE 2012, 7, e44552. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, Y.; Wang, G.Q. Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19. J. Med. Virol. 2020, 92, 726–730. [Google Scholar] [CrossRef]
- Abuohashish, H.M.; Ahmed, M.M.; Sabry, D.; Khattab, M.M.; Al-Rejaie, S.S. The ACE-2/Ang1-7/Mas cascade enhances bone structure and metabolism following angiotensin-II type 1 receptor blockade. Eur. J. Pharmacol. 2017, 807, 44–55. [Google Scholar] [CrossRef] [PubMed]
- de Mello-Aires, M.; Leite-Dellova, D.C.; Castelo-Branco, R.C.; Malnic, G.; Oliveira-Souza, M. ANG II, ANG-(1-7), ALDO and AVP biphasic effects on Na+/H+ transport: The role of cellular calcium. Nephrol. Ren. Dis. 2017, 2, 1–22. [Google Scholar] [CrossRef][Green Version]
- Sharaf-Eldin, W.E.; Abu-Shahba, N.; Mahmoud, M.; El-Badri, N. The modulatory effects of mesenchymal stem cells on osteoclastogenesis. Stem Cells Int. 2016, 2016, 1908365. [Google Scholar] [CrossRef]
- Li, Q.; Xu, R.; Lei, K.; Yuan, Q. Insights into skeletal stem cells. Bone Res. 2022, 10, 61. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.-P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Nagai-Yoshioka, Y.; Yamasaki, R.; Kawamoto, T.; Nishihara, T.; Ariyoshi, W. Mechanisms involved in suppression of osteoclast supportive activity by transforming growth factor-β1 via the ubiquitin-proteasome system. PLoS ONE 2022, 17, e0262612. [Google Scholar] [CrossRef]
- Fischer, V.; Haffner-Luntzer, M. Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. In Seminars in Cell & Developmental Biology; Academic Press: Amsterdam, The Netherlands, 2022; Volume 3, pp. 14–21. [Google Scholar]
- Okman-Kilic, T. Estrogen deficiency and osteoporosis. In Advances in Osteoporosis; IntechOpen: London, UK, 2015. [Google Scholar]
- Weitzmann, M.N.; Pacifici, R. T cells: Unexpected players in the bone loss induced by estrogen deficiency and in basal bone homeostasis. Ann. N. Y. Acad. Sci. 2007, 1116, 360–375. [Google Scholar] [CrossRef]
- Shimizu, H.; Nakagami, H.; Osako, M.K.; Nakagami, F.; Kunugiza, Y.; Tomita, T.; Yoshikawa, H.; Rakugi, H.; Ogihara, T.; Morishita, R. Prevention of osteoporosis by angiotensin-converting enzyme inhibitor in spontaneous hypertensive rats. Hypertens. Res. 2009, 32, 786–790. [Google Scholar] [CrossRef]
- Shimizu, H.; Nakagami, H.; Osako, M.K.; Hanayama, R.; Kunugiza, Y.; Kizawa, T.; Tomita, T.; Yoshikawa, H.; Ogihara, T.; Morishita, R. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008, 22, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-X.; Cummins, C.L. Fresh insights into glucocorticoid-induced diabetes mellitus and new therapeutic directions. Nat. Rev. Endocrinol. 2022, 18, 540–557. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, J.L.; Riddell, M.C. Effects of glucocorticoids and exercise on pancreatic β-cell function and diabetes development. Diabetes/Metab. Res. Rev. 2012, 28, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.; Feng, J.; Zhang, X. Glucocorticoids suppressed osteoblast differentiation by decreasing Sema3A expression via the PIK3/Akt pathway. Exp. Cell Res. 2021, 403, 112595. [Google Scholar] [CrossRef] [PubMed]
- Ohnaka, K.; Tanabe, M.; Kawate, H.; Nawata, H.; Takayanagi, R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem. Biophys. Res. Commun. 2005, 329, 177–181. [Google Scholar] [CrossRef]
- Kasagi, S.; Chen, W. TGF-beta1 on osteoimmunology and the bone component cells. Cell Biosci. 2013, 3, 4. [Google Scholar] [CrossRef]
- Beaupere, C.; Liboz, A.; Fève, B.; Blondeau, B.; Guillemain, G. Molecular mechanisms of glucocorticoid-induced insulin resistance. Int. J. Mol. Sci. 2021, 22, 623. [Google Scholar] [CrossRef]
- Shen, L.; Ma, C.; Shuai, B.; Yang, Y. Effects of 1, 25-dihydroxyvitamin D3 on the local bone renin-angiotensin system in a murine model of glucocorticoid-induced osteoporosis. Exp. Ther. Med. 2017, 13, 3297–3304. [Google Scholar] [CrossRef]
- Yang, N.; Liu, Y. The role of the immune microenvironment in bone regeneration. Int. J. Med. Sci. 2021, 18, 3697. [Google Scholar] [CrossRef]
- Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-β superfamily on osteoclasts/osteoblasts balance in physiological and pathological bone conditions. Int. J. Mol. Sci. 2020, 21, 7597. [Google Scholar] [CrossRef] [PubMed]
- Delgado, B.J.; Lopez-Ojeda, W. Estrogen. In StatPearls [Internet]; StatPearls Publishing: London, UK, 2021. [Google Scholar]
- Lee, H.-R.; Kim, T.-H.; Choi, K.-C. Functions and physiological roles of two types of estrogen receptors, ERα and ERβ, identified by estrogen receptor knockout mouse. Lab. Anim. Res. 2012, 28, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Alemany, M. Estrogens and the regulation of glucose metabolism. World J. Diabetes 2021, 12, 1622. [Google Scholar] [CrossRef]
- Yan, H.; Yang, W.; Zhou, F.; Li, X.; Pan, Q.; Shen, Z.; Han, G.; Newell-Fugate, A.; Tian, Y.; Majeti, R. Estrogen improves insulin sensitivity and suppresses gluconeogenesis via the transcription factor Foxo1. Diabetes 2019, 68, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Merino, B.; García-Arévalo, M. Sexual hormones and diabetes: The impact of estradiol in pancreatic β cell. Int. Rev. Cell Mol. Biol. 2021, 359, 81–138. [Google Scholar]
- Pasquali, R. Sex hormones and the development of type 2 diabetes in women. J. Lab. Precis. Med. 2017, 2, 15. [Google Scholar] [CrossRef]
- Noirrit-Esclassan, E.; Valera, M.-C.; Tremollieres, F.; Arnal, J.-F.; Lenfant, F.; Fontaine, C.; Vinel, A. Critical role of estrogens on bone homeostasis in both male and female: From physiology to medical implications. Int. J. Mol. Sci. 2021, 22, 1568. [Google Scholar] [CrossRef]
- Pollard, K.J.; Daniel, J.M. Nuclear estrogen receptor activation by insulin-like growth factor-1 in Neuro-2A neuroblastoma cells requires endogenous estrogen synthesis and is mediated by mutually repressive MAPK and PI3K cascades. Mol. Cell. Endocrinol. 2019, 490, 68–79. [Google Scholar] [CrossRef]
- Smith, L.C.; Moreno, S.; Robertson, L.; Robinson, S.; Gant, K.; Bryant, A.J.; Sabo-Attwood, T. Transforming growth factor beta1 targets estrogen receptor signaling in bronchial epithelial cells. Respir. Res. 2018, 19, 160. [Google Scholar] [CrossRef]
- Hernández-Vega, A.M.; Camacho-Arroyo, I. Crosstalk between 17β-Estradiol and TGF-β Signaling Modulates Glioblastoma Progression. Brain Sci. 2021, 11, 564. [Google Scholar] [CrossRef]
- Grafe, I.; Alexander, S.; Peterson, J.R.; Snider, T.N.; Levi, B.; Lee, B.; Mishina, Y. TGF-β family signaling in mesenchymal differentiation. Cold Spring Harb. Perspect. Biol. 2018, 10, a022202. [Google Scholar] [CrossRef]
- Khalid, A.B.; Krum, S.A. Estrogen receptors alpha and beta in bone. Bone 2016, 87, 130–135. [Google Scholar] [CrossRef]
- Streicher, C.; Heyny, A.; Andrukhova, O.; Haigl, B.; Slavic, S.; Schüler, C.; Kollmann, K.; Kantner, I.; Sexl, V.; Kleiter, M. Estrogen regulates bone turnover by targeting RANKL expression in bone lining cells. Sci. Rep. 2017, 7, 6460. [Google Scholar] [CrossRef]
- Liedert, A.; Nemitz, C.; Haffner-Luntzer, M.; Schick, F.; Jakob, F.; Ignatius, A. Effects of estrogen receptor and Wnt signaling activation on mechanically induced bone formation in a mouse model of postmenopausal bone loss. Int. J. Mol. Sci. 2020, 21, 8301. [Google Scholar] [CrossRef]
- Ren, Q.; Chen, J.; Liu, Y. LRP5 and LRP6 in Wnt signaling: Similarity and divergence. Front. Cell Dev. Biol. 2021, 9, 670960. [Google Scholar] [CrossRef] [PubMed]
- Marzullo, P.; Mele, C.; Mai, S.; Nardone, A.; Scacchi, M.; Aimaretti, G. Obesity and bone loss at menopause: The role of sclerostin. Diagnostics 2021, 11, 1914. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.S.; Padhi, I.D.; Raisz, L.G.; Lorenzo, J.A. Serum sclerostin levels negatively correlate with parathyroid hormone levels and free estrogen index in postmenopausal women. J. Clin. Endocrinol. Metab. 2010, 95, 1991–1997. [Google Scholar] [CrossRef]
- Matsui, S.; Yasui, T.; Kasai, K.; Keyama, K.; Kato, T.; Uemura, H.; Kuwahara, A.; Matsuzaki, T.; Irahara, M. Increase in circulating sclerostin at the early stage of menopausal transition in Japanese women. Maturitas 2016, 83, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Ito, M.; Fumoto, T.; Fukuhara, R.; Ishida, J.; Fukamizu, A.; Ikeda, K. Physiological function of the angiotensin AT1a receptor in bone remodeling. J. Bone Miner. Res. 2011, 26, 2959–2966. [Google Scholar] [CrossRef]
- Xue, B.; Zhang, Z.; Beltz, T.G.; Guo, F.; Hay, M.; Johnson, A.K. Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H191–H198. [Google Scholar] [CrossRef]
- Wu, Z.; Maric, C.; Roesch, D.M.; Zheng, W.; Verbalis, J.G.; Sandberg, K. Estrogen regulates adrenal angiotensin AT1 receptors by modulating AT1 receptor translation. Endocrinology 2003, 144, 3251–3261. [Google Scholar] [CrossRef]
- Backlund, M.; Paukku, K.; Daviet, L.; De Boer, R.A.; Valo, E.; Hautaniemi, S.; Kalkkinen, N.; Ehsan, A.; Kontula, K.K.; Lehtonen, J.Y. Posttranscriptional regulation of angiotensin II type 1 receptor expression by glyceraldehyde 3-phosphate dehydrogenase. Nucleic Acids Res. 2009, 37, 2346–2358. [Google Scholar] [CrossRef]
- White, M.C.; Miller, A.J.; Loloi, J.; Bingaman, S.S.; Shen, B.; Wang, M.; Silberman, Y.; Lindsey, S.H.; Arnold, A.C. Sex differences in metabolic effects of angiotensin-(1-7) treatment in obese mice. Biol. Sex Differ. 2019, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Chappell, M.C.; Marshall, A.C.; Alzayadneh, E.M.; Shaltout, H.A.; Diz, D.I. Update on the angiotensin converting enzyme 2-angiotensin (1-7)-Mas receptor axis: Fetal programing, sex differences, and intracellular pathways. Front. Endocrinol. 2014, 4, 201. [Google Scholar] [CrossRef] [PubMed]
- Sha, N.-N.; Zhang, J.-L.; Poon, C.C.-W.; Li, W.-X.; Li, Y.; Wang, Y.-F.; Shi, W.; Lin, F.-H.; Lin, W.-P.; Wang, Y.-J. Differential responses of bone to angiotensin II and angiotensin (1-7): Beneficial effects of ANG (1-7) on bone with exposure to high glucose. Am. J. Physiol.-Endocrinol. Metab. 2021, 320, E55–E70. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; Carletti, R.; Ippolito, S.; Villa, I.; Palmisano, B.; Bolamperti, S.; Rubinacci, A.; Zerbini, G.; Meani, M.; Zatti, G. Angiotensin II Modulates Calcium/Phosphate Excretion in Experimental Model of Hypertension: Focus on Bone. Biomedicines 2022, 10, 2928. [Google Scholar] [CrossRef]
- Rajapaksha, I.G.; Gunarathne, L.S.; Asadi, K.; Cunningham, S.C.; Sharland, A.; Alexander, I.E.; Angus, P.W.; Herath, C.B. Liver-targeted angiotensin converting enzyme 2 therapy inhibits chronic biliary fibrosis in multiple drug-resistant gene 2-knockout mice. Hepatol. Commun. 2019, 3, 1656–1673. [Google Scholar] [CrossRef]
- Mills, E.G.; Yang, L.; Nielsen, M.F.; Kassem, M.; Dhillo, W.S.; Comninos, A.N. The relationship between bone and reproductive hormones beyond estrogens and androgens. Endocr. Rev. 2021, 42, 691–719. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.H.Y.; Nano, J.; Cecil, A.; Schederecker, F.; Rathmann, W.; Prehn, C.; Zeller, T.; Lechner, A.; Adamski, J.; Peters, A. Cross-sectional and prospective relationships of endogenous progestogens and estrogens with glucose metabolism in men and women: A KORA F4/FF4 Study. BMJ Open Diabetes Res. Care 2021, 9, e001951. [Google Scholar] [CrossRef]
- Chia, C.W.; Egan, J.M.; Ferrucci, L. Age-related changes in glucose metabolism, hyperglycemia, and cardiovascular risk. Circ. Res. 2018, 123, 886–904. [Google Scholar] [CrossRef]
- Agostini, D.; Donati Zeppa, S.; Lucertini, F.; Annibalini, G.; Gervasi, M.; Ferri Marini, C.; Piccoli, G.; Stocchi, V.; Barbieri, E.; Sestili, P. Muscle and bone health in postmenopausal women: Role of protein and vitamin D supplementation combined with exercise training. Nutrients 2018, 10, 1103. [Google Scholar] [CrossRef]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up-and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef] [PubMed]
- Nwia, S.; Leite, A.P.O.; Li, X.C.; Zhuo, J.L. Sex Differences in The Renin-Angiotensin-Aldosterone System and Their Roles in Hypertension, Cardiovascular, and Kidney Diseases. Front. Cardiovasc. Med. 2023, 10, 1198090. [Google Scholar] [CrossRef]
- White, M.C.; Fleeman, R.; Arnold, A.C. Sex differences in the metabolic effects of the renin-angiotensin system. Biol. Sex Differ. 2019, 10, 31. [Google Scholar] [CrossRef]
- Yanes, L.L.; Romero, D.G.; Iliescu, R.; Zhang, H.; Davis, D.; Reckelhoff, J.F. Postmenopausal hypertension: Role of the renin-angiotensin system. Hypertension 2010, 56, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Gersh, F.L.; O’Keefe, J.H.; Lavie, C.J.; Henry, B.M. The renin-angiotensin-aldosterone system in postmenopausal women: The promise of hormone therapy. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2021; pp. 3130–3141. [Google Scholar]
- Steiner, B.M.; Berry, D.C. The regulation of adipose tissue health by estrogens. Front. Endocrinol. 2022, 13, 889923. [Google Scholar] [CrossRef]
- Mandal, C.C. High cholesterol deteriorates bone health: New insights into molecular mechanisms. Front. Endocrinol. 2015, 6, 165. [Google Scholar] [CrossRef]
- Thapa, S.; Nandy, A.; Rendina-Ruedy, E. Endocrinal metabolic regulation on the skeletal system in post-menopausal women. Front. Physiol. 2022, 13, 1052429. [Google Scholar] [CrossRef]
- Khan, N. Possible protective role of 17β-estradiol against COVID-19. J. Allergy Infect. Dis. 2020, 1, 38. [Google Scholar]
- Ghafar, M.T.A. An overview of the classical and tissue-derived renin-angiotensin-aldosterone system and its genetic polymorphisms in essential hypertension. Steroids 2020, 163, 108701. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Wyne, K. Renin-angiotensin-aldosterone system in diabetes and hypertension. J. Clin. Hypertens. 2011, 13, 224–237. [Google Scholar] [CrossRef]
- Abdulameer, S.A.; Sulaiman, S.A.S.; Hassali, M.A.A.; Subramaniam, K.; Sahib, M.N. Osteoporosis and type 2 diabetes mellitus: What do we know, and what we can do? Patient Prefer. Adherence 2012, 6, 435–448. [Google Scholar] [CrossRef]
- Qiu, J.; Li, C.; Dong, Z.; Wang, J. Is diabetes mellitus a risk factor for low bone density: A systematic review and meta-analysis. BMC Endocr. Disord. 2021, 21, 65. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hu, L.; Yin, X.-L.; Zou, Y.; Fu, H.-Y.; Li, H.-L. Prevalence and Risk Factors of Osteoporosis in Patients with Type 2 Diabetes Mellitus in Nanchang (China): A Retrospective Cohort Study. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 3039–3048. [Google Scholar] [CrossRef]
- Banerjee, D.; Winocour, P.; Chowdhury, T.; De, P.; Wahba, M.; Montero, R.; Fogarty, D.; Frankel, A.; Karalliedde, J.; Mark, P.B. Management of hypertension and renin-angiotensin-aldosterone system blockade in adults with diabetic kidney disease: Association of British Clinical Diabetologists and the Renal Association UK guideline update 2021. BMC Nephrol. 2022, 23, 9. [Google Scholar] [CrossRef] [PubMed]
- Then, C.; Ritzel, K.; Herder, C.; Then, H.; Sujana, C.; Heier, M.; Meisinger, C.; Peters, A.; Koenig, W.; Rathmann, W. Association of renin and aldosterone with glucose metabolism in a Western European population: The KORA F4/FF4 study. BMJ Open Diabetes Res. Care 2022, 10, e002558. [Google Scholar] [CrossRef] [PubMed]
- Mancusi, C.; Izzo, R.; di Gioia, G.; Losi, M.A.; Barbato, E.; Morisco, C. Insulin resistance the hinge between hypertension and type 2 diabetes. High Blood Press. Cardiovasc. Prev. 2020, 27, 515–526. [Google Scholar] [CrossRef]
- Yamout, H.; Lazich, I.; Bakris, G.L. Blood pressure, hypertension, RAAS blockade, and drug therapy in diabetic kidney disease. Adv. Chronic Kidney Dis. 2014, 21, 281–286. [Google Scholar] [CrossRef]
- Kuang, Z.; Hou, N.; Kan, C.; Han, F.; Qiu, H.; Sun, X. The protective effects of SGLT-2 inhibitors, GLP-1 receptor agonists, and RAAS blockers against renal injury in patients with type 2 diabetes. Int. Urol. Nephrol. 2023, 55, 617–629. [Google Scholar] [CrossRef]
- Scheen, A.J.; Delanaye, P. Acute renal injury events in diabetic patients treated with SGLT2 inhibitors: A comprehensive review with a special reference to RAAS blockers. Diabetes Metab. 2022, 48, 101315. [Google Scholar] [CrossRef]
- Carbone, L.D.; Vasan, S.; Prentice, R.; Harshfield, G.; Haring, B.; Cauley, J.; Johnson, K. The renin-angiotensin aldosterone system and osteoporosis: Findings from the Women’s Health Initiative. Osteoporos. Int. 2019, 30, 2039–2056. [Google Scholar] [CrossRef]
- Kao, Y.-T.; Huang, C.-Y.; Fang, Y.-A.; Liu, J.-C. The association between renin angiotensin aldosterone system blockers and future osteoporotic fractures in a hypertensive population–A population-based cohort study in Taiwan. Int. J. Cardiol. 2020, 305, 147–153. [Google Scholar] [CrossRef]
- Wu, J.; Wang, M.; Guo, M.; Du, X.-Y.; Tan, X.-Z.; Teng, F.-Y.; Xu, Y. Angiotensin Receptor Blocker is Associated with a Lower Fracture Risk: An Updated Systematic Review and Meta-Analysis. Int. J. Clin. Pract. 2022, 2022, 7581110. [Google Scholar] [CrossRef] [PubMed]
- Oz, H.; Gavish, D.; Hass, A.; Shargorodsky, M. Effect of angiotensin II receptor blockers, candesartan, on osteoprotegerin level in hypertensive patients: Link between bone and RAAS. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Marre, M. Importance of intensive blood pressure control in type 2 diabetes: Mechanisms, treatments and current guidelines. Diabetes Obes. Metab. 2020, 22, 33–42. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Strawn, W.B. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am. J. Cardiol. 2006, 98, 121–128. [Google Scholar] [CrossRef]
- Lima, M.L.d.S.; Medeiros, C.A.C.X.d.; Guerra, G.C.B.; Santos, R.; Bader, M.; Pirih, F.Q.; Araújo Júnior, R.F.d.; Chan, A.B.; Cruz, L.J.; Brito, G.A.d.C. AT1 and AT2 receptor knockout changed osteonectin and bone density in mice in periodontal inflammation experimental model. Int. J. Mol. Sci. 2021, 22, 5217. [Google Scholar] [CrossRef]
- Weber, A.L.; Randolph, G.; Aksoy, F.G. The thyroid and parathyroid glands: CT and MR imaging and correlation with pathology and clinical findings. Radiol. Clin. North Am. 2000, 38, 1105–1129. [Google Scholar] [CrossRef] [PubMed]
- Milicic Stanic, B.; Ilincic, B.; Zeravica, R.; Milicic Ivanovski, D.; Cabarkapa, V.; Mijovic, R. The Importance of Correlation between Aldosterone and Parathyroid Hormone in Patients with Primary Hyperparathyroidism. Int. J. Endocrinol. 2022, 2022, 3804899. [Google Scholar] [CrossRef]
- Fumoto, T.; Ishii, K.-A.; Ito, M.; Berger, S.; Schütz, G.; Ikeda, K. Mineralocorticoid receptor function in bone metabolism and its role in glucocorticoid-induced osteopenia. Biochem. Biophys. Res. Commun. 2014, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Bencivenga, L.; Liccardo, D.; Elia, A.; Marzano, F.; Gambino, G.; D’Amico, M.L.; Perna, C.; Ferrara, N.; Rengo, G. Aldosterone and mineralocorticoid receptor system in cardiovascular physiology and pathophysiology. Oxidative Med. Cell. Longev. 2018, 2018, 1204598. [Google Scholar] [CrossRef] [PubMed]
- Salpeter, S.; Walsh, J.; Ormiston, T.; Greyber, E.; Buckley, N.; Salpeter, E. Meta-analysis: Effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes. Metab. 2006, 8, 538–554. [Google Scholar] [CrossRef]
- Thethi, T.; Kamiyama, M.; Kobori, H. The link between the renin-angiotensin-aldosterone system and renal injury in obesity and the metabolic syndrome. Curr. Hypertens. Rep. 2012, 14, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Favre, G.A.; Esnault, V.L.; Van Obberghen, E. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am. J. Physiol.-Endocrinol. Metab. 2015, 308, E435–E449. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).