

Repressive Control of Keratinocyte Cytoplasmic Inflammatory Signaling

Abstract
1. Introduction
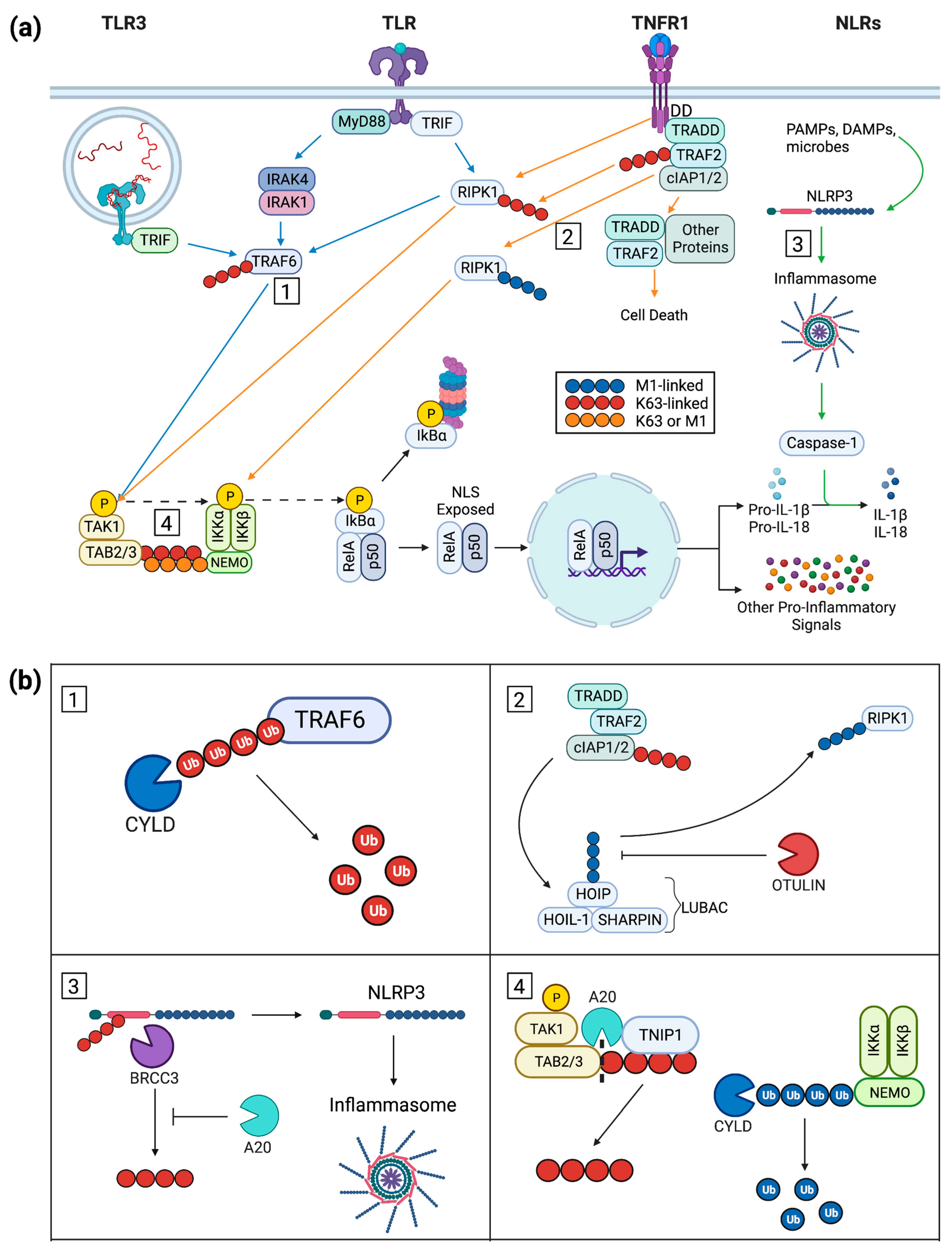
2. Keratinocyte Inflammatory Signal Progression
2.1. Keratinocyte Post-Receptor Inflammatory Signaling
2.2. An Overview of NF-κB Signaling

2.3. Ubiquitin in Cytoplasmic Signaling
3. Inflammation-Repressing Proteins
3.1. LUBAC Modulates Signaling Downstream of TNFR and TLR3
3.2. SHARPIN
3.2.1. Mouse Mutations and Human Disease Analogues
3.2.2. Recombinant Models and Possible Translational Applications
3.3. HOIL-1 and HOIP
3.3.1. Human Pathologies and Protein Function
3.3.2. Recombinant Models
3.4. OTULIN
3.4.1. Human Pathologies and Protein Function

{kind=link}
| Gene # | Alteration * | Keratinocyte/Epidermal or Other Presentation ^ | Refs.$ |
|---|---|---|---|
| Sharpincpdm | mSP: 1 base pair deletion in exon 1 disrupts reading frame generating early stop codon | Chronic proliferative dermatitis; immune-cell accumulation in skin with epidermal hyperplasia and keratinization defects | [55,56,57] |
| Sharpincpdm-Dem | mSP: 14 base pair deletion in exon 1 disrupts reading frame generating early stop codon | Dermatitis, epidermal hyperplasia, and keratinization defects similar to cpdm | [56] |
| Sharpin | mEKO: K14-Cre-mediated deletion | Acanthosis and parakeratosis; apoptotic keratinocytes and dermatitis similar to cpdm | [57,61] |
| SHARPIN | cKCKD | SHARPIN-deficient HaCaT keratinocytes; increased STAT-dependent transcription and increased IL-33 | [60,69] |
| HOIL-1 | HG: mutations resulting in truncation of or frameshift within coding region | Febrile neutrophilic dermatosis; generalized immunodeficiency and hyperinflammation; polyglucosan body myopathy-1 | [83,84] |
| HOIL-1 | mEKO: K14-Cre-mediated deletion | Postnatal epidermal hyperplasia, defective differentiation, immune-cell infiltration to both epidermal and dermal compartments ~2 d postnatal, lethal ~6 d postnatal | [24] |
| HOIP | HG: base pair transition with amino-acid substitution | Nucleotide sequence alteration of undetermined significance, patient displayed autoinflammation and immunodeficiency | [85] |
| HOIP | mEKO: K14-Cre-mediated deletion | Postnatal skin inflammation, epidermal hyperplasia, reduced differentiation, increased expression of multiple interleukins, lethal 4–6 d postnatal | [24,90] |
3.4.2. Recombinant Models
3.5. CYLD
3.5.1. Human Pathologies and Protein Function
3.5.2. Recombinant Models
3.6. A20
3.6.1. Overview and Protein Function
3.6.2. Inflammatory Cell Death
3.6.3. Inflammasome Regulation
3.7. TNIP1
3.7.1. Overview of TNIP1
3.7.2. Protein Function and Interactions
3.7.3. Recombinant Models
3.7.4. Endogenous and Exogenous Control of TNIP1 Protein Levels
| Gene # | Alteration * | Keratinocyte/Epidermal or Other Presentation ^ | Refs.$ |
|---|---|---|---|
| OTULIN | HG: mutations | ORAS/AIPDS; IMD107; systemic inflammation, panniculitis, and rash | [91,92,93] |
| Otulin | mEKO: K14-Cre-mediated deletion | Early postnatal focal cutaneous inflammation, epidermal hyperplasia, reduced differentiation, and wound healing defects; increased cutaneous immune-cell recruitment | [100,101] |
| OTULIN | cKCKD | OTULIN-deficient HaCaTs; hypersensitivity to TNFα-induced necroptosis | [99] |
| CYLD | HG: mutations | Skin appendage neoplasms | [106,107] |
| CYLD | cKCOV: WT protein | WT overexpression in HaCaTs; increased levels of early differentiation markers | [114] |
| CYLD | cKCOV: C601S mutant | Deubiquitinase mutant expression in HaCaT keratinocytes; disorganized in vitro stratification, increased NF-kB signaling, and TNFα production | [109,114] |
| CYLD | mETG: WT protein | Level of late differentiation increased and reduction in NF-κB signaling | [109,116] |
| CYLD | mETG: C601S mutant | Increased phospho-p65, TNFα, and IL-6 with slower phospho-p65 resolution | [109,116] |
| Cyld | mEKO: K14-Cre-mediated deletion | Heightened levels of K63 polyubiquitinated TRAF6 and phospho-IκBα, cylindroma and trichoepithelioma development with DMBA and TPA challenge | [112] |
| TNFAIP3+ | HG: SNPs | Numerous inflammatory diseases (see text) | [120,121,122] |
| TNFAIP3+ | HG: mutations | A20 (see text); skin rash and inflammation in several other organs and tissues | [123] |
| TNFAIP3+ | cKCOV | A20 overexpression in NHEKs; inflammatory genes repressed upon IL-17A or TNFα challenge | [140] |
| TNFAIP3+ | cKCKD | A20-deficient human gingival keratinocytes; increased apoptosis when challenged with both TNFα and cycloheximide | [155] |
| Tnfaip3+ | mEKO: K14-Cre-mediated deletion | Keratinocyte hyperproliferation | [141] |
| Tnfaip3+ | mEKO: K14-Cre-mediated deletion | Enhanced inflammatory gene expression in cultured keratinocytes under basal conditions and IL-17A stimulation | [138] |
| TNIP1 | HG: SNPs | Numerous inflammatory diseases (see text) | [179,180,181,182] |
| TNIP1 | HG: mutations | SLE risk haplotype (P151A), unknown functional consequence | [182] |
| TNIP1 | cKCOV | TNIP1 overexpression in NHEKs; repression of inflammatory genes upon IL-17A and TNFα challenge | [140] |
| TNIP1 | cKCKD | TNIP1-deficient HaCaT keratinocytes; increasedexpression of inflammatory genes upon poly(I:C) exposure | [68,165] |
| Tnip1 | mKO and mEKO: K14-Cre-mediated deletion | Psoriasis-like skin symptoms upon imiquimod exposure | [175] |
4. Conclusions/Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rice, G.; Rompolas, P. Advances in resolving the heterogeneity and dynamics of keratinocyte differentiation. Curr. Opin. Cell Biol. 2020, 67, 92–98. [Google Scholar] [CrossRef]
- Lefèvre-Utile, A.; Braun, C.; Haftek, M.; Aubin, F. Five Functional Aspects of the Epidermal Barrier. Int. J. Mol. Sci. 2021, 22, 11676. [Google Scholar] [CrossRef]
- Samulevich, M.L.; Shamilov, R.; Aneskievich, B.J. Thermostable Proteins from HaCaT Keratinocytes Identify a Wide Breadth of Intrinsically Disordered Proteins and Candidates for Liquid-Liquid Phase Separation. Int. J. Mol. Sci. 2022, 23, 14323. [Google Scholar] [CrossRef]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The diverse contribution of keratinocytes to immune responses in skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef]
- El-Serafi, A.T.; El-Serafi, I.; Steinvall, I.; Sjöberg, F.; Elmasry, M. A Systematic Review of Keratinocyte Secretions: A Regenerative Perspective. Int. J. Mol. Sci. 2022, 23, 7934. [Google Scholar] [CrossRef]
- Piipponen, M.; Li, D.; Landén, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. Int. J. Mol. Sci. 2020, 21, 8790. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2021, 48, 130–139. [Google Scholar] [CrossRef]
- Dikic, I.; Schulman, B.A. An expanded lexicon for the ubiquitin code. Nat. Rev. Mol. Cell Biol. 2023, 24, 273–287. [Google Scholar] [CrossRef]
- Ikeda, F. Protein and nonprotein targets of ubiquitin modification. Am. J. Physiol. Cell Physiol. 2023, 324, c1053–c1060. [Google Scholar] [CrossRef]
- Elliott, P.R. Molecular basis for specificity of the Met1-linked polyubiquitin signal. Biochem. Soc. Trans. 2016, 44, 1581–1602. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Oikawa, D.; Sato, Y.; Ito, H.; Tokunaga, F. Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders. Int. J. Mol. Sci. 2020, 21, 3381. [Google Scholar] [CrossRef]
- Martens, A.; van Loo, G. A20 at the Crossroads of Cell Death, Inflammation, and Autoimmunity. Cold Spring Harb. Perspect. Biol. 2020, 12, a036418. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Lin, M.; Ji, X.; Lv, Y.; Cui, D.; Xie, J. The Roles of TRAF3 in Immune Responses. Dis. Markers 2023, 2023, 7787803. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes. Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Shamilov, R.; Aneskievich, B.J. TNIP1 in Autoimmune Diseases: Regulation of Toll-like Receptor Signaling. J. Immunol. Res. 2018, 2018, 3491269. [Google Scholar] [CrossRef] [PubMed]
- Taraborrelli, L.; Peltzer, N.; Montinaro, A.; Kupka, S.; Rieser, E.; Hartwig, T.; Sarr, A.; Darding, M.; Draber, P.; Haas, T.L.; et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 2018, 9, 3910. [Google Scholar] [CrossRef]
- Fenini, G.; Karakaya, T.; Hennig, P.; Di Filippo, M.; Beer, H.D. The NLRP1 Inflammasome in Human Skin and Beyond. Int. J. Mol. Sci. 2020, 21, 4788. [Google Scholar] [CrossRef] [PubMed]
- Danis, J.; Mellett, M. Nod-Like Receptors in Host Defence and Disease at the Epidermal Barrier. Int. J. Mol. Sci. 2021, 22, 4677. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takeda, K.; Akira, S. TIR domain-containing adaptors define the specificity of TLR signaling. Mol. Immunol. 2004, 40, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, J.; Zhao, Y.; Ma, X.; Yi, H. Toll-like receptor 3 (TLR3) regulation mechanisms and roles in antiviral innate immune responses. J. Zhejiang Univ. Sci. B 2021, 22, 609–632. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Leonard, J.N.; Ghirlando, R.; Askins, J.; Bell, J.K.; Margulies, D.H.; Davies, D.R.; Segal, D.M. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Sakaniwa, K.; Fujimura, A.; Shibata, T.; Shigematsu, H.; Ekimoto, T.; Yamamoto, M.; Ikeguchi, M.; Miyake, K.; Ohto, U.; Shimizu, T.; et al. TLR3 forms a laterally aligned multimeric complex along double-stranded RNA for efficient signal transduction. Nat. Commun. 2023, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Jang, Y.H.; Lee, G.Y.; Han, G.M.; Jeong, H.J.; Kim, J.W.; Lee, J.O. TLR3 forms a highly organized cluster when bound to a poly(I:C) RNA ligand. Nat. Commun. 2022, 13, 6876. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Liu, D.; Zhong, Z.; Karin, M. NF-κB: A Double-Edged Sword Controlling Inflammation. Biomedicines 2022, 10, 1250. [Google Scholar] [CrossRef]
- Jin, H.S.; Park, J.K.; Jo, E.K. Toll-like Receptors and NOD-like Receptors in Innate Immune Defense during Pathogenic Infection. J. Bacteriol. Virol. 2014, 44, 215–225. [Google Scholar] [CrossRef]
- Shen, Y.; Boulton, A.P.R.; Yellon, R.L.; Cook, M.C. Skin manifestations of inborn errors of NF-κB. Front. Pediatr. 2022, 10, 1098426. [Google Scholar] [CrossRef]
- Lorenz, V.N.; Schön, M.P.; Seitz, C.S. c-Rel in Epidermal Homeostasis: A Spotlight on c-Rel in Cell Cycle Regulation. J. Investig. Dermatol. 2016, 136, 1090–1096. [Google Scholar] [CrossRef][Green Version]
- Dittmar, G.; Winklhofer, K.F. Linear Ubiquitin Chains: Cellular Functions and Strategies for Detection and Quantification. Front. Chem. 2019, 7, 915. [Google Scholar] [CrossRef]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell Mol. Biol. Lett. 2021, 26, 1. [Google Scholar] [CrossRef]
- Dziedzic, S.A.; Su, Z.; Jean Barrett, V.; Najafov, A.; Mookhtiar, A.K.; Amin, P.; Pan, H.; Sun, L.; Zhu, H.; Ma, A.; et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat. Cell Biol. 2018, 20, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.D.; Mace, P.D.; Day, C.L. Noncovalent Ubiquitin Interactions Regulate the Catalytic Activity of Ubiquitin Writers. Trends Biochem. Sci. 2016, 41, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Spit, M.; Rieser, E.; Walczak, H. Linear ubiquitination at a glance. J. Cell Sci. 2019, 132, jcs208512. [Google Scholar] [CrossRef]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef]
- Estavoyer, B.; Messmer, C.; Echbicheb, M.; Rudd, C.E.; Milot, E.; Affar, E.B. Mechanisms orchestrating the enzymatic activity and cellular functions of deubiquitinases. J. Biol. Chem. 2022, 298, 102198. [Google Scholar] [CrossRef] [PubMed]
- Aksentijevich, I.; Zhou, Q. NF-κB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 399. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Rittinger, K.; Ikeda, F. Linear ubiquitin chains: Enzymes, mechanisms and biology. Open Biol. 2017, 7, 170026. [Google Scholar] [CrossRef]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-κB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Bakshi, S.; Kelsall, I.R.; Ortiz-Guerrero, J.; Shpiro, N.; Cohen, P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem. Biophys. Res. Commun. 2016, 474, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.; Pedrioli, P.G.; Komander, D.; Cohen, P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252. [Google Scholar] [CrossRef]
- HogenEsch, H.; Gijbels, M.J.; Offerman, E.; van Hooft, J.; van Bekkum, D.W.; Zurcher, C. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol. 1993, 143, 972–982. [Google Scholar]
- Seymour, R.E.; Hasham, M.G.; Cox, G.A.; Shultz, L.D.; Hogenesch, H.; Roopenian, D.C.; Sundberg, J.P. Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes. Immun. 2007, 8, 416–421. [Google Scholar] [CrossRef]
- Sundberg, J.P.; Pratt, C.H.; Goodwin, L.P.; Silva, K.A.; Kennedy, V.E.; Potter, C.S.; Dunham, A.; Sundberg, B.A.; HogenEsch, H. Keratinocyte-specific deletion of SHARPIN induces atopic dermatitis-like inflammation in mice. PLoS ONE 2020, 15, e0235295. [Google Scholar] [CrossRef]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 2014, 3, e03422. [Google Scholar] [CrossRef]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; Lawlor, K.E.; Vanyai, H.; Hall, C.; et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 2014, 3, e03464. [Google Scholar] [CrossRef]
- Tang, L.; Wang, J.; Zhu, J.; Liang, Y. Down-regulated SHARPIN may accelerate the development of atopic dermatitis through activating interleukin-33/ST2 signalling. Exp. Dermatol. 2018, 27, 1328–1335. [Google Scholar] [CrossRef]
- Gijbels, M.J.; Zurcher, C.; Kraal, G.; Elliott, G.R.; HogenEsch, H.; Schijff, G.; Savelkoul, H.F.; Bruijnzeel, P.L. Pathogenesis of skin lesions in mice with chronic proliferative dermatitis (cpdm/cpdm). Am. J. Pathol. 1996, 148, 941–950. [Google Scholar] [PubMed]
- Yoshizawa, Y.; Nomaguchi, H.; Izaki, S.; Kitamura, K. Serum cytokine levels in atopic dermatitis. Clin. Exp. Dermatol. 2002, 27, 225–229. [Google Scholar] [CrossRef]
- Renninger, M.L.; Seymour, R.E.; Whiteley, L.O.; Sundberg, J.P.; Hogenesch, H. Anti-IL5 decreases the number of eosinophils but not the severity of dermatitis in Sharpin-deficient mice. Exp. Dermatol. 2010, 19, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.S.; Wang, Z.; Silva, K.A.; Kennedy, V.E.; Stearns, T.M.; Burzenski, L.; Shultz, L.D.; Hogenesch, H.; Sundberg, J.P. Chronic proliferative dermatitis in Sharpin null mice: Development of an autoinflammatory disease in the absence of B and T lymphocytes and IL4/IL13 signaling. PLoS ONE 2014, 9, e85666. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Wenk, J.; Nenci, A.; Pasparakis, M.; Scharffetter-Kochanek, K.; Smyth, N.; Peters, T.; Kess, D.; Holtkötter, O.; Shephard, P.; et al. Keratin 14 Cre transgenic mice authenticate keratin 14 as an oocyte-expressed protein. Genesis 2004, 38, 176–181. [Google Scholar] [CrossRef]
- Gijbels, M.J.; HogenEsch, H.; Bruijnzeel, P.L.; Elliott, G.R.; Zurcher, C. Maintenance of donor phenotype after full-thickness skin transplantation from mice with chronic proliferative dermatitis (cpdm/cpdm) to C57BL/Ka and nude mice and vice versa. J. Investig. Dermatol. 1995, 105, 769–773. [Google Scholar] [CrossRef]
- Zinngrebe, J.; Rieser, E.; Taraborrelli, L.; Peltzer, N.; Hartwig, T.; Ren, H.; Kovács, I.; Endres, C.; Draber, P.; Darding, M.; et al. LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J. Exp. Med. 2016, 213, 2671–2689. [Google Scholar] [CrossRef]
- Rudraiah, S.; Shamilov, R.; Aneskievich, B.J. TNIP1 reduction sensitizes keratinocytes to post-receptor signalling following exposure to TLR agonists. Cell Signal 2018, 45, 81–92. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Zhu, J.; Liang, Y. Downregulation of SHANK-associated RH domain-interacting protein elevates interleukin-33 expression by stimulating the Janus kinase 2/signal transducer and activator of transcription signalling pathway in HaCaT cells. Clin. Exp. Dermatol. 2021, 46, 880–887. [Google Scholar] [CrossRef]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimäki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in atopic dermatitis: Expression profiles and modulation by triggering factors. J. Investig. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef]
- Imai, Y. Interleukin-33 in atopic dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. [Google Scholar] [CrossRef]
- Gallegos-Alcalá, P.; Jiménez, M.; Cervantes-García, D.; Salinas, E. The Keratinocyte as a Crucial Cell in the Predisposition, Onset, Progression, Therapy and Study of the Atopic Dermatitis. Int. J. Mol. Sci. 2021, 22, 10661. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Moretta, G.; Antonelli, F.; Scala, E.; Abeni, D.; Albanesi, C.; Madonna, S. Multiple Roles for Cytokines in Atopic Dermatitis: From Pathogenic Mediators to Endotype-Specific Biomarkers to Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 2684. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11, eaax2945. [Google Scholar] [CrossRef]
- Iznardo, H.; Puig, L. IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 9479. [Google Scholar] [CrossRef] [PubMed]
- Schuler, C.F., IV; Gudjonsson, J.E. IL-33 antagonism does not improve chronic atopic dermatitis: What can we learn? J. Allergy Clin. Immunol. 2022, 150, 1410–1411. [Google Scholar] [CrossRef]
- Maurer, M.; Cheung, D.S.; Theess, W.; Yang, X.; Dolton, M.; Guttman, A.; Choy, D.F.; Dash, A.; Grimbaldeston, M.A.; Soong, W. Phase 2 randomized clinical trial of astegolimab in patients with moderate to severe atopic dermatitis. J. Allergy Clin. Immunol. 2022, 150, 1517–1524. [Google Scholar] [CrossRef]
- Yang, N.; Chen, Z.; Zhang, X.; Shi, Y. Novel Targeted Biological Agents for the Treatment of Atopic Dermatitis. BioDrugs 2021, 35, 401–415. [Google Scholar] [CrossRef]
- Uppal, S.K.; Kearns, D.G.; Chat, V.S.; Han, G.; Wu, J.J. Review and analysis of biologic therapies currently in phase II and phase III clinical trials for atopic dermatitis. J. Dermatol. Treat. 2022, 33, 626–636. [Google Scholar] [CrossRef]
- Jahan, A.S.; Elbæk, C.R.; Damgaard, R.B. Met1-linked ubiquitin signalling in health and disease: Inflammation, immunity, cancer, and beyond. Cell Death Differ. 2021, 28, 473–492. [Google Scholar] [CrossRef]
- Wu, Q.; Koliopoulos, M.G.; Rittinger, K.; Stieglitz, B. Structural basis for ubiquitylation by HOIL-1. Front. Mol. Biosci. 2022, 9, 1098144. [Google Scholar] [CrossRef]
- Fiil, B.K.; Gyrd-Hansen, M. The Met1-linked ubiquitin machinery in inflammation and infection. Cell Death Differ. 2021, 28, 557–569. [Google Scholar] [CrossRef]
- Krenn, M.; Salzer, E.; Simonitsch-Klupp, I.; Rath, J.; Wagner, M.; Haack, T.B.; Strom, T.M.; Schänzer, A.; Kilimann, M.W.; Schmidt, R.L.J.; et al. Mutations outside the N-terminal part of RBCK1 may cause polyglucosan body myopathy with immunological dysfunction: Expanding the genotype-phenotype spectrum. J. Neurol. 2018, 265, 394–401. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israël, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, N.; Darding, M.; Montinaro, A.; Draber, P.; Draberova, H.; Kupka, S.; Rieser, E.; Fisher, A.; Hutchinson, C.; Taraborrelli, L.; et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018, 557, 112–117. [Google Scholar] [CrossRef]
- Peltzer, N.; Rieser, E.; Taraborrelli, L.; Draber, P.; Darding, M.; Pernaute, B.; Shimizu, Y.; Sarr, A.; Draberova, H.; Montinaro, A.; et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014, 9, 153–165. [Google Scholar] [CrossRef]
- Tang, Y.; Tu, H.; Liu, G.; Zheng, G.; Wang, M.; Li, L.; Zhao, X.; Lin, X. RNF31 Regulates Skin Homeostasis by Protecting Epidermal Keratinocytes from Cell Death. J. Immunol. 2018, 200, 4117–4124. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230.e20. [Google Scholar] [CrossRef]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Elliott, P.R.; Swatek, K.N.; Maher, E.R.; Stepensky, P.; Elpeleg, O.; Komander, D.; Berkun, Y. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 2019, 11, e9324. [Google Scholar] [CrossRef]
- Spaan, A.N.; Neehus, A.L.; Laplantine, E.; Staels, F.; Ogishi, M.; Seeleuthner, Y.; Rapaport, F.; Lacey, K.A.; Van Nieuwenhove, E.; Chrabieh, M.; et al. Human OTULIN haploinsufficiency impairs cell-intrinsic immunity to staphylococcal α-toxin. Science 2022, 376, eabm6380. [Google Scholar] [CrossRef] [PubMed]
- Verboom, L.; Hoste, E.; van Loo, G. OTULIN in NF-κB signaling, cell death, and disease. Trends Immunol. 2021, 42, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Heger, K.; Wickliffe, K.E.; Ndoja, A.; Zhang, J.; Murthy, A.; Dugger, D.L.; Maltzman, A.; de Sousa, E.M.F.; Hung, J.; Zeng, Y.; et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 2018, 559, 120–124. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, S.; Hwang, G.; Song, J. Deubiquitinases: Modulators of Different Types of Regulated Cell Death. Int. J. Mol. Sci. 2021, 22, 4352. [Google Scholar] [CrossRef]
- Rivkin, E.; Almeida, S.M.; Ceccarelli, D.F.; Juang, Y.C.; MacLean, T.A.; Srikumar, T.; Huang, H.; Dunham, W.H.; Fukumura, R.; Xie, G.; et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 2013, 498, 318–324. [Google Scholar] [CrossRef]
- Douglas, T.; Saleh, M. Post-translational Modification of OTULIN Regulates Ubiquitin Dynamics and Cell Death. Cell Rep. 2019, 29, 3652–3663.e5. [Google Scholar] [CrossRef]
- Schünke, H.; Göbel, U.; Dikic, I.; Pasparakis, M. OTULIN inhibits RIPK1-mediated keratinocyte necroptosis to prevent skin inflammation in mice. Nat. Commun. 2021, 12, 5912. [Google Scholar] [CrossRef]
- Hoste, E.; Lecomte, K.; Annusver, K.; Vandamme, N.; Roels, J.; Maschalidi, S.; Verboom, L.; Vikkula, H.K.; Sze, M.; Van Hove, L.; et al. OTULIN maintains skin homeostasis by controlling keratinocyte death and stem cell identity. Nat. Commun. 2021, 12, 5913. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, G.A.; Weusten, B.L.A.; Bogte, A.; Begthel, H.; Brosens, L.A.A.; Clevers, H. Human gastrointestinal epithelia of the esophagus, stomach, and duodenum resolved at single-cell resolution. Cell Rep. 2021, 34, 108819. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Morgan, P.R.; Lane, E.B. Keratin 14 and 19 expression in normal, dysplastic and malignant oral epithelia. A study using in situ hybridization and immunohistochemistry. J. Oral. Pathol. Med. 1996, 25, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Rosin, F.C.P.; Gonsalves, H.; Santos, A.F.; de Paula Novaes, C.; Huang, I.; Deboni, M.C.Z.; Corrêa, L. Keratin expression in gingival tissue and primary cultured gingival keratinocytes: Are there differences? Arch. Oral. Biol. 2020, 117, 104780. [Google Scholar] [CrossRef]
- Chalmers, F.E.; Mogre, S.; Rimal, B.; Son, J.; Patterson, A.D.; Stairs, D.B.; Glick, A.B. The unfolded protein response gene Ire1α is required for tissue renewal and normal differentiation in the mouse tongue and esophagus. Dev. Biol. 2022, 492, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Verhoeft, K.R.; Ngan, H.L.; Lui, V.W.Y. The cylindromatosis (CYLD) gene and head and neck tumorigenesis. Cancers Head. Neck 2016, 1, 10. [Google Scholar] [CrossRef]
- Myers, D.J.F.; Eric, P. Cylindroma; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Alameda, J.P.; Ramírez, Á.; García-Fernández, R.A.; Navarro, M.; Page, A.; Segovia, J.C.; Sanchez, R.; Suárez-Cabrera, C.; Paramio, J.M.; Bravo, A.; et al. Premature aging and cancer development in transgenic mice lacking functional CYLD. Aging 2019, 11, 127–159. [Google Scholar] [CrossRef]
- Cui, Z.; Kang, H.; Grandis, J.R.; Johnson, D.E. CYLD Alterations in the Tumorigenesis and Progression of Human Papillomavirus-Associated Head and Neck Cancers. Mol. Cancer Res. 2021, 19, 14–24. [Google Scholar] [CrossRef]
- Reiley, W.W.; Zhang, M.; Jin, W.; Losiewicz, M.; Donohue, K.B.; Norbury, C.C.; Sun, S.C. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat. Immunol. 2006, 7, 411–417. [Google Scholar] [CrossRef]
- Jin, Y.J.; Wang, S.; Cho, J.; Selim, M.A.; Wright, T.; Mosialos, G.; Zhang, J.Y. Epidermal CYLD inactivation sensitizes mice to the development of sebaceous and basaloid skin tumors. JCI Insight 2016, 1, e86548. [Google Scholar] [CrossRef]
- Miliani de Marval, P.; Lutfeali, S.; Jin, J.Y.; Leshin, B.; Selim, M.A.; Zhang, J.Y. CYLD inhibits tumorigenesis and metastasis by blocking JNK/AP1 signaling at multiple levels. Cancer Prev. Res. 2011, 4, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Alameda, J.P.; Fernández-Aceñero, M.J.; Moreno-Maldonado, R.; Navarro, M.; Quintana, R.; Page, A.; Ramírez, A.; Bravo, A.; Casanova, M.L. CYLD regulates keratinocyte differentiation and skin cancer progression in humans. Cell Death Dis. 2011, 2, e208. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R.; Chmielarska, K.; Hennecke, K.; Pfeifer, A.; Fässler, R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 2006, 125, 665–677. [Google Scholar] [CrossRef]
- Alameda, J.P.; García-García, V.A.; López, S.; Hernando, A.; Page, A.; Navarro, M.; Moreno-Maldonado, R.; Paramio, J.M.; Ramírez, Á.; García-Fernández, R.A.; et al. CYLD Inhibits the Development of Skin Squamous Cell Tumors in Immunocompetent Mice. Int. J. Mol. Sci. 2021, 22, 6736. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Xu, J.; Shen, J.; Li, W.; Tan, F.; Li, C.; Wei, Z.; Liu, Y.; Bai, Y. A novel large deletion of the CYLD gene causes CYLD cutaneous syndrome in a Chinese family. Mol. Genet. Genomic Med. 2020, 8, e1441. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, B.; Carpentier, I.; Beyaert, R. A20: Central gatekeeper in inflammation and immunity. J. Biol. Chem. 2009, 284, 8217–8221. [Google Scholar] [CrossRef]
- Opipari, A.W., Jr.; Boguski, M.S.; Dixit, V.M. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J. Biol. Chem. 1990, 265, 14705–14708. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, L.; Wang, X.; Zhou, L.; Liao, Z.; Xu, L.; Wu, H.; Ren, J.; Li, Z.; Yang, L.; et al. Characteristics of A20 gene polymorphisms and clinical significance in patients with rheumatoid arthritis. J. Transl. Med. 2015, 13, 215. [Google Scholar] [CrossRef]
- Musone, S.L.; Taylor, K.E.; Lu, T.T.; Nititham, J.; Ferreira, R.C.; Ortmann, W.; Shifrin, N.; Petri, M.A.; Kamboh, M.I.; Manzi, S.; et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 1062–1064. [Google Scholar] [CrossRef]
- Tejasvi, T.; Stuart, P.E.; Chandran, V.; Voorhees, J.J.; Gladman, D.D.; Rahman, P.; Elder, J.T.; Nair, R.P. TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J. Investig. Dermatol. 2012, 132 Pt 1, 593–600. [Google Scholar] [CrossRef]
- Yu, M.P.; Xu, X.S.; Zhou, Q.; Deuitch, N.; Lu, M.P. Haploinsufficiency of A20 (HA20): Updates on the genetics, phenotype, pathogenesis and treatment. World J. Pediatr. 2020, 16, 575–584. [Google Scholar] [CrossRef]
- Tsuchida, N.; Kirino, Y.; Soejima, Y.; Onodera, M.; Arai, K.; Tamura, E.; Ishikawa, T.; Kawai, T.; Uchiyama, T.; Nomura, S.; et al. Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behçet’s disease. Arthritis Res. Ther. 2019, 21, 137. [Google Scholar] [CrossRef]
- Wakatsuki, R.; Hatai, Y.; Okamoto, K.; Kaneko, S.; Shimbo, A.; Irabu, H.; Shimizu, M.; Kanegane, H.; Ono, M. An infant with A20 haploinsufficiency presenting with periodic fever syndrome: A case report. Int. J. Rheum. Dis. 2023, 26, 973–976. [Google Scholar] [CrossRef]
- Aeschlimann, F.A.; Batu, E.D.; Canna, S.W.; Go, E.; Gül, A.; Hoffmann, P.; Leavis, H.L.; Ozen, S.; Schwartz, D.M.; Stone, D.L.; et al. A20 haploinsufficiency (HA20): Clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann. Rheum. Dis. 2018, 77, 728–735. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Dainichi, T.; Rathinam, C.V.; Ghosh, S. The deubiquitinase activity of A20 is dispensable for NF-κB signaling. EMBO Rep. 2014, 15, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Polykratis, A.; Martens, A.; Eren, R.O.; Shirasaki, Y.; Yamagishi, M.; Yamaguchi, Y.; Uemura, S.; Miura, M.; Holzmann, B.; Kollias, G.; et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 2019, 21, 731–742. [Google Scholar] [CrossRef]
- Bai, W.; Huo, S.; Li, J.; Shao, J. Advances in the Study of the Ubiquitin-Editing Enzyme A20. Front. Pharmacol. 2022, 13, 845262. [Google Scholar] [CrossRef] [PubMed]
- Skaug, B.; Chen, J.; Du, F.; He, J.; Ma, A.; Chen, Z.J. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol. Cell 2011, 44, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Priem, D.; Devos, M.; Druwé, S.; Martens, A.; Slowicka, K.; Ting, A.T.; Pasparakis, M.; Declercq, W.; Vandenabeele, P.; van Loo, G.; et al. A20 protects cells from TNF-induced apoptosis through linear ubiquitin-dependent and -independent mechanisms. Cell Death Dis. 2019, 10, 692. [Google Scholar] [CrossRef]
- Malynn, B.A.; Ma, A. A20: A multifunctional tool for regulating immunity and preventing disease. Cell Immunol. 2019, 340, 103914. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.V.; Ahmed, M.; Vallejo, A.N.; Ma, A.; Gaffen, S.L. The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci. Signal 2013, 6, ra44. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.; Arvelo, M.B.; Patel, V.I.; Longo, C.R.; Shrikhande, G.; Shukri, T.; Mahiou, J.; Sun, D.W.; Mottley, C.; Grey, S.T.; et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood 2004, 104, 2376–2384. [Google Scholar] [CrossRef]
- Pasquali, L.; Srivastava, A.; Meisgen, F.; Das Mahapatra, K.; Xia, P.; Xu Landén, N.; Pivarcsi, A.; Sonkoly, E. The Keratinocyte Transcriptome in Psoriasis: Pathways Related to Immune Responses, Cell Cycle and Keratinization. Acta Derm. Venereol. 2019, 99, 196–205. [Google Scholar] [CrossRef]
- Devos, M.; Mogilenko, D.A.; Fleury, S.; Gilbert, B.; Becquart, C.; Quemener, S.; Dehondt, H.; Tougaard, P.; Staels, B.; Bachert, C.; et al. Keratinocyte Expression of A20/TNFAIP3 Controls Skin Inflammation Associated with Atopic Dermatitis and Psoriasis. J. Investig. Dermatol. 2019, 139, 135–145. [Google Scholar] [CrossRef]
- Brown, S.J. What Have We Learned from GWAS for Atopic Dermatitis? J. Investig. Dermatol. 2021, 141, 19–22. [Google Scholar] [CrossRef]
- Harirchian, P.; Lee, J.; Hilz, S.; Sedgewick, A.J.; Perez White, B.E.; Kesling, M.J.; Mully, T.; Golovato, J.; Gray, M.; Mauro, T.M.; et al. A20 and ABIN1 Suppression of a Keratinocyte Inflammatory Program with a Shared Single-Cell Expression Signature in Diverse Human Rashes. J. Invest. Dermatol. 2019, 139, 1264–1273. [Google Scholar] [CrossRef]
- Lippens, S.; Lefebvre, S.; Gilbert, B.; Sze, M.; Devos, M.; Verhelst, K.; Vereecke, L.; Mc Guire, C.; Guérin, C.; Vandenabeele, P.; et al. Keratinocyte-specific ablation of the NF-κB regulatory protein A20 (TNFAIP3) reveals a role in the control of epidermal homeostasis. Cell Death Differ. 2011, 18, 1845–1853. [Google Scholar] [CrossRef]
- Aki, A.; Nagasaki, M.; Malynn, B.A.; Ma, A.; Kagari, T. Hypomorphic A20 expression confers susceptibility to psoriasis. PLoS ONE 2017, 12, e0180481. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, E.W. Regulation of NF-κB signaling by the A20 deubiquitinase. Cell Mol. Immunol. 2012, 9, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Wen, G.D.; Yu, C.; Zhao, Z.; Gao, N.; Liu, Z.Y. LncRNA UCA1 negatively regulates NF-kB activity in psoriatic keratinocytes through the miR125a-A20 axis. Kaohsiung J. Med. Sci. 2021, 37, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Xu, F.; Zhuang, Z.; Liu, Z.; Xie, J.; Bai, L. Ebosin Ameliorates Psoriasis-Like Inflammation of Mice via miR-155 Targeting tnfaip3 on IL-17 Pathway. Front. Immunol. 2021, 12, 662362. [Google Scholar] [CrossRef] [PubMed]
- Simanski, M.; Erkens, A.S.; Rademacher, F.; Harder, J. Staphylococcus epidermidis-induced Interleukin-1 Beta and Human Beta-defensin-2 Expression in Human Keratinocytes is Regulated by the Host Molecule A20 (TNFAIP3). Acta Derm. Venereol. 2019, 99, 181–187. [Google Scholar] [CrossRef]
- Saitoh, Y.; Hamano, A.; Mochida, K.; Kakeya, A.; Uno, M.; Tsuruyama, E.; Ichikawa, H.; Tokunaga, F.; Utsunomiya, A.; Watanabe, T.; et al. A20 targets caspase-8 and FADD to protect HTLV-I-infected cells. Leukemia 2016, 30, 716–727. [Google Scholar] [CrossRef]
- Jantaree, P.; Chaithongyot, S.; Sokolova, O.; Naumann, M. USP48 and A20 synergistically promote cell survival in Helicobacter pylori infection. Cell Mol. Life Sci. 2022, 79, 461. [Google Scholar] [CrossRef]
- Moquin, D.M.; McQuade, T.; Chan, F.K. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 2013, 8, e76841. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Bertrand, M.J.; Bogaert, P.; Vandenabeele, P.; Vanden Berghe, T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011, 2, e230. [Google Scholar] [CrossRef]
- Ildefonso, G.V.; Oliver Metzig, M.; Hoffmann, A.; Harris, L.A.; Lopez, C.F. A biochemical necroptosis model explains cell-type-specific responses to cell death cues. Biophys. J. 2023, 122, 817–834. [Google Scholar] [CrossRef]
- Feoktistova, M.; Makarov, R.; Brenji, S.; Schneider, A.T.; Hooiveld, G.J.; Luedde, T.; Leverkus, M.; Yazdi, A.S.; Panayotova-Dimitrova, D. A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes. Cells 2020, 9, 351. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, L.; Sze, M.; Mc Guire, C.; Rogiers, B.; Chu, Y.; Schmidt-Supprian, M.; Pasparakis, M.; Beyaert, R.; van Loo, G. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J. Exp. Med. 2010, 207, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef]
- Li, Y.; Mooney, E.C.; Xia, X.J.; Gupta, N.; Sahingur, S.E. A20 Restricts Inflammatory Response and Desensitizes Gingival Keratinocytes to Apoptosis. Front. Immunol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Kumari, S.; Van, T.M.; Preukschat, D.; Schuenke, H.; Basic, M.; Bleich, A.; Klein, U.; Pasparakis, M. NF-κB inhibition in keratinocytes causes RIPK1-mediated necroptosis and skin inflammation. Life Sci. Alliance 2021, 4, e202000956. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Hu, D.; Sakya, J.; Sun, T.; Wang, D.; Wang, L.; Mao, X.; Su, Z. ABIN-1 is a key regulator in RIPK1-dependent apoptosis (RDA) and necroptosis, and ABIN-1 deficiency potentiates necroptosis-based cancer therapy in colorectal cancer. Cell Death Dis. 2021, 12, 140. [Google Scholar] [CrossRef]
- Verstrepen, L.; Verhelst, K.; van Loo, G.; Carpentier, I.; Ley, S.C.; Beyaert, R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem. Pharmacol. 2010, 80, 2009–2020. [Google Scholar] [CrossRef]
- Gurevich, I.; Zhang, C.; Encarnacao, P.C.; Struzynski, C.P.; Livings, S.E.; Aneskievich, B.J. PPARγ and NF-κB regulate the gene promoter activity of their shared repressor, TNIP1. Biochim. Biophys. Acta 2012, 1819, 1–15. [Google Scholar] [CrossRef]
- Xu, X.; Fan, X.; Wu, X.; Xia, R.; Liang, J.; Gao, F.; Shu, J.; Yang, M.; Sun, W. Luteolin ameliorates necroptosis in Glucocorticoid-induced osteonecrosis of the femoral head via RIPK1/RIPK3/MLKL pathway based on network pharmacology analysis. Biochem. Biophys. Res. Commun. 2023, 661, 108–118. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, Q.; Xi, X.; Cao, X.; Wang, X.; Zhang, M.; Xu, Y.; Deng, T.; Deng, X.; Zhang, G.; et al. KW2449 ameliorates collagen-induced arthritis by inhibiting RIPK1-dependent necroptosis. Front. Immunol. 2023, 14, 1135014. [Google Scholar] [CrossRef]
- Rajamäki, K.; Keskitalo, S.; Seppänen, M.; Kuismin, O.; Vähäsalo, P.; Trotta, L.; Väänänen, A.; Glumoff, V.; Keskitalo, P.; Kaarteenaho, R.; et al. Haploinsufficiency of A20 impairs protein-protein interactome and leads into caspase-8-dependent enhancement of NLRP3 inflammasome activation. RMD Open 2018, 4, e000740. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Shamilov, R.; Ackley, T.W.; Aneskievich, B.J. Enhanced Wound Healing- and Inflammasome-Associated Gene Expression in TNFAIP3-Interacting Protein 1- (TNIP1-) Deficient HaCaT Keratinocytes Parallels Reduced Reepithelialization. Mediators Inflamm. 2020, 2020, 5919150. [Google Scholar] [CrossRef] [PubMed]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17. [Google Scholar] [CrossRef]
- Kattah, M.G.; Shao, L.; Rosli, Y.Y.; Shimizu, H.; Whang, M.I.; Advincula, R.; Achacoso, P.; Shah, S.; Duong, B.H.; Onizawa, M.; et al. A20 and ABIN-1 synergistically preserve intestinal epithelial cell survival. J. Exp. Med. 2018, 215, 1839–1852. [Google Scholar] [CrossRef]
- Zhang, S.; Fukushi, M.; Hashimoto, S.; Gao, C.; Huang, L.; Fukuyo, Y.; Nakajima, T.; Amagasa, T.; Enomoto, S.; Koike, K.; et al. A new ERK2 binding protein, Naf1, attenuates the EGF/ERK2 nuclear signaling. Biochem. Biophys. Res. Commun. 2002, 297, 17–23. [Google Scholar] [CrossRef]
- Gupta, K.; Ott, D.; Hope, T.J.; Siliciano, R.F.; Boeke, J.D. A human nuclear shuttling protein that interacts with human immunodeficiency virus type 1 matrix is packaged into virions. J. Virol. 2000, 74, 11811–11824. [Google Scholar] [CrossRef]
- Lei, Q.; Gu, H.; Li, L.; Wu, T.; Xie, W.; Li, M.; Zhao, N. TNIP1-mediated TNF-α/NF-κB signalling cascade sustains glioma cell proliferation. J. Cell Mol. Med. 2020, 24, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, J.; Han, S.; Li, E. TNIP1 Inhibits Proliferation And Promotes Apoptosis In Clear Cell Renal Carcinoma Through Targeting C/Ebpβ. Onco Targets Ther. 2019, 12, 9861–9871. [Google Scholar] [CrossRef]
- Yin, H.; Karayel, O.; Chao, Y.Y.; Seeholzer, T.; Hamp, I.; Plettenburg, O.; Gehring, T.; Zielinski, C.; Mann, M.; Krappmann, D. A20 and ABIN-1 cooperate in balancing CBM complex-triggered NF-κB signaling in activated T cells. Cell Mol. Life Sci. 2022, 79, 112. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.C.; Walburg, K.V.; van Veen, S.; Wilson, L.G.; Trufen, C.E.M.; Nascimento, I.P.; Ottenhoff, T.H.M.; Leite, L.C.C.; Haks, M.C. Recombinant BCG-LTAK63 Vaccine Candidate for Tuberculosis Induces an Inflammatory Profile in Human Macrophages. Vaccines 2022, 10, 831. [Google Scholar] [CrossRef] [PubMed]
- Ippagunta, S.K.; Gangwar, R.; Finkelstein, D.; Vogel, P.; Pelletier, S.; Gingras, S.; Redecke, V.; Häcker, H. Keratinocytes contribute intrinsically to psoriasis upon loss of Tnip1 function. Proc. Natl. Acad. Sci. USA 2016, 113, E6162–E6171. [Google Scholar] [CrossRef] [PubMed]
- Encarnacao, P.C.; Ramirez, V.P.; Zhang, C.; Aneskievich, B.J. Sp sites contribute to basal and inducible expression of the human TNIP1 (TNFα-inducible protein 3-interacting protein 1) promoter. Biochem. J. 2013, 452, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Nowak, D.E.; Brasier, A.R. A TNF-induced gene expression program under oscillatory NF-kappaB control. BMC Genom. 2005, 6, 137. [Google Scholar] [CrossRef]
- Tian, B.; Nowak, D.E.; Jamaluddin, M.; Wang, S.; Brasier, A.R. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J. Biol. Chem. 2005, 280, 17435–17448. [Google Scholar] [CrossRef]
- Bowes, J.; Orozco, G.; Flynn, E.; Ho, P.; Brier, R.; Marzo-Ortega, H.; Coates, L.; McManus, R.; Ryan, A.W.; Kane, D.; et al. Confirmation of TNIP1 and IL23A as susceptibility loci for psoriatic arthritis. Ann. Rheum. Dis. 2011, 70, 1641–1644. [Google Scholar] [CrossRef]
- Kawasaki, A.; Ito, S.; Furukawa, H.; Hayashi, T.; Goto, D.; Matsumoto, I.; Kusaoi, M.; Ohashi, J.; Graham, R.R.; Matsuta, K.; et al. Association of TNFAIP3 interacting protein 1, TNIP1 with systemic lupus erythematosus in a Japanese population: A case-control association study. Arthritis Res. Ther. 2010, 12, R174. [Google Scholar] [CrossRef]
- Allanore, Y.; Saad, M.; Dieudé, P.; Avouac, J.; Distler, J.H.; Amouyel, P.; Matucci-Cerinic, M.; Riemekasten, G.; Airo, P.; Melchers, I.; et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011, 7, e1002091. [Google Scholar] [CrossRef]
- Adrianto, I.; Wang, S.; Wiley, G.B.; Lessard, C.J.; Kelly, J.A.; Adler, A.J.; Glenn, S.B.; Williams, A.H.; Ziegler, J.T.; Comeau, M.E.; et al. Association of two independent functional risk haplotypes in TNIP1 with systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 3695–3705. [Google Scholar] [CrossRef]
- Nanda, S.K.; Venigalla, R.K.; Ordureau, A.; Patterson-Kane, J.C.; Powell, D.W.; Toth, R.; Arthur, J.S.; Cohen, P. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J. Exp. Med. 2011, 208, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; van den Bedem, H.; Tang, S.; Maslennikov, I.; Wakatsuki, S.; Dikic, I.; Rahighi, S. Molecular Recognition of M1-Linked Ubiquitin Chains by Native and Phosphorylated UBAN Domains. J. Mol. Biol. 2019, 431, 3146–3156. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Carpentier, I.; Rogov, V.; Kreike, M.; Ikeda, F.; Löhr, F.; Wu, C.J.; Ashwell, J.D.; Dötsch, V.; Dikic, I.; et al. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene 2008, 27, 3739–3745. [Google Scholar] [CrossRef]
- Gao, L.; Coope, H.; Grant, S.; Ma, A.; Ley, S.C.; Harhaj, E.W. ABIN1 protein cooperates with TAX1BP1 and A20 proteins to inhibit antiviral signaling. J. Biol. Chem. 2011, 286, 36592–36602. [Google Scholar] [CrossRef]
- Shamilov, R.; Vinogradova, O.; Aneskievich, B.J. The Anti-Inflammatory Protein TNIP1 Is Intrinsically Disordered with Structural Flexibility Contributed by Its AHD1-UBAN Domain. Biomolecules 2020, 10, 1531. [Google Scholar] [CrossRef]
- Mirza, N.; Sowa, A.S.; Lautz, K.; Kufer, T.A. NLRP10 Affects the Stability of Abin-1 To Control Inflammatory Responses. J. Immunol. 2019, 202, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Shinkawa, Y.; Imami, K.; Fuseya, Y.; Sasaki, K.; Ohmura, K.; Ishihama, Y.; Morinobu, A.; Iwai, K. ABIN1 is a signal-induced autophagy receptor that attenuates NF-κB activation by recognizing linear ubiquitin chains. FEBS Lett. 2022, 596, 1147–1164. [Google Scholar] [CrossRef]
- Zhou, J.; Rasmussen, N.L.; Olsvik, H.L.; Akimov, V.; Hu, Z.; Evjen, G.; Kaeser-Pebernard, S.; Sankar, D.S.; Roubaty, C.; Verlhac, P.; et al. TBK1 phosphorylation activates LIR-dependent degradation of the inflammation repressor TNIP1. J. Cell Biol. 2023, 222, e202108144. [Google Scholar] [CrossRef]
- Pang, Y.; Wu, L.; Tang, C.; Wang, H.; Wei, Y. Autophagy-Inflammation Interplay During Infection: Balancing Pathogen Clearance and Host Inflammation. Front. Pharmacol. 2022, 13, 832750. [Google Scholar] [CrossRef]
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: Balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Shin, D.M.; Yuk, J.M.; Shi, G.; Choi, D.K.; Lee, S.H.; Huang, S.M.; Kim, J.M.; Kim, C.D.; Lee, J.H.; et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J. Immunol. 2011, 186, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Le Guerroué, F.; Bunker, E.N.; Rosencrans, W.M.; Nguyen, J.T.; Basar, M.A.; Werner, A.; Chou, T.F.; Wang, C.; Youle, R.J. TNIP1 inhibits selective autophagy via bipartite interaction with LC3/GABARAP and TAX1BP1. Mol. Cell 2023, 83, 927–941.e8. [Google Scholar] [CrossRef] [PubMed]
- Merline, R.; Rödig, H.; Zeng-Brouwers, J.; Poluzzi, C.; Tascher, G.; Michaelis, J.; Lopez-Mosqueda, J.; Rhiner, A.; Huber, L.S.; Diehl, V.; et al. A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1): A novel modulator of mitochondrial autophagy. Am. J. Physiol. Cell Physiol. 2023, 324, c339–c352. [Google Scholar] [CrossRef]
- van der Fits, L.; Mourits, S.; Voerman, J.S.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef]
- Callahan, J.A.; Hammer, G.E.; Agelides, A.; Duong, B.H.; Oshima, S.; North, J.; Advincula, R.; Shifrin, N.; Truong, H.A.; Paw, J.; et al. Cutting edge: ABIN-1 protects against psoriasis by restricting MyD88 signals in dendritic cells. J. Immunol. 2013, 191, 535–539. [Google Scholar] [CrossRef]
- Yamamoto, M.; Gohda, J.; Akiyama, T.; Inoue, J.I. TNF receptor-associated factor 6 (TRAF6) plays crucial roles in multiple biological systems through polyubiquitination-mediated NF-κB activation. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2021, 97, 145–160. [Google Scholar] [CrossRef]
- Dainichi, T.; Matsumoto, R.; Mostafa, A.; Kabashima, K. Immune Control by TRAF6-Mediated Pathways of Epithelial Cells in the EIME (Epithelial Immune Microenvironment). Front. Immunol. 2019, 10, 1107. [Google Scholar] [CrossRef]
- Chen, Y.; Yan, H.; Song, Z.; Chen, F.; Wang, H.; Niu, J.; Shi, X.; Zhang, D.; Zhang, N.; Zhai, Z.; et al. Downregulation of TNIP1 Expression Leads to Increased Proliferation of Human Keratinocytes and Severer Psoriasis-Like Conditions in an Imiquimod-Induced Mouse Model of Dermatitis. PLoS ONE 2015, 10, e0127957. [Google Scholar] [CrossRef]
- Brady, M.P.; Korte, E.A.; Caster, D.J.; Powell, D.W. TNIP1/ABIN1 and lupus nephritis: Review. Lupus Sci. Med. 2020, 7, e000437. [Google Scholar] [CrossRef]
- Cruz, J.A.; Childs, E.E.; Amatya, N.; Garg, A.V.; Beyaert, R.; Kane, L.P.; Aneskievich, B.J.; Ma, A.; Gaffen, S.L. Interleukin-17 signaling triggers degradation of the constitutive NF-κB inhibitor ABIN-1. Immunohorizons 2017, 1, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, N.L.; Zhou, J.; Olsvik, H.; Kaeser-Pebernard, S.; Lamark, T.; Dengjel, J.; Johansen, T. The inflammation repressor TNIP1/ABIN-1 is degraded by autophagy following TBK1 phosphorylation of its LIR. Autophagy 2023, 1–3. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, J.; Kim, M.S.; Kwon, Y.; Shin, S.; Yi, H.; Kim, H.; Chang, M.J.; Chang, C.B.; Kang, S.B.; et al. Coordinate regulation of the senescent state by selective autophagy. Dev. Cell 2021, 56, 1512–1525.e7. [Google Scholar] [CrossRef] [PubMed]
- Erdei, L.; Bolla, B.S.; Bozó, R.; Tax, G.; Urbán, E.; Kemény, L.; Szabó, K. TNIP1 Regulates Cutibacterium acnes-Induced Innate Immune Functions in Epidermal Keratinocytes. Front. Immunol. 2018, 9, 2155. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, I.; Zhang, C.; Francis, N.; Struzynsky, C.P.; Livings, S.E.; Aneskievich, B.J. Human TNFα-induced protein 3-interacting protein 1 (TNIP1) promoter activation is regulated by retinoic acid receptors. Gene 2013, 515, 42–48. [Google Scholar] [CrossRef][Green Version]
- Katsuya, K.; Hori, Y.; Oikawa, D.; Yamamoto, T.; Umetani, K.; Urashima, T.; Kinoshita, T.; Ayukawa, K.; Tokunaga, F.; Tamaru, M. High-Throughput Screening for Linear Ubiquitin Chain Assembly Complex (LUBAC) Selective Inhibitors Using Homogenous Time-Resolved Fluorescence (HTRF)-Based Assay System. SLAS Discov. 2018, 23, 1018–1029. [Google Scholar] [CrossRef]
- Johansson, H.; Isabella Tsai, Y.C.; Fantom, K.; Chung, C.W.; Kümper, S.; Martino, L.; Thomas, D.A.; Eberl, H.C.; Muelbaier, M.; House, D.; et al. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141, 2703–2712. [Google Scholar] [CrossRef]
- Oikawa, D.; Sato, Y.; Ohtake, F.; Komakura, K.; Hanada, K.; Sugawara, K.; Terawaki, S.; Mizukami, Y.; Phuong, H.T.; Iio, K.; et al. Molecular bases for HOIPINs-mediated inhibition of LUBAC and innate immune responses. Commun. Biol. 2020, 3, 163. [Google Scholar] [CrossRef]
- Yuan, W.; Chen, Y.; Zhou, Y.; Bao, K.; Yu, X.; Xu, Y.; Zhang, Y.; Zheng, J.; Jiang, G.; Hong, M.; et al. Formononetin attenuates atopic dermatitis by upregulating A20 expression via activation of G protein-coupled estrogen receptor. J. Ethnopharmacol. 2021, 266, 113397. [Google Scholar] [CrossRef]
| Disease | Pathophysiology |
|---|---|
| Psoriasis (plaque) | Raised, scaly, erythematous regions; over-production of and hyperresponsiveness to cytokines (e.g., TNFα, IL-23, and IL-17); increased numbers of immune cells; keratinocyte hyperproliferation and altered differentiation |
| Atopic dermatitis | Epidermal barrier defect, increased permeability of irritants; Th1 overactivity causes chronic lesions and pruritus; scratching skin stimulates keratinocyte release of inflammatory cytokines (e.g., TNFα and IL-1, IL-6) |
| Systemic lupus erythematosus | Cutaneous features: immune-cell infiltration; hyperkeratosis; autoinflammation and vasculopathy presenting as a malar (facial) rash, frequent photosensitivity; keratinocyte DAMP release and interferon production |
| Systemic sclerosis | Vascular insult progresses to chronic tissue hypoxia concurrent with increased pro-fibrotic cytokines (e.g., TGF-β, IL-5, and IL-13) and decreased anti-inflammatory cytokine (e.g., IFN-γ) production; autoimmunity |
| Cylindromatosis | Skin-appendage-derived tumors including cylindromas, trichoepitheliomas, and spiradenomas; usually benign but can progress to malignancy |
| Haploinsufficiency A20 | Clinically variable presentation similar to Behcet’s disease; hyperkeratosis; pustular rash, acne, dermal abscesses, and oral and genital ulcers; hyperresponsive to stimuli for production of cytokines, e.g., IL-6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carman, L.E.; Samulevich, M.L.; Aneskievich, B.J. Repressive Control of Keratinocyte Cytoplasmic Inflammatory Signaling. Int. J. Mol. Sci. 2023, 24, 11943. https://doi.org/10.3390/ijms241511943
Carman LE, Samulevich ML, Aneskievich BJ. Repressive Control of Keratinocyte Cytoplasmic Inflammatory Signaling. International Journal of Molecular Sciences. 2023; 24(15):11943. https://doi.org/10.3390/ijms241511943
Chicago/Turabian StyleCarman, Liam E., Michael L. Samulevich, and Brian J. Aneskievich. 2023. "Repressive Control of Keratinocyte Cytoplasmic Inflammatory Signaling" International Journal of Molecular Sciences 24, no. 15: 11943. https://doi.org/10.3390/ijms241511943
APA StyleCarman, L. E., Samulevich, M. L., & Aneskievich, B. J. (2023). Repressive Control of Keratinocyte Cytoplasmic Inflammatory Signaling. International Journal of Molecular Sciences, 24(15), 11943. https://doi.org/10.3390/ijms241511943