
Resistin-like Molecule α and Pulmonary Vascular Remodeling: A Multi-Strain Murine Model of Antigen and Urban Ambient Particulate Matter Co-Exposure

 , , , and
, , , and 
Abstract

1. Introduction
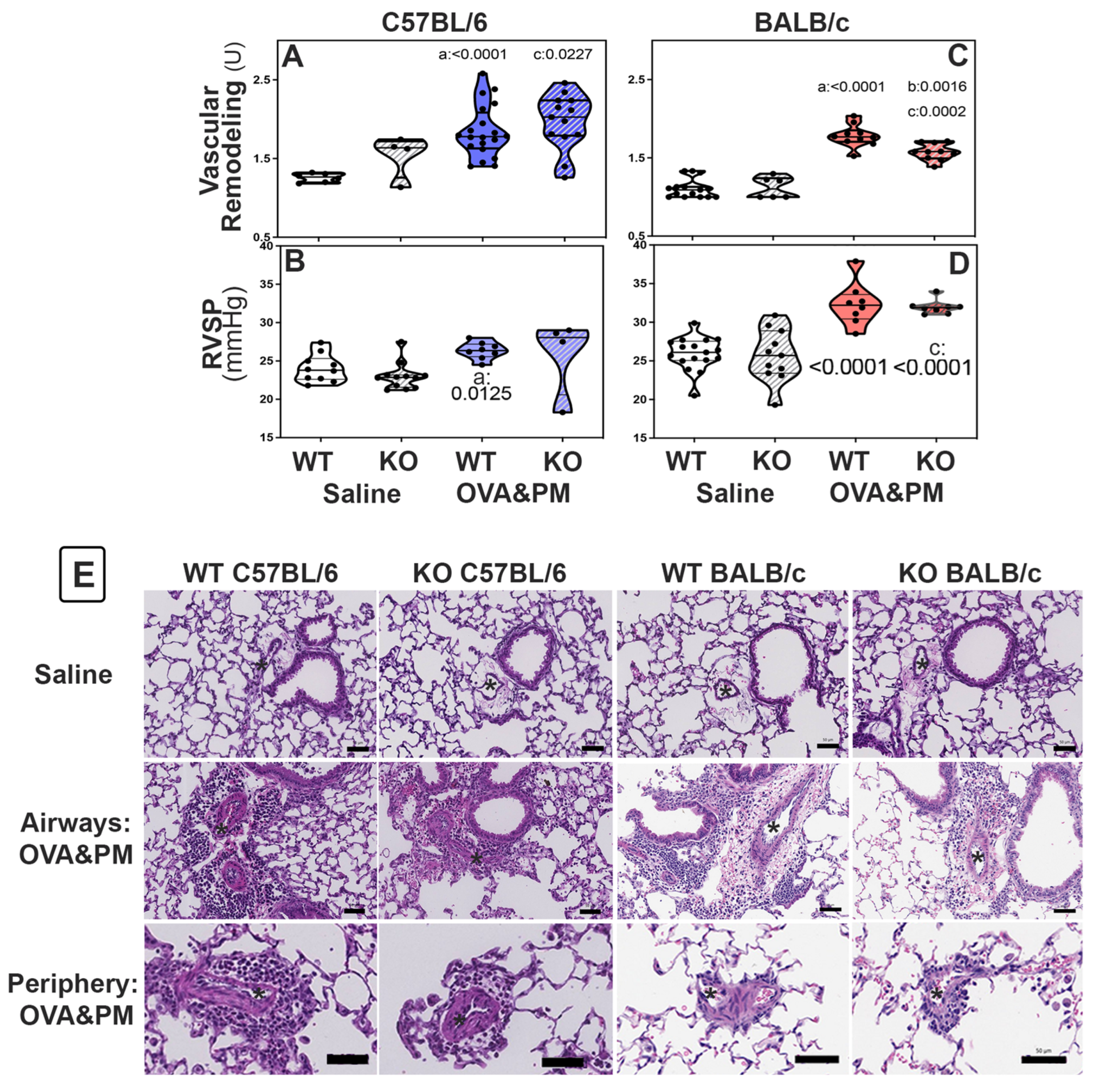
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daley, E.; Emson, C.; Guignabert, C.; Malefyt, R.D.W.; Louten, J.; Kurup, V.P.; Hogaboam, C.; Taraseviciene-Stewart, L.; Voelkel, N.F.; Rabinovitch, M.; et al. Pulmonary arterial remodeling induced by a Th2 immune response. J. Exp. Med. 2008, 205, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Cogan, J.D.; Pauciulo, M.W.; Batchman, A.P.; Prince, M.A.; Robbins, I.M.; Hedges, L.K.; Stanton, K.C.; Wheeler, L.A.; Phillips, J.A.; Loyd, J.E.; et al. High Frequency of BMPR2 Exonic Deletions/Duplications in Familial Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 590–598. [Google Scholar] [CrossRef]
- Davies, R.J.; Morrell, N.W. Molecular mechanisms of pulmonary arterial hypertension: Role of mutations in the bone morphogenetic protein type II receptor. Chest 2008, 134, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Sztrymf, B.; Coulet, F.; Girerd, B.; Yaici, A.; Jais, X.; Sitbon, O.; Montani, D.; Souza, R.; Simonneau, G.; Soubrier, F.; et al. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am. J. Respir. Crit. Care Med. 2008, 177, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Crosswhite, P.; Sun, Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. J. Hypertens. 2010, 28, 201–212. [Google Scholar] [CrossRef]
- Aldred, M.A.; Machado, R.D.; James, V.; Morrell, N.W.; Trembath, R.C. Characterization of the BMPR2 5′-Untranslated Region and a Novel Mutation in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2007, 176, 819–824. [Google Scholar] [CrossRef]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2008, 118, 2372–2379. [Google Scholar] [CrossRef]
- Chan, S.Y.; Loscalzo, J. Pathogenic mechanisms of pulmonary arterial hypertension. J. Mol. Cell Cardiol. 2008, 44, 14–30. [Google Scholar] [CrossRef]
- Strange, J.W.; Wharton, J.; Phillips, P.G.; Wilkins, M.R. Recent insights into the pathogenesis and therapeutics of pulmonary hypertension. Clin. Sci. 2002, 102, 253–268. [Google Scholar] [CrossRef]
- Grunig, G.; Marsh, L.M.; Esmaeil, N.; Jackson, K.; Gordon, T.; Reibman, J.; Kwapiszewska, G.; Park, S.-H. Perspective: Ambient Air Pollution: Inflammatory Response and Effects on the Lung’s Vasculature. Pulm. Circ. 2014, 4, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Chen, W.-C.; Durmus, N.; Bleck, B.; Reibman, J.; Riemekasten, G.; Grunig, G. The Effects of Antigen-Specific IgG1 Antibody for the Pulmonary-Hypertension-Phenotype and B Cells for Inflammation in Mice Exposed to Antigen and Fine Particles from Air Pollution. PLoS ONE 2015, 10, e0129910. [Google Scholar] [CrossRef]
- Park, S.H.; Chen, W.C.; Esmaeil, N.; Lucas, B.; Marsh, L.M.; Reibman, J.L.; Grunig, G. IL-13 and IL-17A induced pulmonary-hypertension-phenotype due to inhalation of antigen and fine particles from air pollution. Pulm. Circ. 2014, 4, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Kolosova, I.A.; Fan, C.; Skinner, J.T.; Yamaji-Kegan, K.; Collector, M.; Sharkis, S.J.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELM alpha) recruits bone marrow-derived cells to the murine pulmonary vasculature. PLoS ONE 2010, 5, e11251. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Skinner, J.T.; Champion, H.C.; Crow, M.T.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am. J. Physiol. Lung. Cell Mol. Physiol. 2009, 296, L582–L593. [Google Scholar] [CrossRef]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Skinner, J.T.; Poloczek, A.; El-Haddad, H.; Cheadle, C.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) in chronic hypoxia- and antigen-mediated pulmonary vascular remodeling. Respir. Res. 2013, 14, 1–16. [Google Scholar] [CrossRef]
- Teng, X.; Li, D.; Champion, H.C.; Johns, R.A. FIZZ1/RELMα, a Novel Hypoxia-Induced Mitogenic Factor in Lung With Vasoconstrictive and Angiogenic Properties. Circ. Res. 2003, 92, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Champion, H.C.; Johns, R.A. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am. J. Physiol. Cell Mol. Physiol. 2006, 291, L1159–L1168. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Tan, H.; Irwin, D.M. Evolution of the Vertebrate Resistin Gene Family. PLoS ONE 2015, 10, e0130188. [Google Scholar] [CrossRef]
- Hue, I.; Capilla, E.; Rosell-Moll, E.; Balbuena-Pecino, S.; Goffette, V.; Gabillard, J.-C.; Navarro, I. Recent advances in the crosstalk between adipose, muscle and bone tissues in fish. Front. Endocrinol. 2023, 14, 1155202. [Google Scholar] [CrossRef]
- Holcomb, I.N.; Kabakoff, R.C.; Chan, B.; Baker, T.W.; Gurney, A.; Henzel, W.; Nelson, C.; Lowman, H.B.; Wright, B.D.; Skelton, N.J.; et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000, 19, 4046–4055. [Google Scholar] [CrossRef]
- Dong, L.; Wang, S.-J.; Camoretti-Mercado, B.; Li, H.-J.; Chen, M.; Bi, W.-X. FIZZ1 Plays a Crucial Role in Early Stage Airway Remodeling of OVA-Induced Asthma. J. Asthma 2008, 45, 648–653. [Google Scholar] [CrossRef]
- Munitz, A.; Waddell, A.; Seidu, L.; Cole, E.T.; Ahrens, R.; Hogan, S.P.; Rothenberg, M.E. Resistin-like molecule α enhances myeloid cell activation and promotes colitis. J. Allergy Clin. Immunol. 2008, 122, 1200–1207.e1. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.G.; Du, Y.; Perrigoue, J.G.; Zaph, C.; Taylor, J.J.; Goldschmidt, M.; Swain, G.P.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.; et al. Alternatively activated macrophage-derived RELM-{alpha} is a negative regulator of type 2 inflammation in the lung. J. Exp. Med. 2009, 206, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Johns, R.A.; Takimoto, E.; Meuchel, L.W.; Elsaigh, E.; Zhang, A.; Heller, N.M.; Semenza, G.L.; Yamaji-Kegan, K. Hypoxia-Inducible Factor 1alpha Is a Critical Downstream Mediator for Hypoxia-Induced Mitogenic Factor (FIZZ1/RELMalpha)-Induced Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 134–144. [Google Scholar] [CrossRef]
- Renigunta, A.; Hild, C.; Rose, F.; Klepetko, W.; Grimminger, F.; Seeger, W.; Hänze, J. Faculty Opinions recommendation of Human RELMbeta is a mitogenic factor in lung cells and induced in hypoxia. FEBS Lett. 2006, 580, 900–903. [Google Scholar] [CrossRef]
- Su, Q.; Zhou, Y.; Johns, R.A. Bruton’s tyrosine kinase (BTK) is a binding partner for hypoxia induced mitogenic factor (HIMF/FIZZ1) and mediates myeloid cell chemotaxis. FASEB J. 2007, 21, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Su, Q.; Li, Y.; Liang, L.; Angelini, D.J.; Guggino, W.B.; Johns, R.A.; Yadav, V.R.; Song, T.; Mei, L.; et al. Hypoxia-induced mitogenic factor/FIZZ1 induces intracellular calcium release through the PLC-IP3 pathway. Am. J. Physiol. Cell Mol. Physiol. 2009, 297, L263–L270. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Fu, Z.; Su, Q.; Angelini, D.J.; Van Eyk, J.; Johns, R.A. S100A11 Mediates Hypoxia-induced Mitogenic Factor (HIMF)-induced Smooth Muscle Cell Migration, Vesicular Exocytosis, and Nuclear Activation. Mol. Cell Proteom. 2011, 10, M110.000901. [Google Scholar] [CrossRef]
- Patel, S.D.; Rajala, M.W.; Rossetti, L.; Scherer, P.E.; Shapiro, L. Disulfide-Dependent Multimeric Assembly of Resistin Family Hormones. Science 2004, 304, 1154–1158. [Google Scholar] [CrossRef]
- Gerstmayer, B.; Küsters, D.; Gebel, S.; Müller, T.; Van Miert, E.; Hofmann, K.; Bosio, A. Identification of RELMγ, a novel resistin-like molecule with a distinct expression pattern☆. Genomics 2003, 81, 588–595. [Google Scholar] [CrossRef]
- Steppan, C.M.; Brown, E.J.; Wright, C.M.; Bhat, S.; Banerjee, R.R.; Dai, C.Y.; Enders, G.H.; Silberg, D.G.; Wen, X.; Wu, G.D.; et al. A family of tissue-specific resistin-like molecules. Proc. Natl. Acad. Sci. USA 2001, 98, 502–506. [Google Scholar] [CrossRef]
- Munitz, A.; Seidu, L.; Cole, E.T.; Ahrens, R.; Hogan, S.P.; Rothenberg, M.E. Resistin-Like Molecule α Decreases Glucose Tolerance during Intestinal Inflammation. J. Immunol. 2009, 182, 2357–2363. [Google Scholar] [CrossRef]
- Pesce, J.T.; Ramalingam, T.R.; Wilson, M.S.; Mentink-Kane, M.M.; Thompson, R.W.; Cheever, A.W.; Urban, J.F.; Wynn, T.A. Retnla (Relmα/Fizz1) Suppresses Helminth-Induced Th2-Type Immunity. PLOS Pathog. 2009, 5, e1000393. [Google Scholar] [CrossRef]
- Munitz, A.; Cole, E.T.; Karo-Atar, D.; Finkelman, F.D.; Rothenberg, M.E. Resistin-Like Molecule–α Regulates IL-13–Induced Chemokine Production but Not Allergen-Induced Airway Responses. Am. J. Respir. Cell Mol. Biol. 2012, 46, 703–713. [Google Scholar] [CrossRef]
- Weatherald, J.; Boucly, A.; Peters, A.; Montani, D.; Prasad, K.; Psotka, M.A.; Zannad, F.; Gomberg-Maitland, M.; McLaughlin, V.; Simonneau, G.; et al. The evolving landscape of pulmonary arterial hypertension clinical trials. Lancet 2022, 400, 1884–1898. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.J.; Johnston, B.; Issekutz, A.; Kubes, P. Endothelin-1 causes P-selectin-dependent leukocyte rolling and adhesion within rat mesenteric microvessels. Am. J. Physiol. Circ. Physiol. 1999, 277, H1823–H1830. [Google Scholar] [CrossRef] [PubMed]
- Czopek, A.; Moorhouse, R.; Gallacher, P.J.; Pugh, D.; Ivy, J.R.; Farrah, T.E.; Godden, E.; Hunter, R.W.; Webb, D.J.; Tharaux, P.-L.; et al. Endothelin blockade prevents the long-term cardiovascular and renal sequelae of acute kidney injury in mice. Sci. Transl. Med. 2022, 14, eabf5074. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Haczku, A.; Lee, J.J.; Irvin, C.G.; Gelfand, E.W. Strain dependence of airway hyperresponsiveness reflects differences in eosinophil localization in the lung. Am. J. Physiol. Cell Mol. Physiol. 2001, 281, L394–L402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lamm, W.J.; Albert, R.K.; Chi, E.Y.; Henderson, W.R.; Lewis, D.B. Influence of the route of allergen administration and genetic background on the murine allergic pulmonary response. Am. J. Respir. Crit. Care Med. 1997, 155, 661–669. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Watanabe, H.; Numata, K.; Ito, T.; Takagi, K.; Matsukawa, A. Innate immune response in Th1- and Th2-dominant mouse strains. Shock 2004, 22, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Chen, W.-C.; Hoffman, C.; Marsh, L.M.; West, J.; Grunig, G. Modification of Hemodynamic and Immune Responses to Exposure with a Weak Antigen by the Expression of a Hypomorphic BMPR2 Gene. PLoS ONE 2013, 8, e55180. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.C.; Kelley, K.B.; Merisko-Liversidge, E.M.; Kennedy, J.; Klein, R.M. Developmental Pattern of Ventricular Atrial Natriuretic Peptide (ANP) Expression in Chronically Hypoxic Rats as an Indicator of the Hypertrophic Process. J. Mol. Cell Cardiol. 1994, 26, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Sanada, S.; Hakuno, D.; Higgins, L.J.; Schreiter, E.R.; McKenzie, A.N.; Lee, R.T. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J. Clin. Investig. 2007, 117, 1538–1549. [Google Scholar] [CrossRef]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Lin, Q.; Fan, C.; Gomez-Arroyo, J.; Van Raemdonck, K.; Meuchel, L.W.; Skinner, J.T.; Everett, A.D.; Fang, X.; Macdonald, A.A.; Yamaji-Kegan, K.; et al. HIMF (Hypoxia-Induced Mitogenic Factor) Signaling Mediates the HMGB1 (High Mobility Group Box 1)-Dependent Endothelial and Smooth Muscle Cell Crosstalk in Pulmonary Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 2505–2519. [Google Scholar] [CrossRef]
- Lin, Q.; Fan, C.; Skinner, J.T.; Hunter, E.N.; Macdonald, A.A.; Illei, P.B.; Yamaji-Kegan, K.; Johns, R.A. RELMα Licenses Macrophages for Damage-Associated Molecular Pattern Activation to Instigate Pulmonary Vascular Remodeling. J. Immunol. 2019, 203, 2862–2871. [Google Scholar] [CrossRef]
- Tarkowski, A.; Bjersing, J.; Shestakov, A.; Bokarewa, M.I. Resistin competes with lipopolysaccharide for binding to toll-like receptor 4. J. Cell Mol. Med. 2009, 14, 1419–1431. [Google Scholar] [CrossRef]
- Jang, J.C.; Li, J.; Gambini, L.; Batugedara, H.M.; Sati, S.; Lazar, M.A.; Fan, L.; Pellecchia, M.; Nair, M.G. Human resistin protects against endotoxic shock by blocking LPS–TLR4 interaction. Proc. Natl. Acad. Sci. USA 2017, 114, E10399–E10408. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Fukudome, K.; Takao, S.; Tsuneyoshi, N.; Ohta, S.; Nagai, Y.; Ihara, H.; Miyake, K.; Ikeda, Y.; Kimoto, M. Reduced Surface Expression of TLR4 by a V254I Point Mutation Accounts for the Low Lipopolysaccharide Responder Phenotype of BALB/c B Cells. J. Immunol. 2013, 190, 195–204. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef]
- Joshi, S.R.; Liu, J.; Bloom, T.; Atabay, E.K.; Kuo, T.-H.; Lee, M.; Belcheva, E.; Spaits, M.; Grenha, R.; Maguire, M.C.; et al. Sotatercept analog suppresses inflammation to reverse experimental pulmonary arterial hypertension. Sci. Rep. 2022, 12, 7803. [Google Scholar] [CrossRef]
- Grunig, G.; Eichstaedt, C.A.; Verweyen, J.; Durmus, N.; Saxer, S.; Krafsur, G.; Stenmark, K.; Ulrich, S.; Grünig, E.; Pylawka, S. Circulating MicroRNA Markers for Pulmonary Hypertension in Supervised Exercise Intervention and Nightly Oxygen Intervention. Front. Physiol. 2018, 9, 955. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Beck, G.J.; Newman, J.H.; Abidov, A.; Aldred, M.A.; Barnard, J.; Rosenzweig, E.B.; Borlaug, B.A.; Chung, W.K.; Comhair, S.A.A. PVDOMICS: A Multi-Center Study to Improve Understanding of Pulmonary Vascular Disease Through Phenomics. Circ. Res. 2017, 121, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Leopold, J.A.; Radeva, M.K.; Beck, G.J.; Abidov, A.; Aldred, M.A.; Barnard, J.; Rosenzweig, E.B.; Borlaug, B.A.; Chung, W.K.; et al. Clinical Characteristics and Transplant-Free Survival Across the Spectrum of Pulmonary Vascular Disease. J. Am. Coll. Cardiol. 2022, 80, 697–718. [Google Scholar] [CrossRef] [PubMed]
- Louis, R.; Harrison, T.W.; Chanez, P.; Menzella, F.; Philteos, G.; Cosio, B.G.; Lugogo, N.L.; de Luiz, G.; Burden, A.; Adlington, T.; et al. Severe Asthma Standard-of-Care Background Medication Reduction With Benralizumab: ANDHI in Practice Substudy. J. Allergy Clin. Immunol. Pract. 2023, 11, 1759–1770.e7. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Johns, R.A. Resistin family proteins in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L422–L434. [Google Scholar] [CrossRef]
- Gordon, T. A centrifugal particle concentrator for use in inhalation toxicology. Inhal. Toxicol. 1999, 11, 71–87. [Google Scholar] [CrossRef]
- Gordon, T. Linking Health Effects to PM Components, Size, and Sources. Inhal. Toxicol. 2007, 19, 3–6. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Piggott, D.A.; Huleatt, J.W.; Visintin, I.; Herrick, C.A.; Bottomly, K. Lipopolysaccharide-enhanced, toll-like receptor 4–dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 2002, 196, 1645–1651. [Google Scholar] [CrossRef]
- Chen, W.C.; Park, S.H.; Hoffman, C.; Philip, C.; Robinson, L.; West, J.; Grunig, G. Right ventricular systolic pressure measurements in combination with harvest of lung and immune tissue samples in mice. J. Vis. Exp. 2013, 71, e50023. [Google Scholar]
- Hoffman, C.; Park, S.-H.; Daley, E.; Emson, C.; Louten, J.; Sisco, M.; Malefyt, R.d.W.; Grunig, G. Interleukin-19: A Constituent of the Regulome That Controls Antigen Presenting Cells in the Lungs and Airway Responses to Microbial Products. PLoS ONE 2011, 6, e27629. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Daley, E.; Chow, A.; Robinson, K.; Parthasarathi, K.; McKenzie, A.N.J.; Tschernig, T.; Kurup, V.P.; Donaldson, D.D.; Grunig, G. IL-13 Regulates the Immune Response to Inhaled Antigens. J. Immunol. 2005, 174, 8097–8105. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.G.; Rennick, D.; Donaldson, D.D.; Venkayya, R.; McArthur, C.; Hansell, E.; Kurup, V.P.; Warnock, M.; Grunig, G. Il-13 and IFN-gamma: Interactions in lung inflammation. J. Immunol. 2001, 167, 1769–1777. [Google Scholar] [CrossRef]
- Grunig, G.; Warnock, M.; Wakil, A.E.; Venkayya, R.; Brombacher, F.; Rennick, D.M.; Sheppard, D.; Mohrs, M.; Donaldson, D.D.; Locksley, R.M.; et al. Requirement for IL-13 Independently of IL-4 in Experimental Asthma. Science 1998, 282, 2261–2263. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Severely Remodeled Pulmonary Artery (OVA-PM Exposed) | C57BL/6 | BALB/c | ||||||
|---|---|---|---|---|---|---|---|---|
| Median | Quartiles | Group n | p-Value | Median | Quartiles | Group n | p-Value | |
| Wild Type | 12.03 | 1.315, 50.460 | 8 | 0.1919 | 7.275 | 0.00, 10.390 | 10 | 0.0126 |
| RELMα−/− | 39.13 | 19.790, 41.800 | 9 | 0.000 | 0.00, 3.846 | 10 | ||
| Target | Gene Name | Sequence (5’ to 3’) |
|---|---|---|
| ANP-F 1 | nppa | TACAGTGCGGTGTCCAACACAG |
| ANP-R | nppa | TGCTTCCTCAGTCTGCTCACTC |
| BNP-F | nppb | TCCTAGCCAGTCTCCAGAGCAA |
| BNP-R | nppb | GGTCCTTCAAGAGCTGTCTCTG |
| IL-33-F | il33 | ACTGCATGAGACTCCGTTCTG |
| IL-33-R | il33 | CCTAGAATCCCGTGGATAGGC |
| ST2 F | il1rl1 | GGATTGAGGTTGCTCTGTTCTGG |
| ST2 R | il1rl1 | TCGGGCAGAGTGTGGTGAACAA |
| β-actin-F | actb | GGCTGTATTCCCCTCCATCG |
| β-actin-R | actb | CCAGTTGGTAACAATGCCATGT |
| RELMα (TaqMan) | retnla | CTTGCCAATCCAGCTAACTATCCCT |
| RELMβ (TaqMan) | retnlb | GGAAGCTCTCAGTCGTCAAGAGCCT |
| RELMγ (TaqMan) | retnlg | AAACCTGGCTCATATCCCATTGATG |
| Actin, β (TaqMan) | actb | ACTGAGCTGCGTTTTACACCCTTTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durmus, N.; Chen, W.-C.; Park, S.-H.; Marsh, L.M.; Kwon, S.; Nolan, A.; Grunig, G. Resistin-like Molecule α and Pulmonary Vascular Remodeling: A Multi-Strain Murine Model of Antigen and Urban Ambient Particulate Matter Co-Exposure. Int. J. Mol. Sci. 2023, 24, 11918. https://doi.org/10.3390/ijms241511918
Durmus N, Chen W-C, Park S-H, Marsh LM, Kwon S, Nolan A, Grunig G. Resistin-like Molecule α and Pulmonary Vascular Remodeling: A Multi-Strain Murine Model of Antigen and Urban Ambient Particulate Matter Co-Exposure. International Journal of Molecular Sciences. 2023; 24(15):11918. https://doi.org/10.3390/ijms241511918
Chicago/Turabian StyleDurmus, Nedim, Wen-Chi Chen, Sung-Hyun Park, Leigh M. Marsh, Sophia Kwon, Anna Nolan, and Gabriele Grunig. 2023. "Resistin-like Molecule α and Pulmonary Vascular Remodeling: A Multi-Strain Murine Model of Antigen and Urban Ambient Particulate Matter Co-Exposure" International Journal of Molecular Sciences 24, no. 15: 11918. https://doi.org/10.3390/ijms241511918
APA StyleDurmus, N., Chen, W.-C., Park, S.-H., Marsh, L. M., Kwon, S., Nolan, A., & Grunig, G. (2023). Resistin-like Molecule α and Pulmonary Vascular Remodeling: A Multi-Strain Murine Model of Antigen and Urban Ambient Particulate Matter Co-Exposure. International Journal of Molecular Sciences, 24(15), 11918. https://doi.org/10.3390/ijms241511918