
The Electronic Effects of 3-Methoxycarbonylcoumarin Substituents on Spectral, Antioxidant, and Protein Binding Properties

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
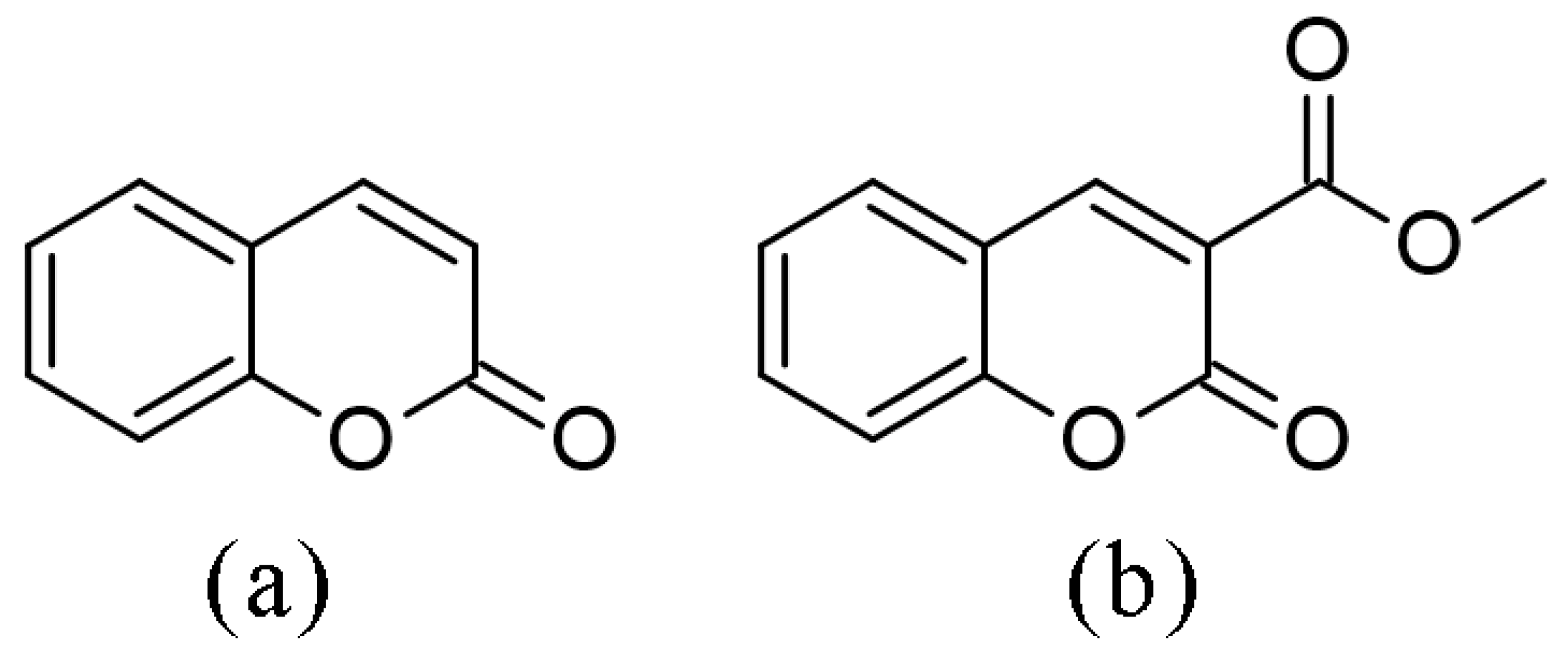
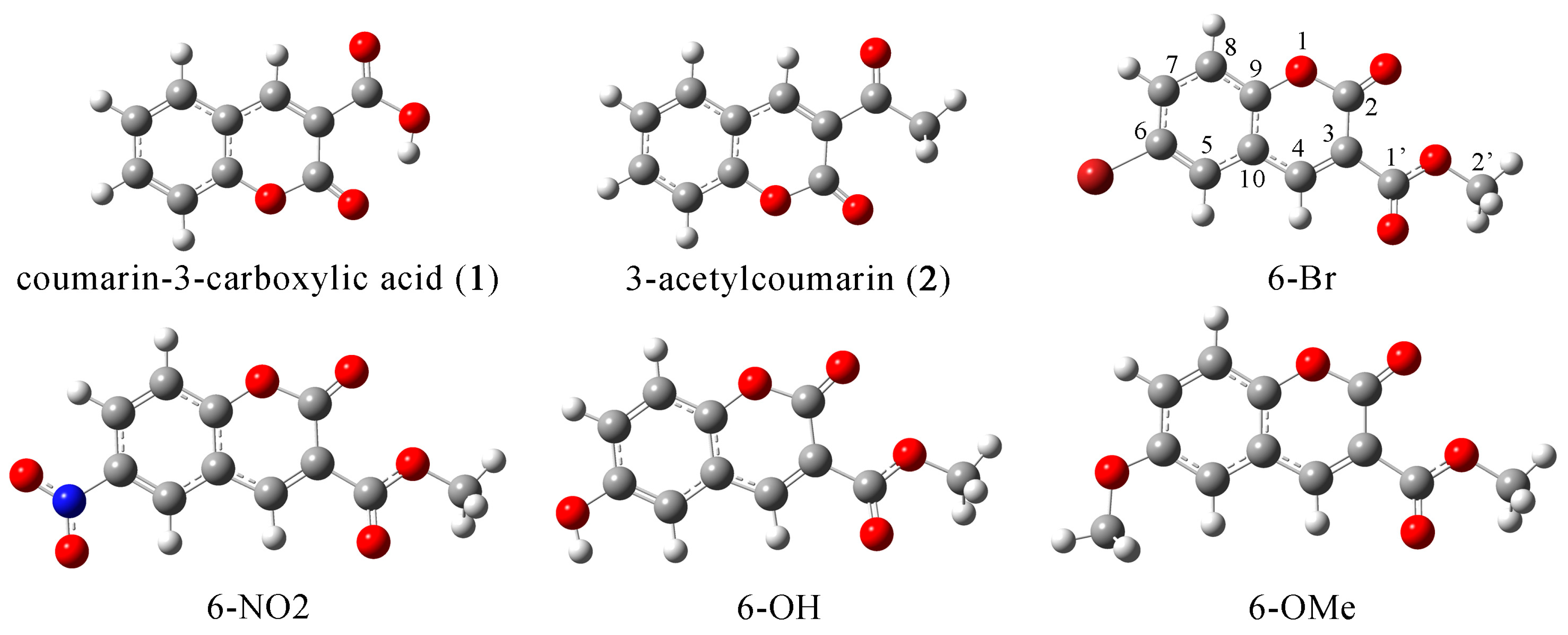
2.1. Structural Optimization, NBO, and QTAIM Analysis
2.2. Experimental and Theoretical NMR Spectra
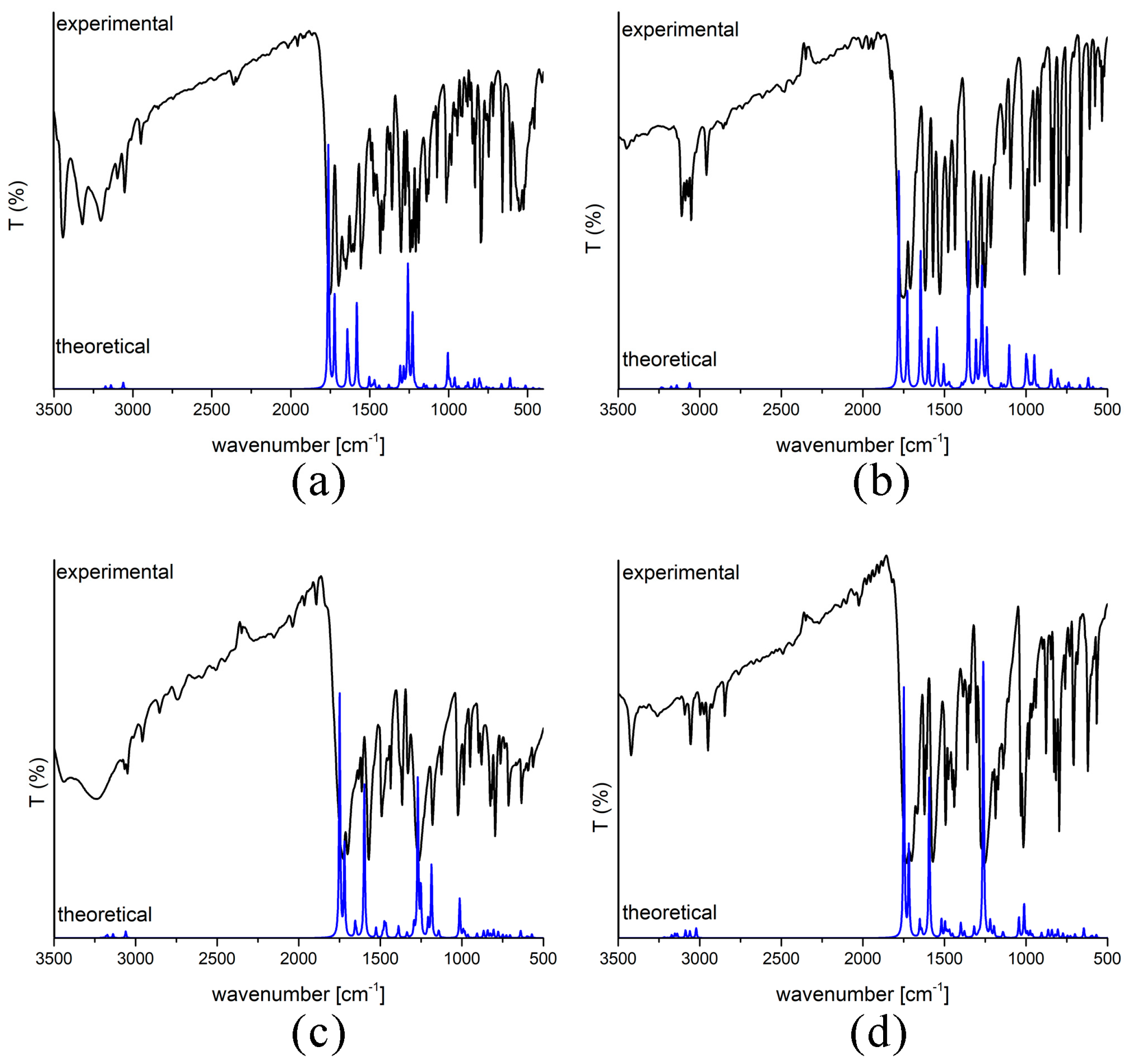
2.3. Experimental and Theoretical IR Spectra
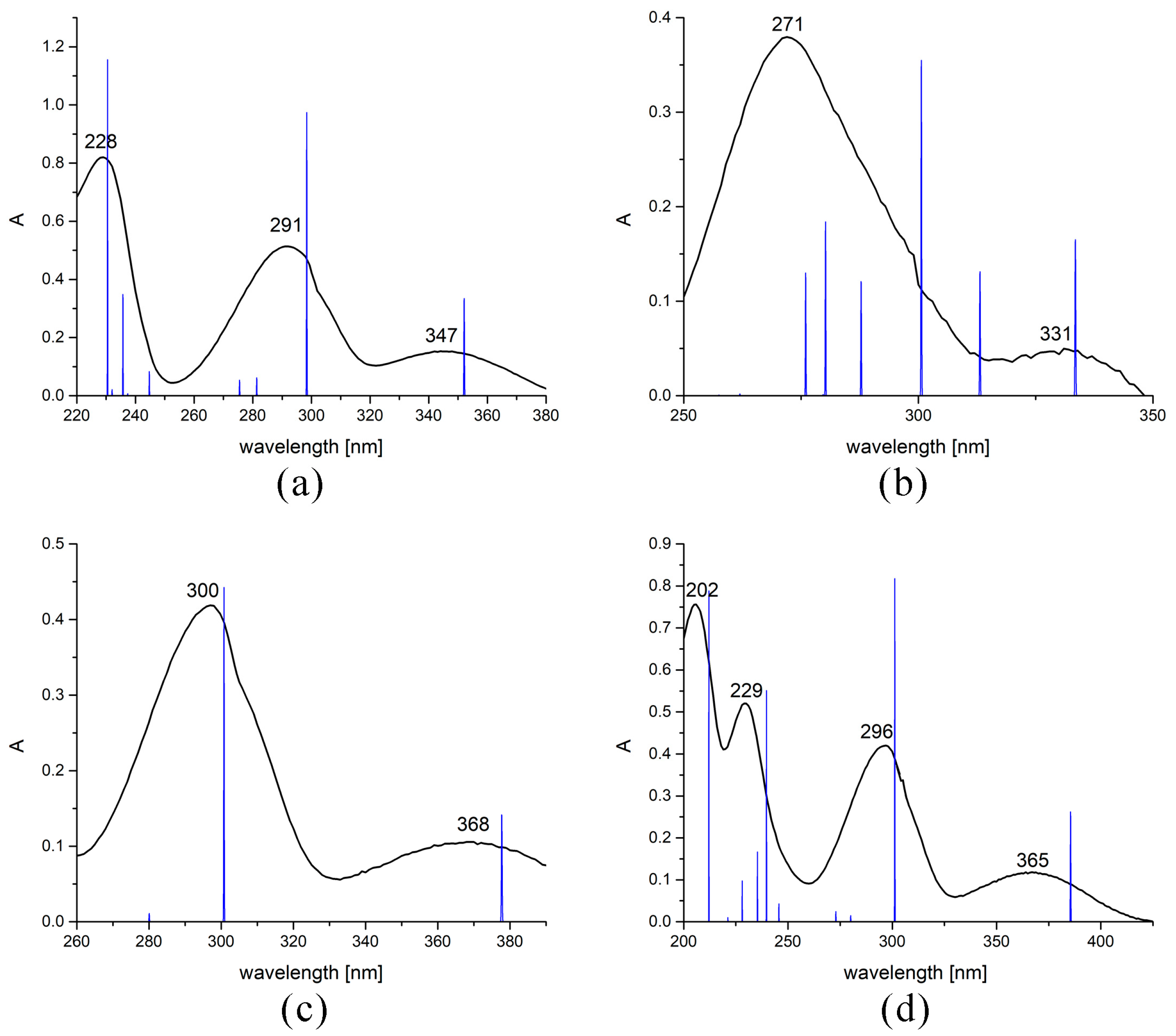
2.4. Experimental and Theoretical UV-Vis Spectra
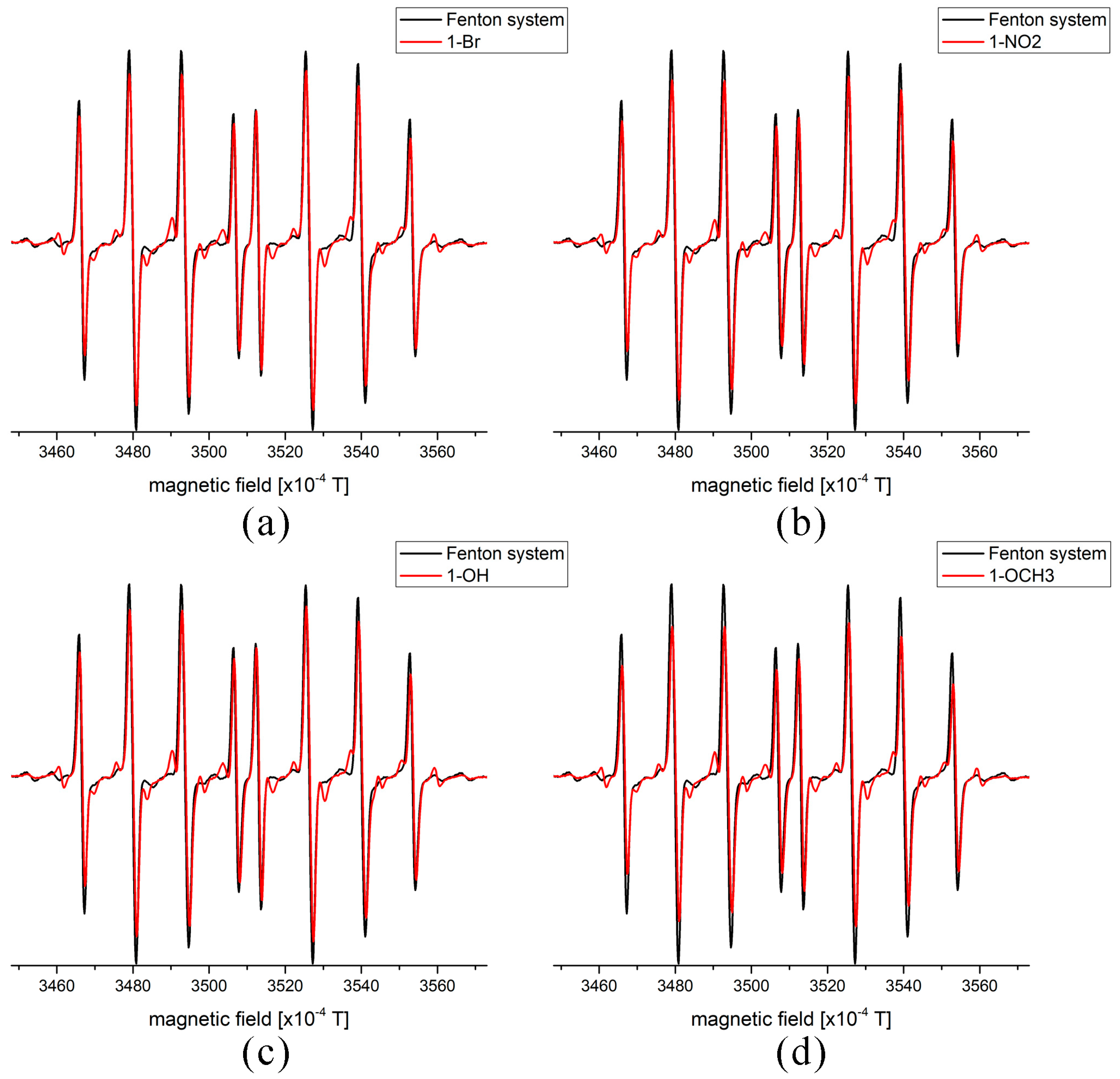
2.5. EPR Measurements of Antioxidant Activity
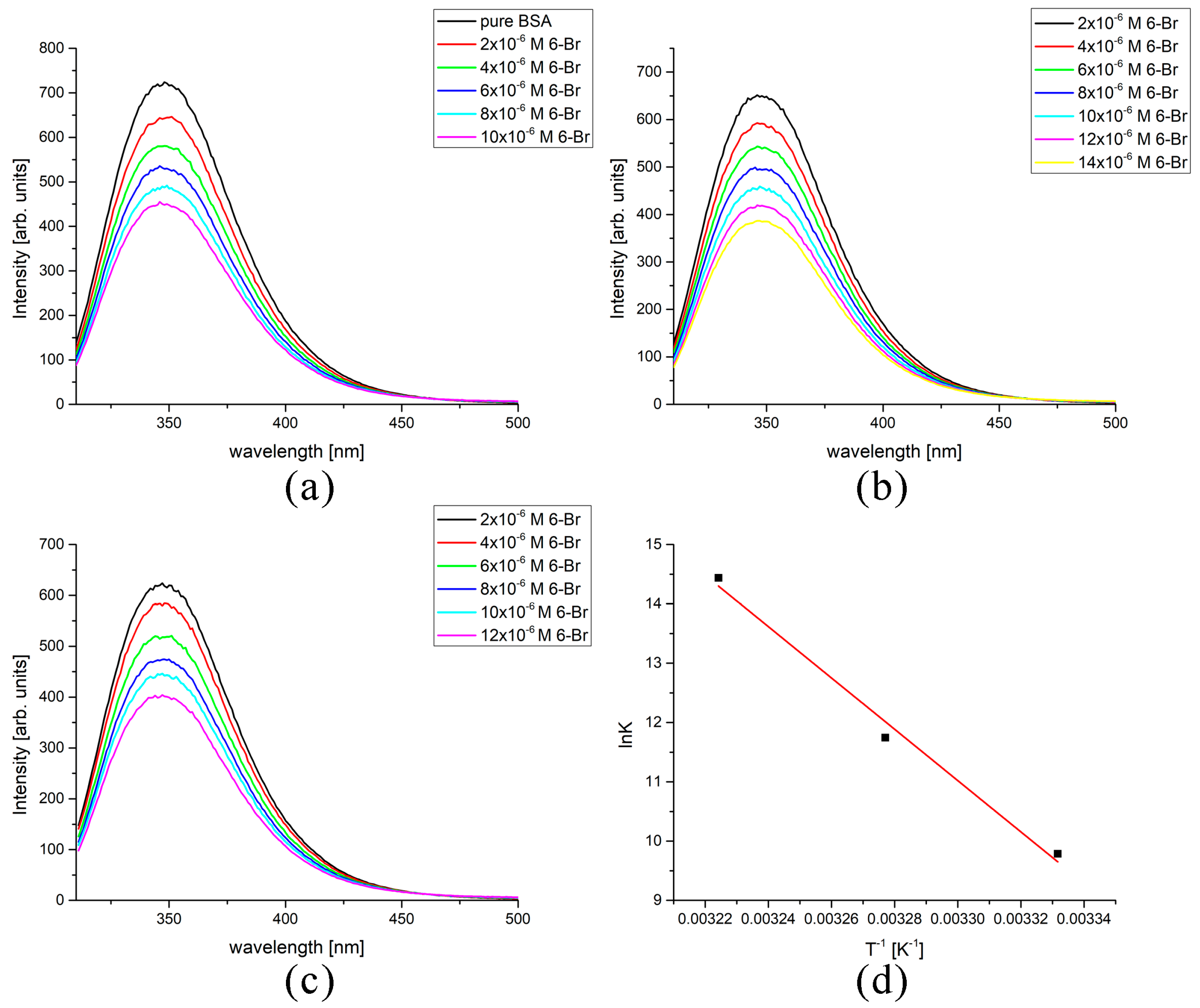
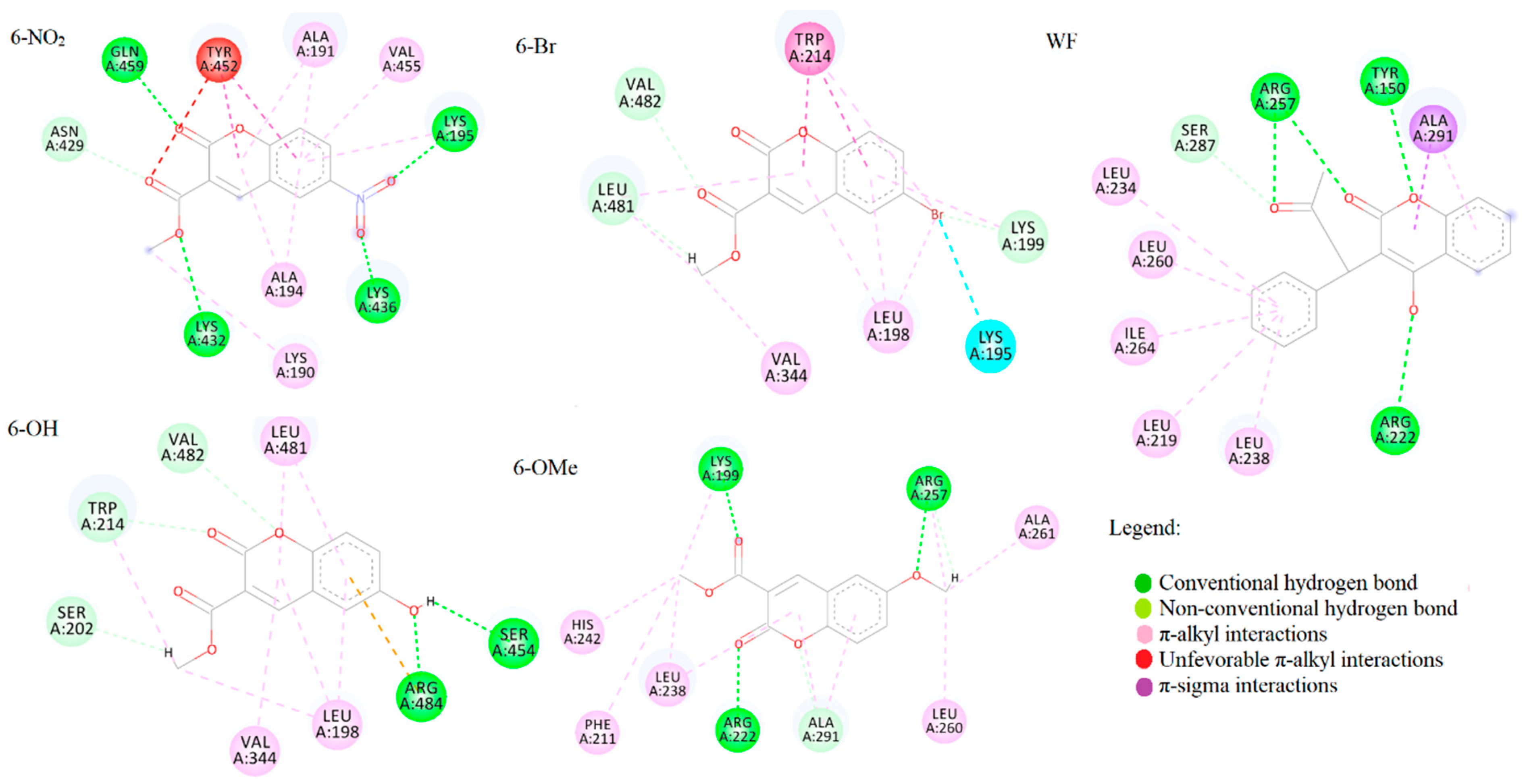
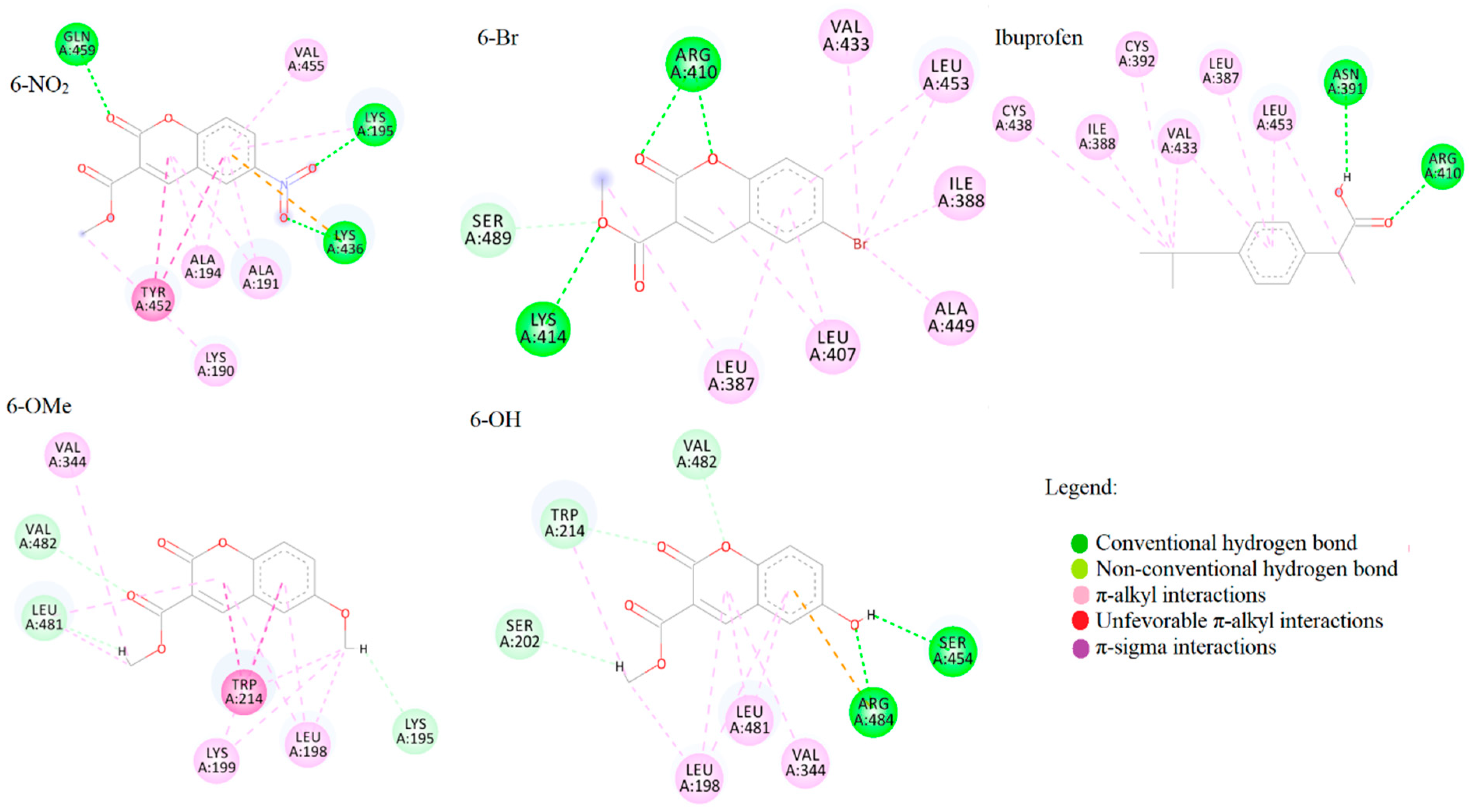
2.6. Spectrofluorometric and Molecular Docking Investigation of Binding to BSA
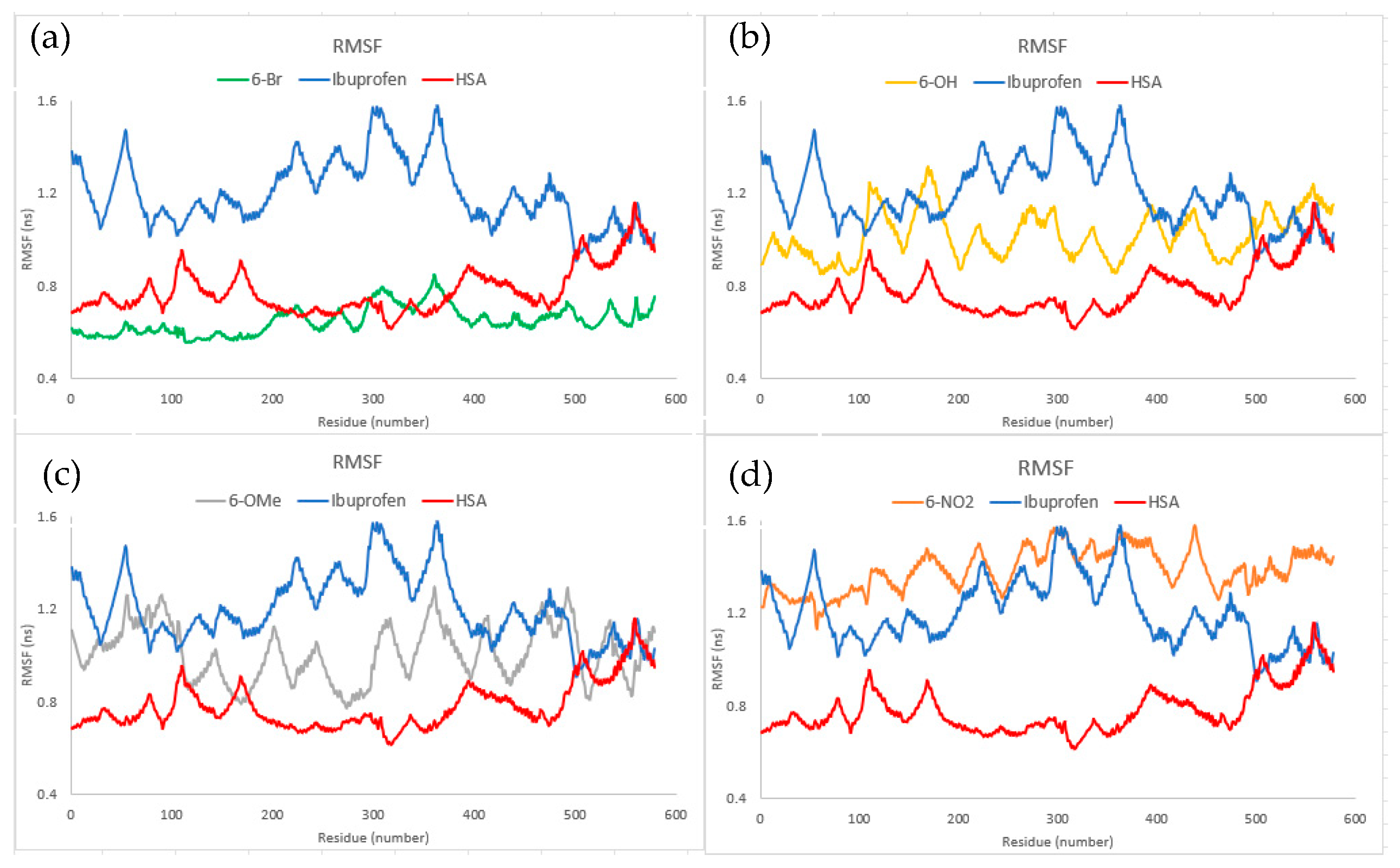
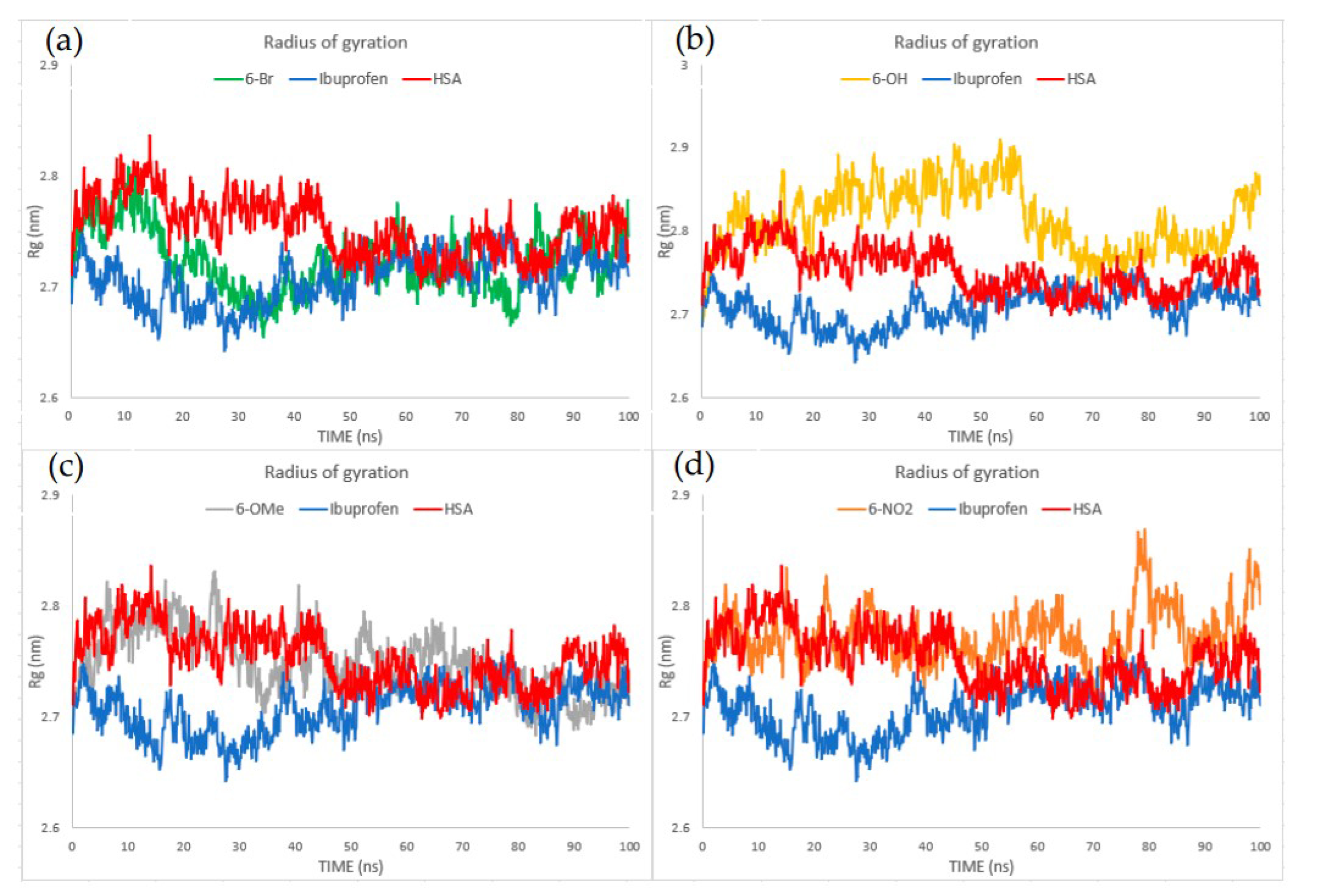
2.7. Molecular Dynamics
3. Materials and Methods
3.1. Chemicals
3.2. Spectroscopic Analysis
3.3. Spectrofluorimetric Analysis of BSA Protein Binding
3.4. Electron Paramagnetic Resonance Spectroscopy (EPR) Analysis of Radical Scavenging Activity
3.5. Theoretical Methods
3.6. Molecular Docking Analysis
3.7. Molecular Dynamics Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abernethy, J.L. The historical and current interest in coumarin. J. Chem. Educ. 1969, 46, 561. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Rashmi, V.; Odhav, B. Review on natural coumarin lead compounds for their pharmacological activity. Biomed. Res. Int. 2013, 2013, 963248. [Google Scholar] [CrossRef]
- Hatano, T.; Yasuhara, T.; Fukuda, T.; Noro, T.; Okuda, T. Phenolic constituents of licorice. II. Structures of licopyranocoumarin, licoarylcoumarin and glisoflavone, and inhibitory effects of licorice phenolics on xanthine oxidase. Chem. Pharm. Bull. 1989, 37, 3005–3009. [Google Scholar] [CrossRef] [PubMed]
- Häser, K.; Wenk, H.H.; Schwab, W. Biocatalytic production of dihydrocoumarin from coumarin by Saccharomyces cerevisiae. J. Agric. Food Chem. 2006, 54, 6236–6240. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.; O’kennedy, R.; Moran, E.; Cox, D.; Prosser, E.; Thornes, R.D. The pharmacology, metabolism, analysis, and applications of coumarin and coumarin-related compounds. Drug Metab. Rev. 1990, 22, 503–529. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An overview of coumarin as a versatile and readily accessible scaffold with broad-ranging biological activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, Y.; Li, G.; Chen, L.; Nie, M.; Wang, Z.; Ji, H. Coumarin sulfonamides and amides derivatives: Design, synthesis, and antitumor activity in vitro. Molecules 2021, 26, 786. [Google Scholar] [CrossRef]
- Wei, J.-N.; Jia, Z.-D.; Zhou, Y.-Q.; Chen, P.-H.; Li, B.; Zhang, N.; Hao, X.-Q.; Xu, Y.; Zhang, B. Synthesis, characterization and antitumor activity of novel ferrocene-coumarin conjugates. J. Organomet. Chem. 2019, 902, 120968. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, H.-R.; Liu, H.-S.; Cheng, M.; Xia, P.; Qian, K.; Wu, P.-C.; Lai, C.-Y.; Xia, Y.; Yang, Z.-Y.; et al. Antitumor agents 292. Design, synthesis and pharmacological study of S- and O-substituted 7-mercapto- or hydroxy-coumarins and chromones as potent cytotoxic agents. Eur. J. Med. Chem. 2012, 49, 74–85. [Google Scholar] [CrossRef]
- Dimić, D.S.; Marković, Z.S.; Saso, L.; Avdović, E.H.; Đorović, J.R.; Petrović, I.P.; Stanisavljević, D.; Stevanović, M.J.; Potočňák, I.; Samoľová, E.; et al. Synthesis and characterization of 3-(1-((3,4-dihydroxyphenethyl)amino)ethylidene)-chroman-2,4-dione as potentional anti-tumor agent. Oxid. Med. Cell. Longev. 2019, 2019, 2069250. [Google Scholar] [CrossRef]
- Marshall, M.E.; Mendelsohn, L.; Butler, K.; Riley, L.; Cantrell, J.; Wiseman, C.; Taylor, R.; Macdonald, J.S. Treatment of metastatic renal cell carcinoma with coumarin (1,2-benzopyrone) and cimetidine: A pilot study. J. Clin. Oncol. 1987, 5, 862–866. [Google Scholar] [CrossRef]
- Avdović, E.H.; Milenković, D.; Dimitrić Marković, J.M.; Đorović, J.; Vuković, N.; Vukić, M.D.; Jevtić, V.V.; Trifunović, S.R.; Potočňák, I.; Marković, Z. Synthesis, spectroscopic characterization (FT-IR, FT-Raman, and NMR), quantum chemical studies and molecular docking of 3-(1-(phenylamino)ethylidene)-chroman-2,4-dione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 195, 31–40. [Google Scholar] [CrossRef]
- Patil, S.A.; Unki, S.N.; Badami, P.S. In vitro antibacterial, antifungal, and DNA cleavage studies of coumarin Schiff bases and their metal complexes: Synthesis and spectral characterization. Med. Chem. Res. 2012, 21, 4017–4027. [Google Scholar] [CrossRef]
- Refat, M.S.; El-Deen, I.M.; Anwer, Z.M.; El-Ghol, S. Bivalent transition metal complexes of coumarin-3-yl thiosemicarbazone derivatives: Spectroscopic, antibacterial activity and thermogravimetric studies. J. Mol. Struct. 2009, 920, 149–162. [Google Scholar] [CrossRef]
- Kontogiorgis, C.A.; Hadjipavlou-Litina, D.J. Synthesis and anti-inflammatory activity of coumarin derivatives. Proc. J. Med. Chem. 2005, 48, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Kontogiorgis, C.; Hadjipavlou-Litina, D. Biological evaluation of several coumarin derivatives designed as possible anti-inflammatory/antioxidant agents. J. Enzyme Inhib. Med. Chem. 2003, 18, 63–69. [Google Scholar] [CrossRef]
- Skalicka-Woźniak, K.; Orhan, I.E.; Cordell, G.A.; Nabavi, S.M.; Budzyńska, B. Implication of coumarins towards central nervous system disorders. Pharmacol. Res. 2016, 103, 188–203. [Google Scholar] [CrossRef]
- Hoult, J.R.S.; Payá, M. Pharmacological and biochemical actions of simple coumarins: Natural products with therapeutic potential. Gen. Pharmacol. Vasc. Syst. 1996, 27, 713–722. [Google Scholar] [CrossRef]
- Kontogiorgis, C.A.; Savvoglou, K.; Hadjipavlou-Litina, D.J. Anti-inflammatory and antioxidant evaluation of novel coumarin derivatives. J. Enzyme Inhib. Med. Chem. 2006, 21, 21–29. [Google Scholar] [CrossRef]
- Kostova, I.; Bhatia, S.; Grigorov, P.; Balkansky, S.; Parmar, S.V.; Prasad, A.K.; Saso, L. Coumarins as Antioxidants. Curr. Med. Chem. 2011, 18, 3929–3951. [Google Scholar] [CrossRef] [PubMed]
- Küpeli Akkol, E.; Genç, Y.; Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Zhu, J.; Jiang, J. Pharmacological and nutritional effects of natural coumarins and their structure—Activity relationships. Mol. Nutr. Food Res. 2018, 62, 1701073. [Google Scholar] [CrossRef] [PubMed]
- Musawwir, A.; Farhat, A.; Khera, R.A.; Ayub, A.R.; Iqbal, J. Theoretical and computational study on electronic effect caused by electron withdrawing/electron-donating groups upon the coumarin thiourea derivatives. Comput. Theor. Chem. 2021, 1201, 113271. [Google Scholar] [CrossRef]
- Cisse, L.; Djande, A.; Capo-Chichi, M.; Khonté, A.; Bakhoum, J.; Delattre, F.; Yoda, J.; Saba, A.; Tine, A.; Aaron, J. Quantitative study of the substituent effects on the electronic absorption and fluorescence spectra of coumarins. J. Phys. Org. Chem. 2020, 33, e4014. [Google Scholar] [CrossRef]
- Vrakas, D.; Panderi, I.; Hadjipavlou-Litina, D.; Tsantili-Kakoulidou, A. Investigation of the relationships between logP and various chromatographic indices for a series of substituted coumarins. Evaluation of their similarity/dissimilarity using multivariate statistics. QSAR Comb. Sci. 2005, 24, 254–260. [Google Scholar] [CrossRef]
- Vrakas, D.; Tsantili-Kakoulidou, A.; Hadjipavlou-Litina, D. Exploring the consistency of logP estimation for substituted coumarins. QSAR Comb. Sci. 2003, 22, 622–629. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef]
- Avdović, E.H.; Milanović, Ž.B.; Živanović, M.N.; Šeklić, D.S.; Radojević, I.D.; Čomić, L.R.; Trifunović, S.R.; Amić, A.; Marković, Z.S. Synthesis, spectroscopic characterization, biological activity, DFT and molecular docking study of novel 4-hydroxycoumarine derivatives and corresponding palladium(II) complexes. Inorganica Chim. Acta 2020, 504, 119465. [Google Scholar] [CrossRef]
- Milanovic, Ž.B.; Markovic, Z.S.; Dimic, D.S.; Klisuric, O.R.; Radojevic, I.D.; Šeklic, D.S.; Živanovic, M.N.; Markovic, J.D.; Radulovic, M.; Avdovic, E.H. Synthesis, structural characterization, biological activity and molecular docking study of 4,7-dihydroxycoumarinmodified by aminophenol derivatives. Comptes Rendus Chim. 2021, 24, 215–232. [Google Scholar] [CrossRef]
- Milenković, D.; Avdović, E.; Dimić, D.; Sudha, S.; Ramarajan, D.; Milanović, Ž.; Trifunović, S.; Marković, Z.S. Vibrational and Hirshfeld surface analyses, quantum chemical calculations, and molecular docking studies of coumarin derivative 3-(1-m-toluidinoethylidene)-chromane-2,4-dione and its corresponding palladium(II) complex. J. Mol. Struct. 2020, 1209, 127935. [Google Scholar] [CrossRef]
- Avdović, E.H.; Milanović, Ž.B.; Molčanov, K.; Roca, S.; Vikić-Topić, D.; Mrkalić, E.M.; Jelić, R.M.; Marković, Z.S. Synthesis, characterization and investigating the binding mechanism of novel coumarin derivatives with human serum albumin: Spectroscopic and computational approach. J. Mol. Struct. 2022, 1254, 132366. [Google Scholar] [CrossRef]
- Avdović, E.H.; Antonijević, M.; Simijonović, D.; Roca, S.; Topić, D.V.; Grozdanić, N.; Stanojković, T.; Radojević, I.; Vojinović, R.; Marković, Z. Synthesis and Cytotoxicity Evaluation of Novel Coumarin–Palladium(II) Complexes against Human Cancer Cell Lines. Pharmaceuticals 2022, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.; Ramesh, G.; Naik, J.L.; Ojha, J.K.; Reddy, B.V.; Rao, G.R. Molecular Structure, Vibrational Analysis and First Order Hyperpolarizability of 4-Methyl-3-Nitrobenzoic Acid Using Density Functional Theory. Opt. Photonics J. 2015, 5, 91–107. [Google Scholar] [CrossRef]
- Clarkson, J.; Smith, W.E.; Batchelder, D.N.; Smith, D.A.; Coats, A.M. A theoretical study of the structure and vibrations of 2,4,6-trinitrotolune. J. Mol. Struct. 2003, 648, 203–214. [Google Scholar] [CrossRef]
- Avdović, E.H.; Milenković, D.; Dimitrić-Marković, J.M.; Vuković, N.; Trifunović, S.R.; Marković, Z. Structural, spectral and NBO analysis of 3-(1-(3-hydroxypropylamino)ethylidene)chroman-2,4-dione. J. Mol. Struct. 2017, 1147, 69–75. [Google Scholar] [CrossRef]
- Murata, C.; Masuda, T.; Kamochi, Y.; Todoroki, K.; Yoshida, H.; Nohta, H.; Yamaguchi, M.; Takadate, A. Improvement of fluorescence characteristics of coumarins: Syntheses and fluorescence properties of 6-methoxycoumarin and benzocoumarin ferivatives as novel fluorophores emitting in the longer wavelength region and their application to analytical reagents. Chem. Pharm. Bull. 2005, 53, 750–758. [Google Scholar] [CrossRef]
- Dimić, D.S.; Milenković, D.A.; Avdović, E.H.; Nakarada, Đ.J.; Dimitrić Marković, J.M.; Marković, Z.S. Advanced oxidation processes of coumarins by hydroperoxyl radical: An experimental and theoretical study, and ecotoxicology assessment. Chem. Eng. J. 2021, 424, 130331. [Google Scholar] [CrossRef]
- Milenković, D.A.; Dimić, D.S.; Avdović, E.H.; Amić, A.D.; Dimitrić Marković, J.M.; Marković, Z.S. Advanced oxidation process of coumarins by hydroxyl radical: Towards the new mechanism leading to less toxic products. Chem. Eng. J. 2020, 395, 124971. [Google Scholar] [CrossRef]
- Milanović, Ž.; Dimić, D.; Antonijević, M.; Žižić, M.; Milenković, D.; Avdović, E.; Marković, Z. Influence of acid-base equilibria on the rate of the chemical reaction in the advanced oxidation processes: Coumarin derivatives and hydroxyl radical. Chem. Eng. J. 2023, 453, 139648. [Google Scholar] [CrossRef]
- Dimitrić Marković, J.M.; Milenković, D.; Amić, D.; Mojović, M.; Pašti, I.; Marković, Z.S. The preferred radical scavenging mechanisms of fisetin and baicalein towards oxygen-centred radicals in polar protic and polar aprotic solvents. RSC Adv. 2014, 4, 32228–32236. [Google Scholar] [CrossRef]
- Lončarić, M.; Sušjenka, M.; Molnar, M. An Extensive study of coumarin synthesis via Knoevenagel condensation in choline chloride based deep eutectic solvents. Curr. Org. Synth. 2020, 17, 98–108. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density functional with dpherical atom dispersion terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Roy, D.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Zieliński, R.; Szymusiak, H. Application of Dft B3Lyp/Giao and B3Lyp/Csgt Methods for interpretation of nmr Spectra of Flavonoids. Pol. J. Food Nutr. Sci. 2003, 12, 157–162. [Google Scholar]
- Bohmann, J.A.; Weinhold, F.; Farrar, T.C. Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge-including atomic orbital calculations. J. Chem. Phys. 1997, 107, 1173. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Keith, T.A. TK Gristmill Software; (Version 19.10.12); AIMAll: Overland Park, KS, USA, 2019. [Google Scholar]
- Dimic, D.; Petkovic, M. Control of a photoswitching chelator by metal ions: DFT, NBO, and QTAIM analysis. Int. J. Quantum Chem. 2016, 116, 27–34. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural Basis of the Drug-binding Specificity of Human Serum Albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systèmes, Discovery Studio 2016; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Milanović, Ž.B.; Antonijević, M.R.; Amić, A.D.; Avdović, E.H.; Dimić, D.S.; Milenković, D.A.; Marković, Z.S. Inhibitory activity of quercetin, its metabolite, and standard antiviral drugs towards enzymes essential for SARS-CoV-2: The role of acid–base equilibria. RSC Adv. 2021, 11, 2838–2847. [Google Scholar] [CrossRef]
- Amić, A.; Dimitrić Marković, J.M.; Marković, Z.; Milenković, D.; Milanović, Ž.; Antonijević, M.; Mastiľák Cagardová, D.; Rodríguez-Guerra Pedregal, J. Theoretical Study of Radical Inactivation, LOX Inhibition, and Iron Chelation: The Role of Ferulic Acid in Skin Protection against UVA Induced Oxidative Stress. Antioxidants 2021, 10, 1303. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jovanović, J.Đ.; Manojlović, N.; Marković, Z. Usnic Acid as a Potential Free Radical Scavenger and its Inhibitory Activity Toward SARS-CoV-2 Proteins. J. Comput. Biophys. Chem. 2021, 20, 655–666. [Google Scholar] [CrossRef]
- Jovanović, J.Đ.; Antonijević, M.; Vojinović, R.; Filipović, N.D.; Marković, Z. In silico study of inhibitory capacity of sacubitril/valsartan toward neprilysin and angiotensin receptor. RSC Adv. 2022, 12, 29719–29726. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | ||||||
|---|---|---|---|---|---|---|---|
| APFD | B3LYP-D3BJ | M06-2X | APFD | B3LYP-D3BJ | M06-2X | ||
| Bond lengths | R | 0.982 | 0.981 | 0.981 | 0.995 | 0.996 | 0.998 |
| MAE [Å] | 0.014 | 0.015 | 0.014 | 0.007 | 0.006 | 0.007 | |
| Bond angles | R | 0.945 | 0.943 | 0.936 | 0.982 | 0.983 | 0.979 |
| MAE [o] | 1.05 | 1.01 | 1.15 | 0.46 | 0.43 | 0.48 | |
| 1H Chemical Shifts [ppm] | 13C Chemical Shifts [ppm] | ||||
|---|---|---|---|---|---|
| H Atom | Experimental | Theoretical | C Atom | Experimental | Theoretical |
| C2′-H | 3.85 | 3.88 | C2’ | 53 | 55 |
| C8-H | 7.40 | 7.46 | C3 | 117 | 118 |
| C7-H | 7.90 | 7.84 | C8 | 119 | 119 |
| C5-H | 8.20 | 7.95 | C10 | 120 | 121 |
| C4-H | 8.70 | 8.82 | C5 | 132 | 133 |
| R | 0.999 | C6 | 137 | 137 | |
| MAE [ppm] | 0.10 | C7 | 148 | 140 | |
| C4 | 154 | 152 | |||
| C2 | 156 | 157 | |||
| C9 | 160 | 157 | |||
| C1’ | 163 | 167 | |||
| R | 0.995 | ||||
| MAE [ppm] | 2.0 | ||||
| Derivative | T [K] | lnK | R | ΔH [kJ mol−1] | ΔS [kJ mol−1 K−1] | ΔG [kJ mol−1] |
|---|---|---|---|---|---|---|
| 6-Br | 300 | 9.78 | 0.98 | 360 | 1278 | −22.9 |
| 305 | 11.74 | 0.98 | −30.3 | |||
| 310 | 14.44 | 0.99 | −36.7 | |||
| 6-NO2 | 300 | 9.03 | 0.99 | 556 | 1924 | −21.8 |
| 305 | 12.16 | 0.99 | −31.4 | |||
| 310 | 16.21 | 0.99 | −41.0 | |||
| 6-OH | 300 | 5.39 | 0.99 | −254 | −740 | −32.0 |
| 305 | 4.84 | 0.98 | −28.3 | |||
| 310 | 3.96 | 0.99 | −24.6 | |||
| 6-OMe | 300 | 12.62 | 0.99 | −236 | −685 | −30.5 |
| 305 | 9.95 | 0.98 | −27.1 | |||
| 310 | 9.58 | 0.98 | −23.7 |
| Compound | Sudlow I | Sudlow II | ||
|---|---|---|---|---|
| ΔGbind | Ki | ΔGbind | Ki | |
| 6-Br | −28.5 | 9.87 | −30.1 | 5.18 |
| 6-NO2 | −31.4 | 3.17 | −30.9 | 3.85 |
| 6-OH | −28.8 | 9.06 | −28.7 | 9.19 |
| 6-OMe | −26.3 | 24.27 | −28.1 | 11.59 |
| Warfarin/Ibuprofen | −34.3 | 0.95 | −28.3 | 10.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasić, J.; Dimić, D.; Antonijević, M.; Avdović, E.H.; Milenković, D.; Nakarada, Đ.; Dimitrić Marković, J.; Molnar, M.; Lončarić, M.; Bešlo, D.; et al. The Electronic Effects of 3-Methoxycarbonylcoumarin Substituents on Spectral, Antioxidant, and Protein Binding Properties. Int. J. Mol. Sci. 2023, 24, 11820. https://doi.org/10.3390/ijms241411820
Vasić J, Dimić D, Antonijević M, Avdović EH, Milenković D, Nakarada Đ, Dimitrić Marković J, Molnar M, Lončarić M, Bešlo D, et al. The Electronic Effects of 3-Methoxycarbonylcoumarin Substituents on Spectral, Antioxidant, and Protein Binding Properties. International Journal of Molecular Sciences. 2023; 24(14):11820. https://doi.org/10.3390/ijms241411820
Chicago/Turabian StyleVasić, Jelena, Dušan Dimić, Marko Antonijević, Edina H. Avdović, Dejan Milenković, Đura Nakarada, Jasmina Dimitrić Marković, Maja Molnar, Melita Lončarić, Drago Bešlo, and et al. 2023. "The Electronic Effects of 3-Methoxycarbonylcoumarin Substituents on Spectral, Antioxidant, and Protein Binding Properties" International Journal of Molecular Sciences 24, no. 14: 11820. https://doi.org/10.3390/ijms241411820
APA StyleVasić, J., Dimić, D., Antonijević, M., Avdović, E. H., Milenković, D., Nakarada, Đ., Dimitrić Marković, J., Molnar, M., Lončarić, M., Bešlo, D., & Marković, Z. (2023). The Electronic Effects of 3-Methoxycarbonylcoumarin Substituents on Spectral, Antioxidant, and Protein Binding Properties. International Journal of Molecular Sciences, 24(14), 11820. https://doi.org/10.3390/ijms241411820