
Building Up a Piperazine Ring from a Primary Amino Group via Catalytic Reductive Cyclization of Dioximes


Abstract
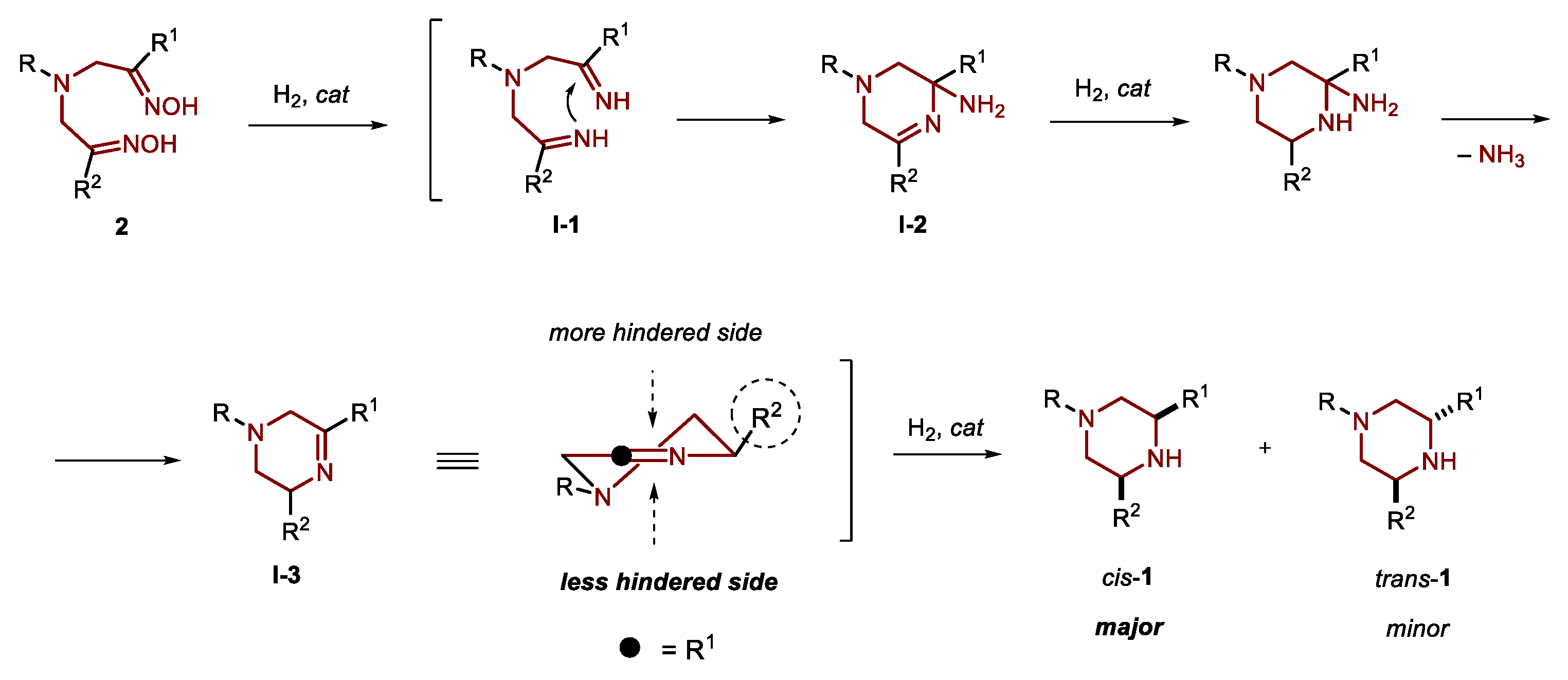
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedures
3.3. Characterization of Final Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.; Shin, H.-S.; Patel, R.V. Piperazine derivatives for therapeutic use: A patent review (2010-present). Expert Opin. Ther. Pat. 2016, 26, 777–797. [Google Scholar] [CrossRef] [PubMed]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, S.; Yu, L.; Wang, Y.; Chi, Y.R.; Wu, J. Piperazine: Its role in the discovery of pesticides. Chin. Chem. Lett. 2023, 34, 108123. [Google Scholar] [CrossRef]
- Kant, R.; Maji, S. Recent advances in the synthesis of piperazine based ligands and metal complexes and their applications. Dalton Trans. 2021, 50, 785–800. [Google Scholar] [CrossRef]
- Park, D.J.; Choi, J.H.; Kim, Y.E.; Nam, S.C.; Lee, K.B.; Yoon, Y.I. Chemical Absorption of Carbon Dioxide Using Aqueous Piperidine Derivatives. Chem. Eng. Technol. 2017, 40, 2266–2273. [Google Scholar] [CrossRef]
- Guo, W.; Fu, X.; Chen, J. Supramolecular adducts of mesocyclic diamines with various carboxylic acids: Charge-assisted hydrogen-bonding in molecular recognition. J. Saudi Chem. Soc. 2020, 24, 885–895. [Google Scholar] [CrossRef]
- Lesnikov, V.K.; Nelyubina, Y.V.; Sukhorukov, A.Y. Piperazine-1,4-diol (PipzDiol): Synthesis, stereodynamics and assembly of supramolecular hydrogen-bonded 2D networks. New J. Chem. 2022, 46, 20386–20394. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Zeng, G.; Yang, S.; Liao, X.; Sun, D. Antibacterial activity and mechanism of piperazine polymer. J. Appl. Polym. Sci. 2021, 138, 50451. [Google Scholar] [CrossRef]
- Jalageri, M.D.; Nagaraja, A.; Puttaiahgowda, Y.M. Piperazine based antimicrobial polymers: A review. RSC Adv. 2021, 11, 15213–15230. [Google Scholar] [CrossRef]
- Gettys, K.E.; Ye, Z.; Dai, M. Recent Advances in Piperazine Synthesis. Synthesis 2017, 49, 2589–2604. [Google Scholar] [CrossRef]
- Magriotis, P.A. Recent progress toward the asymmetric synthesis of carbon-substituted piperazine pharmacophores and oxidative related heterocycles. RSC Med. Chem. 2020, 11, 745–759. [Google Scholar] [CrossRef]
- Nishiyama, M.; Yamamoto, T.; Koie, Y. Synthesis of N-arylpiperazines from aryl halides and piperazine under a palladium tri-tert-butylphosphine catalyst. Tetrahedron Lett. 1998, 39, 617–620. [Google Scholar] [CrossRef]
- Thamban Chandrika, N.; Shrestha, S.K.; Ngo, H.X.; Tsodikov, O.V.; Howard, K.C.; Garneau-Tsodikova, S. Alkylated Piperazines and Piperazine-Azole Hybrids as Antifungal Agents. J. Med. Chem. 2018, 61, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fitzgerald, A.E.; Mani, N.S. Reductive Amination by Continuous-Flow Hydrogenation: Direct and Scalable Synthesis of a Benzylpiperazine. Synthesis 2012, 44, 2469–2473. [Google Scholar] [CrossRef]
- Srinivas, V.; Mohan, C.D.; Baburajeev, C.P.; Rangappa, S.; Jagadish, S.; Fuchs, J.E.; Sukhorukov, A.Y.; Chandra; Mason, D.J.; Sharath Kumar, K.S.; et al. Synthesis and characterization of novel oxazines and demonstration that they specifically target cyclooxygenase 2. Bioorg. Med. Chem. Lett. 2015, 25, 2931–2936. [Google Scholar] [CrossRef]
- Durand, C.; Szostak, M. Recent Advances in the Synthesis of Piperazines: Focus on C–H Functionalization. Organics 2021, 2, 337–347. [Google Scholar] [CrossRef]
- Firth, J.D.; O’Brien, P.; Ferris, L. Synthesis of Enantiopure Piperazines via Asymmetric Lithiation–Trapping of N-Boc Piperazines: Unexpected Role of the Electrophile and Distal N-Substituent. J. Am. Chem. Soc. 2016, 138, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-X.; Liu, L.-J.; Wu, B.; Feng, G.-S.; Wang, B.; Zhou, Y.-G. Synthesis of Chiral Piperazines via Hydrogenation of Pyrazines Activated by Alkyl Halides. Org. Lett. 2016, 18, 3082–3085. [Google Scholar] [CrossRef] [PubMed]
- Trinchera, P.; Musio, B.; Degennaro, L.; Moliterni, A.; Falcicchio, A.; Luisi, R. One-pot preparation of piperazines by regioselective ring-opening of non-activated arylaziridines. Org. Biomol. Chem. 2012, 10, 1962–1965. [Google Scholar] [CrossRef]
- Halimehjani, A.Z.; Badali, E. DABCO bond cleavage for the synthesis of piperazine derivatives. RSC Adv. 2019, 9, 36386–36409. [Google Scholar] [CrossRef] [PubMed]
- Ruider, S.A.; Müller, S.; Carreira, E.M. Ring Expansion of 3-Oxetanone-Derived Spirocycles: Facile Synthesis of Saturated Nitrogen Heterocycles. Angew. Chem. Int. Ed. 2013, 52, 11908–11911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-H.; Guo, H.-Y.; Deng, H.; Li, J.; Quan, Z.-S. Piperazine skeleton in the structural modification of natural products: A review. J. Enzym. Inhib. Med. Chem. 2021, 36, 1165–1197. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Huh, K.T.; Ohsugi, Y.; Watanabe, Y. Ruthenium complex catalyzed N-heterocyclization. Syntheses of N-substituted piperidines, morpholines, and piperazines from amines and 1,5-diols. J. Org. Chem. 1985, 50, 1365–1370. [Google Scholar] [CrossRef]
- Huang, J.; Xu, W.; Xie, H.; Li, S. One-Step Cyclization: Synthesis of N-Heteroalkyl-N′-tosylpiperazines. J. Org. Chem. 2012, 77, 7506–7511. [Google Scholar] [CrossRef]
- Pospelov, E.V.; Boyko, Y.D.; Ioffe, S.L.; Sukhorukov, A.Y. Synthesis of Bis(β-Oximinoalkyl)malonates and Their Catalytic Reductive Cyclization to Piperidines. Adv. Synth. Catal. 2022, 364, 2557–2564. [Google Scholar] [CrossRef]
- Boyko, Y.D.; Dorokhov, V.S.; Sukhorukov, A.Y.; Ioffe, S.L. Conjugated nitrosoalkenes as Michael acceptors in carbon–carbon bond forming reactions: A review and perspective. Beilstein J. Org. Chem. 2017, 13, 2214–2234. [Google Scholar] [CrossRef]
- Lopes, S.M.M.; Cardoso, A.L.; Lemos, A.; Pinho e Melo, T.M.V.D. Recent Advances in the Chemistry of Conjugated Nitrosoalkenes and Azoalkenes. Chem. Rev. 2018, 118, 11324–11352. [Google Scholar] [CrossRef]
- Gilchrist, T. Nitroso-alkenes and Nitroso-alkynes. Chem. Soc. Rev. 1983, 12, 53–73. [Google Scholar] [CrossRef]
- Naumovich, Y.A.; Ioffe, S.L.; Sukhorukov, A.Y. Michael Addition of P-Nucleophiles to Conjugated Nitrosoalkenes. J. Org. Chem. 2019, 84, 7244–7254. [Google Scholar] [CrossRef]
- Feger, H.; Simchen, G. Reaktionen der Trialkylsilyl-trifluormethansulfonate, V. Synthese und Reaktionen von N,N-Bis(trialkylsiloxy)-1-alken-1-aminen. Liebigs Ann. Chem. 1986, 1986, 1456–1465. [Google Scholar] [CrossRef]
- Semakin, A.N.; Sukhorukov, A.Y.; Lesiv, A.V.; Khomutova, Y.A.; Ioffe, S.L.; Lyssenko, K.A. A Convenient Method for the Synthesis of Poly(β-hydroxyiminoalkyl)amines from Aliphatic Nitro Compounds. Synthesis 2007, 2007, 2862–2866. [Google Scholar] [CrossRef]
- Semakin, A.N.; Sukhorukov, A.Y.; Ioffe, S.L.; Tartakovsky, V.A. A General Procedure for the Synthesis of Unsymmetrically Substituted Tris(β-oximinoalkyl)amines. Synthesis 2011, 2011, 1403–1412. [Google Scholar]
- Makarenkova, L.M.; Bliznets, I.V.; Ioffe, S.L.; Strelenko, Y.A.; Tartakovsky, V.A. The chemistry of N,N-bis (trialkylsilyloxy) enamines. 2. Alkylation of primary amines with N,N-bis(trimethylsilyloxy) enamines. Russ. Chem. Bull. 2000, 49, 1261–1269. [Google Scholar] [CrossRef]
- Fraser, R.R.; Bresse, M. The effect of stereochemistry on 1JC—H at the sp2 carbons of oxime, hydrazone, and imine derivatives of aldehydes. Can. J. Chem. 1983, 61, 576–578. [Google Scholar] [CrossRef]
- Redina, E.A.; Ivanova, I.I.; Arkhipova, N.Y.; Kustov, L.M. Heterogeneous Catalysis as an Efficient Tool for Selective Hydrogenation of Oximes to Amines and Hydroxylamines. Catalysts 2022, 12, 1614. [Google Scholar] [CrossRef]
- Nagaraj, M.; Boominathan, M.; Muthusubramanian, S.; Bhuvanesh, N. Synthesis of novel N-hydroxy heterocycles via intramolecular reductive cyclization of diketoximes by NaBH3CN. Org. Biomol. Chem. 2011, 9, 4642–4652. [Google Scholar] [CrossRef]
- Johnson, J.K.; Skoda, E.M.; Zhou, J.; Parrinello, E.; Wang, D.; O’Malley, K.; Eyer, B.R.; Kazancioglu, M.; Eisermann, K.; Johnston, P.A.; et al. Small Molecule Antagonists of the Nuclear Androgen Receptor for the Treatment of Castration-Resistant Prostate Cancer. ACS Med. Chem. Lett. 2016, 7, 785–790. [Google Scholar] [CrossRef]
- Di Fabio, R.; Alvaro, G.; Braggio, S.; Carletti, R.; Gerrard, P.A.; Griffante, C.; Marchioro, C.; Pozzan, A.; Melotto, S.; Poffe, A.; et al. Identification, biological characterization and pharmacophoric analysis of a new potent and selective NK1 receptor antagonist clinical candidate. Bioorg. Med. Chem. 2013, 21, 6264–6273. [Google Scholar] [CrossRef]
- Atkinson, B.N.; Steadman, D.; Mahy, W.; Zhao, Y.; Sipthorp, J.; Bayle, E.D.; Svensson, F.; Papageorgiou, G.; Jeganathan, F.; Frew, S.; et al. Scaffold-hopping identifies furano[2,3-d]pyrimidine amides as potent Notum inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126751. [Google Scholar] [CrossRef]
- Manetti, D.; Ghelardini, C.; Bartolini, A.; Bellucci, C.; Dei, S.; Galeotti, N.; Gualtieri, F.; Romanelli, M.N.; Scapecchi, S.; Teodori, E. Design, Synthesis, and Preliminary Pharmacological Evaluation of 1,4-Diazabicyclo[4.3.0]nonan-9-ones as a New Class of Highly Potent Nootropic Agents. J. Med. Chem. 2000, 43, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Scapecchi, S.; Martini, E.; Manetti, D.; Ghelardini, C.; Martelli, C.; Dei, S.; Galeotti, N.; Guandalini, L.; Novella Romanelli, M.; Teodori, E. Structure–activity relationship studies on unifiram (DM232) and sunifiram (DM235), two novel and potent cognition enhancing drugs. Bioorg. Med. Chem. 2004, 12, 71–85. [Google Scholar] [CrossRef]
- Chandrika, N.T.; Shrestha, S.K.; Ngo, H.X.; Garneau-Tsodikova, S. Synthesis and investigation of novel benzimidazole derivatives as antifungal agents. Bioorg. Med. Chem. 2016, 24, 3680–3686. [Google Scholar] [CrossRef]
- Arutla, V.; Leal, J.; Liu, X.; Sokalingam, S.; Raleigh, M.; Adaralegbe, A.; Liu, L.; Pentel, P.R.; Hecht, S.M.; Chang, Y. Prescreening of Nicotine Hapten Linkers in Vitro To Select Hapten-Conjugate Vaccine Candidates for Pharmacokinetic Evaluation in Vivo. ACS Comb. Sci. 2017, 19, 286–298. [Google Scholar] [CrossRef]
- Dilman, A.D.; Tishkov, A.A.; Lyapkalo, I.M.; Ioffe, S.L.; Strelenko, Y.A.; Tartakovsky, V.A. Novel Convenient Method for the Synthesis of N,N-Bis(trimethylsilyloxy)enamines. Synthesis 1998, 1998, 181–185. [Google Scholar] [CrossRef]
- Dilman, A.D.; Ioffe, S.L.; Mayr, H. Determination of the Nucleophilicities of N,N-Bis(silyloxy)enamines. J. Org. Chem. 2001, 66, 3196–3200. [Google Scholar] [CrossRef] [PubMed]
- Sukhorokov, A.Y.; Bliznets, I.V.; Lesiv, A.V.; Khomutova, Y.A.; Strelenko, Y.A.; Ioffe, S.L. The Chemistry of N,N-Bis(siloxy)enamines. Part 8. A General Method for the Preparation of α-Azido Oximes from Aliphatic Nitro Compounds. Synthesis 2005, 2005, 1077–1082. [Google Scholar] [CrossRef]
- Dilman, A.D.; Tishkov, A.A.; Lyapkalo, I.M.; Ioffe, S.L.; Kachala, V.V.; Strelenko, Y.A.; Tartakovsky, V.A. Synthesis of N,N-bis(silyloxy)enamines with a functionalized double bond. J. Chem. Soc. Perkin Trans. 1 2000, 2926–2929. [Google Scholar] [CrossRef]
- Walton, M.C.; Yang, Y.-F.; Hong, X.; Houk, K.N.; Overman, L.E. Ligand-Controlled Diastereoselective 1,3-Dipolar Cycloadditions of Azomethine Ylides with Methacrylonitrile. Org. Lett. 2015, 17, 6166–6169. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, L.; Bortoluzzi, M.; Ferretti, E.; Hayatifar, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S. The reactions of α-amino acids and α-amino acid esters with high valent transition metal halides: Synthesis of coordination complexes, activation processes and stabilization of α-ammonium acylchloride cations. RSC Adv. 2017, 7, 10158–10174. [Google Scholar] [CrossRef]
- Toma, L.; Cignarella, G.; Barlocco, D.; Ronchetti, F. Molecular mechanics and 1H NMR conformational study of 3,8-diazabicyclo[3,2,1] octanes and related cis-2,6-dimethylpiperazines active on opioid receptors. Tetrahedron 1992, 48, 159–166. [Google Scholar] [CrossRef]
- Crosignani, S.; Gonzalez, J.; Swinnen, D. Polymer-Supported Mukaiyama Reagent: A Useful Coupling Reagent for the Synthesis of Esters and Amides. Org. Lett. 2004, 6, 4579–4582. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kadow, J.F.; Zhang, Z.; Yin, Z.; Gao, Q.; Wu, D.; Parker, D.D.; Yang, Z.; Zadjura, L.; Robinson, B.A.; et al. Inhibitors of HIV-1 attachment. Part 4: A study of the effect of piperazine substitution patterns on antiviral potency in the context of indole-based derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 5140–5145. [Google Scholar] [CrossRef] [PubMed]
- Wodtke, R.; Steinberg, J.; Köckerling, M.; Löser, R.; Mamat, C. NMR-based investigations of acyl-functionalized piperazines concerning their conformational behavior in solution. RSC Adv. 2018, 8, 40921–40933. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.; Hoevelmann, S.; Martin, E.; Hentsch, B.; Gassen, M.; Kraus, J.; Krauss, R.; Vincek, A.-S. Novel compounds as histone deacetylase inhibitors. U.S. Patent 10/624,571, 11 August 2005. (4SC AG and G2M Cancer Drugs AG). [Google Scholar]
- Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der Organischen Chemie; Georg Thieme Verlag: Stuttgart, Germany; New York, NY, USA, 2005; p. 200. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pospelov, E.V.; Sukhorukov, A.Y. Building Up a Piperazine Ring from a Primary Amino Group via Catalytic Reductive Cyclization of Dioximes. Int. J. Mol. Sci. 2023, 24, 11794. https://doi.org/10.3390/ijms241411794
Pospelov EV, Sukhorukov AY. Building Up a Piperazine Ring from a Primary Amino Group via Catalytic Reductive Cyclization of Dioximes. International Journal of Molecular Sciences. 2023; 24(14):11794. https://doi.org/10.3390/ijms241411794
Chicago/Turabian StylePospelov, Evgeny V., and Alexey Yu. Sukhorukov. 2023. "Building Up a Piperazine Ring from a Primary Amino Group via Catalytic Reductive Cyclization of Dioximes" International Journal of Molecular Sciences 24, no. 14: 11794. https://doi.org/10.3390/ijms241411794
APA StylePospelov, E. V., & Sukhorukov, A. Y. (2023). Building Up a Piperazine Ring from a Primary Amino Group via Catalytic Reductive Cyclization of Dioximes. International Journal of Molecular Sciences, 24(14), 11794. https://doi.org/10.3390/ijms241411794