
DNA Methylation in the Fields of Prenatal Diagnosis and Early Detection of Cancers

 , , , ,
, , , , 
Abstract
1. DNA Methylation: Looking Back over the Century
“Genes are equivalent to blueprints; epigenetics is the contractor. They change the assembly, the structure.”—Bruce Lipton.
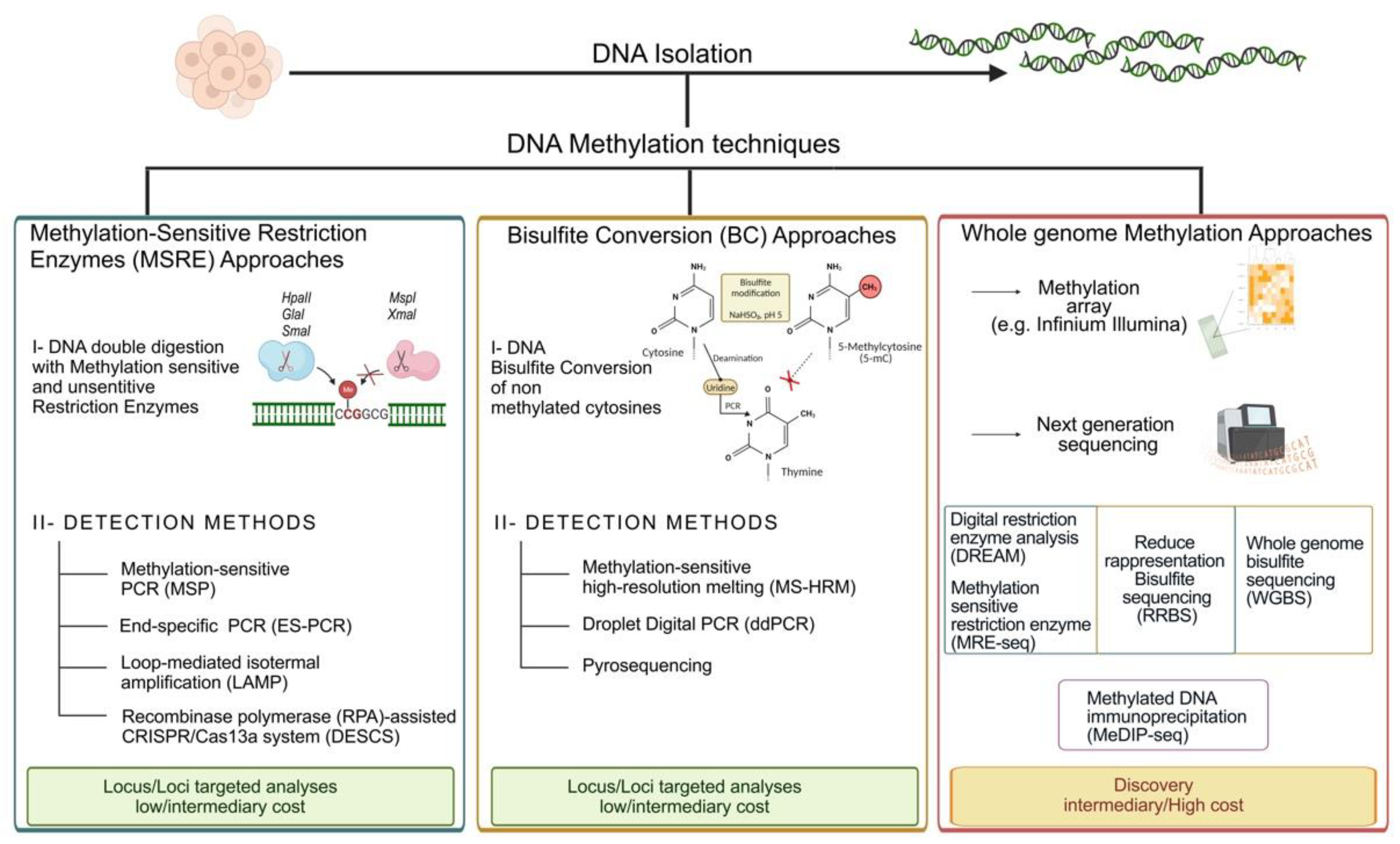
2. DNA Methylation Techniques
2.1. Methods Based on Methylation-Sensitive or -Dependent Restriction Enzymes (MSRE/MSDE)
2.2. Bisulfite-Based Methods
2.3. Whole Genome Methylation Approaches
3. DNA Methylation in Prenatal Diagnosis
3.1. DNA Methylation as a Biomarker for Fetal DNA Enrichment and Non-Invasive Prenatal Testing (NIPT)
3.2. DNA Methylation in Imprinting Disorders Diagnosis and Assisted Reproductive Technology (ART) Impact
4. DNA Methylation Test in Early Cancer Diagnosis
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, T.B.; Coghill, R.D. Researches on pyrimidines. Cl 11. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the Tubercle bacillus. J. Am. Chem. Soc. 1925, 47, 2838–2844. [Google Scholar] [CrossRef]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA methylation: A historical perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.D. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem. 1948, 175, 315–332. [Google Scholar] [CrossRef]
- Wyatt, G.R. Occurrence of 5-methylcytosine in nucleic acids. Nature 1950, 166, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, G.R. Recognition and estimation of 5-methylcytosine in nucleic acids. Biochem. J. 1951, 48, 581–584. [Google Scholar] [CrossRef]
- Deichmann, U. Epigenetics: The origins and evolution of a fashionable topic. Dev. Biol. 2016, 416, 249–254. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Compere, S.J.; Palmiter, R.D. DNA methylation controls the inducibility of the mouse metallothionein-I gene lymphoid cells. Cell 1981, 25, 233–240. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef]
- Canovas, S.; Ross, P.J.; Kelsey, G.; Coy, P. DNA methylation in embryo development: Epigenetic impact of ART (Assisted Reproductive Technologies). BioEssays News Rev. Mol. Cell. Dev. Biol. 2017, 39, 1700106. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarasimhan, R.; Liang, G. The role of DNA methylation in cancer. In DNA Methyltransferases—Role and Function; Jeltsch, A., Jurkowska, R.Z., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 945, pp. 151–172. ISBN 978-3-319-43622-7. [Google Scholar]
- Pappalardo, X.G.; Barra, V. Losing DNA methylation at repetitive elements and breaking bad. Epigenetics Chromatin 2021, 14, 25. [Google Scholar] [CrossRef]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin 2015, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Robertson, K.D. DNA Methyltransferases, DNA damage repair, and cancer. In Epigenetic Alterations in Oncogenesis; Karpf, A.R., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2013; Volume 754, pp. 3–29. ISBN 978-1-4419-9966-5. [Google Scholar]
- Kuo, K.C.; McCune, R.A.; Gehrke, C.W.; Midgett, R.; Ehrlich, M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res. 1980, 8, 4763–4776. [Google Scholar] [CrossRef]
- Khalil, H.M.; Abdel Ghaffar, F.M.; Bishara, S.A.; Soffar, S.A.; Khalil, N.M.; Riad, S.M. Evaluation of some direct diagnostic methods in intestinal parasitic infections. J. Egypt. Soc. Parasitol. 1990, 20, 789–792. [Google Scholar] [PubMed]
- Gupta, R.; Nagarajan, A.; Wajapeyee, N. Advances in genome-wide DNA methylation analysis. BioTechniques 2010, 49, iii–xi. [Google Scholar] [CrossRef]
- Chenarani, N.; Emamjomeh, A.; Allahverdi, A.; Mirmostafa, S.; Afsharinia, M.H.; Zahiri, J. Bioinformatic tools for DNA methylation and histone modification: A survey. Genomics 2021, 113, 1098–1113. [Google Scholar] [CrossRef]
- Cedar, H.; Solage, A.; Glaser, G.; Razin, A. Direct detection of methylated cytosine in DNA by use of the restriction enzyme MspI. Nucleic Acids Res. 1979, 6, 2125–2132. [Google Scholar] [CrossRef]
- Redshaw, N.; Huggett, J.F.; Taylor, M.S.; Foy, C.A.; Devonshire, A.S. Quantification of epigenetic biomarkers: An evaluation of established and emerging methods for DNA methylation analysis. BMC Genomics 2014, 15, 1174. [Google Scholar] [CrossRef]
- Hambalek, J.A.; Kong, J.E.; Brown, C.; Munoz, H.E.; Horn, T.; Bogumil, M.; Quick, E.; Ozcan, A.; Di Carlo, D. Methylation-sensitive loop-mediated isothermal amplification (LAMP): Nucleic acid methylation detection through LAMP with mobile fluorescence readout. ACS Sens. 2021, 6, 3242–3252. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, S.; Chu, C.; Yang, M.; Huo, D.; Hou, C. Dual methylation-sensitive restriction endonucleases coupling with an RPA-assisted CRISPR/Cas13a system (DESCS) for highly sensitive analysis of DNA methylation and its application for point-of-care detection. ACS Sens. 2021, 6, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, J.; Lee, J.T.; Cesaroni, M.; Madzo, J.; Liang, S.; Lu, Y.; Issa, J.-P.J. Digital restriction enzyme analysis of methylation (DREAM). In DNA Methylation Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1708, pp. 247–265. [Google Scholar] [CrossRef]
- Yang, H.; Qiu, J.; Zhen, L.; Huang, Y.; Ren, W.; Gu, H.; Xu, H.; Xu, G. Sensitive GlaI digestion and terminal transferase PCR for DNA methylation detection. Talanta 2022, 247, 123616. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.N.; Young, G.P.; Ho, T.; Molloy, P.L. Sensitive and selective amplification of methylated DNA sequences using helper-dependent chain reaction in combination with a methylation-dependent restriction enzymes. Nucleic Acids Res. 2013, 41, e15. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, Y.; Tian, W.; Liu, C.; Gao, K.; Li, Z. A novel restriction endonuclease GlaI for rapid and highly sensitive detection of DNA methylation coupled with isothermal exponential amplification reaction. Chem. Sci. 2018, 9, 1344–1351. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Husseiny, M.I.; Fahmy, A.; Du, W.; Gu, A.; Garcia, P.; Ferreri, K.; Kandeel, F. Development of quantitative methylation-specific droplet digital PCR (DdMSP) for assessment of natural tregs. Front. Genet. 2020, 11, 300. [Google Scholar] [CrossRef]
- Olmedillas-López, S.; Olivera-Salazar, R.; García-Arranz, M.; García-Olmo, D. Current and emerging applications of droplet digital PCR in oncology: An updated review. Mol. Diagn. Ther. 2022, 26, 61–87. [Google Scholar] [CrossRef]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef]
- Quillien, V.; Lavenu, A.; Ducray, F.; Joly, M.-O.; Chinot, O.; Fina, F.; Sanson, M.; Carpentier, C.; Karayan-Tapon, L.; Rivet, P.; et al. Validation of the high-performance of pyrosequencing for clinical MGMT Testing on a cohort of glioblastoma patients from a prospective dedicated multicentric trial. Oncotarget 2016, 7, 61916–61929. [Google Scholar] [CrossRef]
- Ben Maamar, M.; Sadler-Riggleman, I.; Beck, D.; Skinner, M.K. Genome-wide mapping of DNA methylation 5mC by methylated DNA immunoprecipitation (MeDIP)-sequencing. In DNA Modifications; Methods in Molecular Biology; Humana: New York, NY, USA, 2021; Volume 2198, pp. 301–310. [Google Scholar] [CrossRef]
- Beck, D.; Ben Maamar, M.; Skinner, M.K. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons. Epigenetics 2022, 17, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Yasuda, K.; Takeda, N.; Miura, K. Growth hormone response to arginine administration in diabetics—With special reference to the multiple regression analysis in association with diabetic retinopathy. Nihon Naibunpi Gakkai Zasshi 1989, 65, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Fang, F.; Miller, D.F.; Pilrose, J.M.; Matei, D.; Huang, T.H.-M.; Nephew, K.P. Global DNA methylation profiling technologies and the ovarian cancer methylome. In Cancer Epigenetics; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1238, pp. 653–675. [Google Scholar] [CrossRef]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jerónimo, C. Profiling DNA methylation based on next-generation sequencing approaches: New insights and clinical applications. Genes 2018, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Ost, T.W.B.; Beraldi, D.; Bell, N.M.; Branco, M.R.; Reik, W.; Balasubramanian, S. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat. Protoc. 2013, 8, 1841–1851. [Google Scholar] [CrossRef]
- Philpott, M.; Cribbs, A.P.; Brown, T.; Brown, T.; Oppermann, U. Advances and challenges in epigenomic single-cell sequencing applications. Curr. Opin. Chem. Biol. 2020, 57, 17–26. [Google Scholar] [CrossRef]
- Soto, J.; Rodriguez-Antolin, C.; Vallespín, E.; de Castro Carpeño, J.; Ibanez de Caceres, I. The impact of next-generation sequencing on the DNA methylation-based translational cancer research. Transl. Res. J. Lab. Clin. Med. 2016, 169, 1–18.e1. [Google Scholar] [CrossRef]
- Wong, F.C.K.; Lo, Y.M.D. Prenatal diagnosis innovation: Genome sequencing of maternal plasma. Annu. Rev. Med. 2016, 67, 419–432. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Chiu, R.W.K. Genomic analysis of fetal nucleic acids in maternal blood. Annu. Rev. Genomics Hum. Genet. 2012, 13, 285–306. [Google Scholar] [CrossRef]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet Lond. Engl. 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Chan, K.C.A.; Zhang, J.; Hui, A.B.Y.; Wong, N.; Lau, T.K.; Leung, T.N.; Lo, K.-W.; Huang, D.W.S.; Lo, Y.M.D. Size distributions of maternal and fetal DNA in maternal plasma. Clin. Chem. 2004, 50, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zimmermann, B.; Rusterholz, C.; Kang, A.; Holzgreve, W.; Hahn, S. Size separation of circulatory DNA in maternal plasma permits ready detection of fetal DNA polymorphisms. Clin. Chem. 2004, 50, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Kamataki, A.; Yamaki, J.; Homma, Y. Characterization of circulating DNA in healthy human plasma. Clin. Chim. Acta Int. J. Clin. Chem. 2008, 387, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Rafi, I.; Hill, M.; Hayward, J.; Chitty, L.S. Non-invasive prenatal testing: Use of cell-free fetal DNA in down syndrome screening. Br. J. Gen. Pract. J. R. Coll. Gen. Pract. 2017, 67, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.M.; Quezada, M.S.; Revello, R.; Akolekar, R.; Nicolaides, K.H. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: Updated meta-analysis. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2015, 45, 249–266. [Google Scholar] [CrossRef]
- Poon, L.L.M.; Leung, T.N.; Lau, T.K.; Chow, K.C.K.; Lo, Y.M.D. Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin. Chem. 2002, 48, 35–41. [Google Scholar] [CrossRef]
- Chim, S.S.C.; Tong, Y.K.; Chiu, R.W.K.; Lau, T.K.; Leung, T.N.; Chan, L.Y.S.; Oudejans, C.B.M.; Ding, C.; Lo, Y.M.D. Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc. Natl. Acad. Sci. USA 2005, 102, 14753–14758. [Google Scholar] [CrossRef]
- Chim, S.S.C.; Jin, S.; Lee, T.Y.H.; Lun, F.M.F.; Lee, W.S.; Chan, L.Y.S.; Jin, Y.; Yang, N.; Tong, Y.K.; Leung, T.Y.; et al. Systematic search for placental DNA-methylation markers on chromosome 21: Toward a maternal plasma-based epigenetic test for fetal Trisomy 21. Clin. Chem. 2008, 54, 500–511. [Google Scholar] [CrossRef]
- Tong, Y.K.; Jin, S.; Chiu, R.W.K.; Ding, C.; Chan, K.C.A.; Leung, T.Y.; Yu, L.; Lau, T.K.; Lo, Y.M.D. Noninvasive prenatal detection of Trisomy 21 by an epigenetic-genetic chromosome-dosage approach. Clin. Chem. 2010, 56, 90–98. [Google Scholar] [CrossRef]
- Grunau, C.; Clark, S.J.; Rosenthal, A. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001, 29, E65. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, E.A.; Fiegler, H.; Rakyan, V.; Beck, S.; Hulten, M.; Lamnissou, K.; Carter, N.P.; Patsalis, P.C. Sites of differential DNA methylation between placenta and peripheral blood: Molecular markers for noninvasive prenatal diagnosis of aneuploidies. Am. J. Pathol. 2009, 174, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Tsui, D.W.Y.; Lam, Y.M.D.; Lee, W.S.; Leung, T.Y.; Lau, T.K.; Lau, E.T.; Tang, M.H.Y.; Akolekar, R.; Nicolaides, K.H.; Chiu, R.W.K.; et al. Systematic Identification of placental epigenetic signatures for the noninvasive prenatal detection of Edwards syndrome. PLoS ONE 2010, 5, e15069. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhang, J.; Li, Q.; Zhou, X.; Wang, T.; Xu, M.; Xia, S.; Xing, Q.; Wang, L.; He, L.; et al. DNA Methylome profiling of maternal peripheral blood and placentas reveal potential fetal DNA markers for non-invasive prenatal testing. Mol. Hum. Reprod. 2014, 20, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Schübeler, D.; Roloff, T.-C. Methylated DNA immunoprecipitation (MeDIP). In DNA Methylation; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2009; Volume 507, pp. 55–64. [Google Scholar] [CrossRef]
- Keravnou, A.; Ioannides, M.; Loizides, C.; Tsangaras, K.; Achilleos, A.; Mina, P.; Kypri, E.; Hadjidaniel, M.D.; Neofytou, M.; Kyriacou, S.; et al. MeDIP combined with in-solution targeted enrichment followed by NGS: Inter-individual methylation variability of fetal-specific biomarkers and their implementation in a proof of concept study for NIPT. PLoS ONE 2018, 13, e0199010. [Google Scholar] [CrossRef]
- Peng, X.L.; Jiang, P. Bioinformatics approaches for fetal DNA fraction estimation in noninvasive prenatal testing. Int. J. Mol. Sci. 2017, 18, 453. [Google Scholar] [CrossRef]
- Duboc, V.; Pratella, D.; Milanesio, M.; Boudjarane, J.; Descombes, S.; Paquis-Flucklinger, V.; Bottini, S. NiPTUNE: An automated pipeline for noninvasive prenatal testing in an accurate, integrative and flexible framework. Brief. Bioinform. 2022, 23, bbab380. [Google Scholar] [CrossRef]
- FDA Warns of Risks Associated with Non-Invasive Prenatal Screening Tests. U.S. Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-warns-risks-associated-non-invasive-prenatal-screening-tests (accessed on 5 July 2023).
- Raj, H.; Yelne, P. Cell-free fetal deoxyribonucleic acid (CffDNA) analysis as a remarkable method of non-invasive prenatal screening. Cureus 2022, 14, e29965. [Google Scholar] [CrossRef]
- Hui, L.; Ellis, K.; Mayen, D.; Pertile, M.D.; Reimers, R.; Sun, L.; Vermeesch, J.; Vora, N.L.; Chitty, L.S. Position statement from the international society for prenatal diagnosis on the use of non-invasive prenatal testing for the detection of fetal chromosomal conditions in singleton pregnancies. Prenat. Diagn. 2023, 43, 814–828. [Google Scholar] [CrossRef]
- Niemitz, E.L.; Feinberg, A.P. Epigenetics and assisted reproductive technology: A call for investigation. Am. J. Hum. Genet. 2004, 74, 599–609. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Niemitz, E.L.; Feinberg, A.P. Association of in vitro fertilization with beckwith-wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 2003, 72, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Katalinic, A.; Gross, S.; Sutcliffe, A.; Varon, R.; Horsthemke, B. Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J. Med. Genet. 2005, 42, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Hiura, H.; Kitamura, A.; Miyauchi, N.; Kobayashi, N.; Takahashi, S.; Okae, H.; Kyono, K.; Kagami, M.; Ogata, T.; et al. Association of four imprinting disorders and ART. Clin. Epigenetics 2019, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Hiura, H.; Okae, H.; Chiba, H.; Miyauchi, N.; Sato, F.; Sato, A.; Arima, T. Imprinting methylation errors in ART. Reprod. Med. Biol. 2014, 13, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Uyar, A.; Seli, E. The impact of assisted reproductive technologies on genomic imprinting and imprinting disorders. Curr. Opin. Obstet. Gynecol. 2014, 26, 210–221. [Google Scholar] [CrossRef]
- Chiba, H.; Hiura, H.; Okae, H.; Miyauchi, N.; Sato, F.; Sato, A.; Arima, T. DNA methylation errors in imprinting disorders and assisted reproductive technology. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2013, 55, 542–549. [Google Scholar] [CrossRef]
- Anvar, Z.; Chakchouk, I.; Demond, H.; Sharif, M.; Kelsey, G.; Van den Veyver, I.B. DNA methylation dynamics in the female germline and maternal-effect mutations that disrupt genomic imprinting. Genes 2021, 12, 1214. [Google Scholar] [CrossRef]
- Ochoa, E.; Lee, S.; Lan-Leung, B.; Dias, R.P.; Ong, K.K.; Radley, J.A.; Pérez de Nanclares, G.; Martinez, R.; Clark, G.; Martin, E.; et al. ImprintSeq, a novel tool to interrogate DNA methylation at human imprinted regions and diagnose multilocus imprinting disturbance. Genet. Med. Off. J. Am. Coll. Med. Genet. 2022, 24, 463–474. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. TIG 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Quiñonez-Silva, G.; Dávalos-Salas, M.; Recillas-Targa, F.; Ostrosky-Wegman, P.; Aranda, D.A.; Benítez-Bribiesca, L. Monoallelic germline methylation and sequence variant in the promoter of the RB1 gene: A possible constitutive epimutation in hereditary retinoblastoma. Clin. Epigenetics 2016, 8, 1. [Google Scholar] [CrossRef]
- Pinto, D.; Pinto, C.; Guerra, J.; Pinheiro, M.; Santos, R.; Vedeld, H.M.; Yohannes, Z.; Peixoto, A.; Santos, C.; Pinto, P.; et al. Contribution of MLH1 constitutional methylation for lynch syndrome diagnosis in patients with tumor MLH1 downregulation. Cancer Med. 2018, 7, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Azzollini, J.; Pesenti, C.; Pizzamiglio, S.; Fontana, L.; Guarino, C.; Peissel, B.; Plebani, M.; Tabano, S.; Sirchia, S.M.; Colapietro, P.; et al. Constitutive BRCA1 promoter hypermethylation can be a predisposing event in isolated early-onset breast cancer. Cancers 2019, 11, 58. [Google Scholar] [CrossRef]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.-S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zeng, Q.; Wang, Z.; Li, C.; Xu, Y.; Cui, P.; Zhu, X.; Lu, H.; Wang, G.; Cai, S.; et al. Circulating cell-free DNA for cancer early detection. Innov. Camb. Mass 2022, 3, 100259. [Google Scholar] [CrossRef] [PubMed]
- Ned, R.M.; Melillo, S.; Marrone, M. Fecal DNA testing for colorectal cancer screening: The ColoSureTM test. PLoS Curr. 2011, 3, RRN1220. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef]
- Han, Y.D.; Oh, T.J.; Chung, T.-H.; Jang, H.W.; Kim, Y.N.; An, S.; Kim, N.K. Early detection of colorectal cancer based on presence of methylated syndecan-2 (SDC2) in stool DNA. Clin. Epigenetics 2019, 11, 51. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Wang, H.; Zheng, L.; Zhou, C.; Li, G.; Huang, R.; Wang, H.; Li, C.; Fan, X.; et al. Robust performance of a novel stool DNA test of methylated SDC2 for colorectal cancer detection: A multicenter clinical study. Clin. Epigenetics 2020, 12, 162. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Zhang, D.; Xiong, X.; Hao, T.; Zhong, L.; Zhao, Y. Diagnostic accuracy of DNA-based SDC2 methylation test in colorectal cancer screening: A meta-analysis. BMC Gastroenterol. 2022, 22, 314. [Google Scholar] [CrossRef]
- Wang, Z.; Shang, J.; Zhang, G.; Kong, L.; Zhang, F.; Guo, Y.; Dou, Y.; Lin, J. Evaluating the clinical performance of a dual-target stool DNA test for colorectal cancer detection. J. Mol. Diagn. JMD 2022, 24, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhao, G.; Yang, J.; Zhou, X.; Xiong, S.; Lu, X.; Gao, L.; Wu, J.; Xu, Z.; Fei, S.; et al. A simplified multiplex methylated DNA testing for early detection of colorectal cancer in stool DNA. BMC Gastroenterol. 2022, 22, 428. [Google Scholar] [CrossRef]
- Lamb, Y.N.; Dhillon, S. Epi ProColon® 2.0 CE: A blood-based screening test for colorectal cancer. Mol. Diagn. Ther. 2017, 21, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Roberts, W.L.; Fang, J.C.; Samowitz, W.S.; Heichman, K.A. Septin 9 Methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Oussalah, A.; Rischer, S.; Bensenane, M.; Conroy, G.; Filhine-Tresarrieu, P.; Debard, R.; Forest-Tramoy, D.; Josse, T.; Reinicke, D.; Garcia, M.; et al. Plasma MSEPT9: A novel circulating cell-free DNA-Based epigenetic biomarker to diagnose hepatocellular carcinoma. EBioMedicine 2018, 30, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Lin, Y.; Xu, J.; Liu, D.; Li, D.; Meng, H.; Gallant, M.A.; Kubota, N.; Roy, D.; Li, J.S.; et al. A multi-analyte cell-free DNA-based blood test for early detection of hepatocellular carcinoma. Hepatol. Commun. 2022, 6, 1753–1763. [Google Scholar] [CrossRef]
- Cheishvili, D.; Wong, C.; Karim, M.M.; Kibria, M.G.; Jahan, N.; Das, P.C.; Yousuf, M.A.K.; Islam, M.A.; Das, D.C.; Noor-E-Alam, S.M.; et al. A high-throughput test enables specific detection of hepatocellular carcinoma. Nat. Commun. 2023, 14, 3306. [Google Scholar] [CrossRef]
- Dietrich, D.; Kneip, C.; Raji, O.; Liloglou, T.; Seegebarth, A.; Schlegel, T.; Flemming, N.; Rausch, S.; Distler, J.; Fleischhacker, M.; et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the Aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int. J. Oncol. 2012, 40, 825–832. [Google Scholar] [CrossRef]
- Ilse, P.; Biesterfeld, S.; Pomjanski, N.; Fink, C.; Schramm, M. SHOX2 DNA methylation is a tumour marker in pleural effusions. Cancer Genom. Proteom. 2013, 10, 217–223. [Google Scholar]
- Weiss, G.; Schlegel, A.; Kottwitz, D.; König, T.; Tetzner, R. Validation of the SHOX2/PTGER4 DNA methylation marker panel for plasma-based discrimination between patients with malignant and nonmalignant lung disease. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 77–84. [Google Scholar] [CrossRef]
- Gaga, M.; Chorostowska-Wynimko, J.; Horváth, I.; Tammemagi, M.C.; Shitrit, D.; Eisenberg, V.H.; Liang, H.; Stav, D.; Levy Faber, D.; Jansen, M.; et al. Validation of lung EpiCheck, a novel methylation-based blood assay, for the detection of lung cancer in European and Chinese high-risk individuals. Eur. Respir. J. 2021, 57, 2002682. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Chen, Z.; Li, C.; Liu, J.; Tao, J.; Liu, X.; Zhao, D.; Yin, W.; Chen, H.; Cheng, C.; et al. Accurate diagnosis of pulmonary nodules using a noninvasive DNA methylation test. J. Clin. Investig. 2021, 131, e145973. [Google Scholar] [CrossRef] [PubMed]
- Wojno, K.J.; Costa, F.J.; Cornell, R.J.; Small, J.D.; Pasin, E.; Van Criekinge, W.; Bigley, J.W.; Van Neste, L. Reduced rate of repeated prostate biopsies observed in ConfirmMDx clinical utility field study. Am. Health Drug Benefits 2014, 7, 129–134. [Google Scholar] [PubMed]
- O’Reilly, E.; Tuzova, A.V.; Walsh, A.L.; Russell, N.M.; O’Brien, O.; Kelly, S.; Dhomhnallain, O.N.; DeBarra, L.; Dale, C.M.; Brugman, R.; et al. EpiCaPture: A urine DNA methylation test for early detection of aggressive prostate cancer. JCO Precis. Oncol. 2019, 2019, 1–18. [Google Scholar] [CrossRef]
- van Kessel, K.E.M.; Van Neste, L.; Lurkin, I.; Zwarthoff, E.C.; Van Criekinge, W. Evaluation of an epigenetic profile for the detection of bladder cancer in patients with hematuria. J. Urol. 2016, 195, 601–607. [Google Scholar] [CrossRef] [PubMed]
- van Kessel, K.E.M.; Beukers, W.; Lurkin, I.; Ziel-van der Made, A.; van der Keur, K.A.; Boormans, J.L.; Dyrskjøt, L.; Márquez, M.; Ørntoft, T.F.; Real, F.X.; et al. Validation of a DNA methylation-mutation urine assay to select patients with hematuria for cystoscopy. J. Urol. 2017, 197, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Feber, A.; Dhami, P.; Dong, L.; de Winter, P.; Tan, W.S.; Martínez-Fernández, M.; Paul, D.S.; Hynes-Allen, A.; Rezaee, S.; Gurung, P.; et al. UroMark-a urinary biomarker assay for the detection of bladder cancer. Clin. Epigenetics 2017, 9, 8. [Google Scholar] [CrossRef]
- Piatti, P.; Chew, Y.C.; Suwoto, M.; Yamada, T.; Jara, B.; Jia, X.-Y.; Guo, W.; Ghodoussipour, S.; Daneshmand, S.; Ahmadi, H.; et al. Clinical evaluation of bladder CARE, a new epigenetic test for bladder cancer detection in urine samples. Clin. Epigenetics 2021, 13, 84. [Google Scholar] [CrossRef]
- Ruan, W.; Chen, X.; Huang, M.; Wang, H.; Chen, J.; Liang, Z.; Zhang, J.; Yu, Y.; Chen, S.; Xu, S.; et al. A urine-based DNA methylation assay to facilitate early detection and risk stratification of bladder cancer. Clin. Epigenetics 2021, 13, 91. [Google Scholar] [CrossRef]
- Schmitz, M.; Wunsch, K.; Hoyer, H.; Scheungraber, C.; Runnebaum, I.B.; Hansel, A.; Dürst, M. Performance of a methylation specific real-time PCR assay as a triage test for HPV-positive women. Clin. Epigenetics 2017, 9, 118. [Google Scholar] [CrossRef]
- De Strooper, L.M.A.; Verhoef, V.M.J.; Berkhof, J.; Hesselink, A.T.; de Bruin, H.M.E.; van Kemenade, F.J.; Bosgraaf, R.P.; Bekkers, R.L.M.; Massuger, L.F.A.G.; Melchers, W.J.G.; et al. Validation of the FAM19A4/Mir124-2 DNA methylation test for both lavage- and brush-based self-samples to detect cervical (pre)cancer in HPV-positive women. Gynecol. Oncol. 2016, 141, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, A.; Takács, T.; Jeney, C.; Schaff, Z.; Koiss, R.; Járay, B.; Sobel, G.; Pap, K.; Székely, I.; Ferenci, T.; et al. Performance of a new HPV and biomarker assay in the management of HrHPV positive women: Subanalysis of the ongoing multicenter TRACE clinical trial (n > 6,000) to evaluate POU4F3 methylation as a potential biomarker of cervical precancer and cancer. Int. J. Cancer 2017, 140, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Kan, Y.-Y.; Liou, Y.-L.; Wang, H.-J.; Chen, C.-Y.; Sung, L.-C.; Chang, C.-F.; Liao, C.-I. PAX1 methylation as a potential biomarker for cervical cancer screening. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2014, 24, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-J.; Chang, C.-F.; Lee, J.-J.; Chen, H.-M.; Wang, H.-J.; Liou, Y.-L.; Yen, C.; Chiang, C.-P. Hypermethylated ZNF582 and PAX1 are effective biomarkers for detection of oral dysplasia and oral cancer. Oral Oncol. 2016, 62, 34–43. [Google Scholar] [CrossRef]
- Moinova, H.R.; LaFramboise, T.; Lutterbaugh, J.D.; Chandar, A.K.; Dumot, J.; Faulx, A.; Brock, W.; De la Cruz Cabrera, O.; Guda, K.; Barnholtz-Sloan, J.S.; et al. Identifying DNA methylation biomarkers for non-endoscopic detection of Barrett’s esophagus. Sci. Transl. Med. 2018, 10, eaao5848. [Google Scholar] [CrossRef]
- Moran, S.; Martínez-Cardús, A.; Sayols, S.; Musulén, E.; Balañá, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.C.; et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Stackpole, M.L.; Zeng, W.; Li, S.; Liu, C.-C.; Zhou, Y.; He, S.; Yeh, A.; Wang, Z.; Sun, F.; Li, Q.; et al. Cost-effective methylome sequencing of cell-free DNA for accurately detecting and locating cancer. Nat. Commun. 2022, 13, 5566. [Google Scholar] [CrossRef]
- Liang, N.; Li, B.; Jia, Z.; Wang, C.; Wu, P.; Zheng, T.; Wang, Y.; Qiu, F.; Wu, Y.; Su, J.; et al. Ultrasensitive detection of circulating tumour DNA via deep methylation sequencing aided by machine learning. Nat. Biomed. Eng. 2021, 5, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Kim, N.; Moon, Y.; Kim, M.S.; Hoehn, B.D.; Park, C.H.; Kim, T.S.; Kim, N.K.; Chung, H.C.; An, S. Genome-wide identification and validation of a novel methylation biomarker, SDC2, for blood-based detection of colorectal cancer. J. Mol. Diagn. JMD 2013, 15, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.-Q.; Zhao, H.; Wang, Y.; Song, K.; Yang, J.; He, W.; Miao, D.-Z.; Wang, Q.; Jia, Y.-H. SEPTIN9-SDC2-VIM methylation signature as a biomarker for the early diagnosis of colorectal cancer. Am. J. Cancer Res. 2022, 12, 3128–3140. [Google Scholar] [PubMed]
- Zhang, L.; Li, D.; Gao, L.; Fu, J.; Sun, S.; Huang, H.; Zhang, D.; Jia, C.; Zheng, T.; Cui, B.; et al. Promoter methylation of QKI as a potential specific biomarker for early detection of colorectal cancer. Front. Genet. 2022, 13, 928150. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Bañobre, J.; Rodriguez-Casanova, A.; Costa-Fraga, N.; Bao-Caamano, A.; Alvarez-Castro, A.; Carreras-Presas, M.; Brozos-Vazquez, E.; Vidal-Insua, Y.; Vazquez-Rivera, F.; Candamio-Folgar, S.; et al. Noninvasive early detection of colorectal cancer by hypermethylation of the LINC00473 promoter in plasma cell-free DNA. Clin. Epigenetics 2022, 14, 86. [Google Scholar] [CrossRef]
- Tang, Q.; Cheng, J.; Cao, X.; Surowy, H.; Burwinkel, B. Blood-based DNA methylation as biomarker for breast cancer: A systematic review. Clin. Epigenetics 2016, 8, 115. [Google Scholar] [CrossRef]
- Aubele, M.; Schmitt, M.; Napieralski, R.; Paepke, S.; Ettl, J.; Absmaier, M.; Magdolen, V.; Martens, J.; Foekens, J.A.; Wilhelm, O.G.; et al. The predictive value of PITX2 DNA methylation for high-risk breast cancer therapy: Current guidelines, medical needs, and challenges. Dis. Markers 2017, 2017, 4934608. [Google Scholar] [CrossRef]
- Ghoreifi, A.; Ladi-Seyedian, S.-S.; Piatti, P.; Chew, Y.C.; Jara, B.; Sanossian, L.; Bhasin, J.M.; Yamada, T.; Fuchs, G.; Bhanvadia, S.; et al. A urine-based DNA methylation marker test to detect upper tract urothelial carcinoma: A prospective cohort study. J. Urol. 2023, 209, 854–862. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L. Consortium for top-down proteomics the human proteoform project: Defining the human proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef]
- Magzoub, M.M.; Prunello, M.; Brennan, K.; Gevaert, O. The impact of DNA methylation on the cancer proteome. PLoS Comput. Biol. 2019, 15, e1007245. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of Cancer | Sample | Technique | Epigenetic Alteration | Test Name | Sensitivity/Specificity 1 | Phase | Reference |
|---|---|---|---|---|---|---|---|
| Colorectal cancer | Stool | MSP | VIM methylation | ColoSureTM | 72–77% Se 83–94% Sp | CLIA | [83] |
| Colorectal cancer | Stool | qMSP | BMP3 and NDRG4 methylation | ColoGuard (Exact Sciences Co.) | 92.3% Se 86.6% Sp | FDA approved | [84] |
| Colorectal cancer | Stool | qMSP | SDC2 methylation | EarlyTect-Colon Cancer (Genomictree, Inc.) | 90.2% Se 90% Sp | Korean Food and Drug Administration | [85] |
| Colorectal cancer | Stool | qMSP | SDC2 methylation | Colosafe (Creative Biosciences) | 83.8–87% Se 98% Sp | China National Medical Products Administration | [86] |
| Colorectal cancer | Stool | Multiplex PCR | SDC2 and TFPI2 methylation | iColocomf (Wuhan Ammunition Life-tech Co.) | 95.31% Se 90.3% Sp | Trademark registered | [87,88] |
| Colorectal cancer | Stool and plasma | qMSP | SDC2 and SEPT9 methylation | ColoDefense (VersaBio Technologies, Inc.) | 90.8% Se in stool; 45–88% Se in plasma 92.9% Sp | Trademark registered | [89] |
| Colorectal cancer | Plasma | qMSP | SEPT9 methylation | Epi proColon (Epigenomics, Inc.) | 75–81% Se 96–99% Sp | FDA approved | [90] |
| Colorectal cancer | Plasma | qMSP | SEPT9 methylation | ColoVantage | 90% Se 88% Sp | CLIA | [91] |
| Hepatocellular carcinoma | Plasma | rtPCR | SEPT9 | HCCBloodTest (Epigenomics, Inc.) | 85–94% Se 84–91% Sp | CE marked | [92] |
| Hepatocellular carcinoma | Blood | Targeted capture assay | 77 CpG sites in 28 genes | HelioLiver | 85% Sp 91% Se | CLIA | [93] |
| Hepatocellular carcinoma | Plasma | Bisulfite next-generation sequencing | CHFR, VASH2, CCNJ, GRID2IP and F12 genes | epiLiver | 95% Sp 84.5 Se | In development | [94] |
| Lung cancer | Bronchial aspirates and pleural effusion samples | qMSP | SHOX2 methylation | Epi proLung BL Reflex Assay (Epigenomics, Inc.) | 78–96% Se/96% Sp in bronchial aspirates 39.5% Se/96.2% Sp in pleural effusion | CE marked | [95,96] |
| Lung cancer | Plasma | MethyLight-based assay | SHOX2 and PTGER4 | Epi proLung® blood-based version (Epigenomics, Inc.) | 67–90% Se 73–90% Sp | CE marked | [97] |
| Lung cancer | Plasma | MSRE-qPCR | Methylation of six genes (names of the gene not available) | Lung EpiCheck (Nucleix) | 56.7–87.2% Se 64.2–100% Sp | In development | [98] |
| Lung cancer | Plasma | Bisulfite next-generation sequencing | Over 100,000 CpG sites | PulmoSeek | 41% Sp 96% Se | CE marked | [99] |
| Prostate cancer | Primary tissue biopsy samples | MSP | GSTP1, APC and RASSF1 methylation | ConfirmMDX (mdxhealth) | NA | CLIA | [100] |
| Prostate cancer | Urine | qMSP | GSTP1, SFRP2, IGFBP3, IGFBP7, APC, and PTGS2 | epiCaPture | 73% Se 76% Sp | In development | [101] |
| Bladder cancer | Urine | MSP | TWIST1, ONECUT2 and OTX1 methylation | AssureMDX (MDxHealth) | 93% Se 86% Sp | Laboratory-developed test | [102,103] |
| Bladder cancer | Urine | Bisulfite next-generation sequencing | 150 CpG loci biomarker panel | UroMark | 98% Se 97% Sp | Laboratory-developed test | [104] |
| Bladder cancer | Urine | qMSP | TRNA-Cys, SIM2, and NKX1-1 | Bladder CARE | 96.2% Sp 93.5% Se | CLIA | [105] |
| Bladder and urothelial cancers | Urine | Multiplex qPCR | ONECUT2 and VIM | UriFind Bladder Cancer Detection Kit | 85.7–89.7% Sp 88.1–91.2% Se | CE marked | [106] |
| Cervical cancer | Cervical scrapes | MSP | ASTN1, DLX1, ITGA4, RXFP3, SOX17, and ZNF671 | GynTect® | 65% Se 95% Sp | CE marked | [107] |
| Cervical cancer | Cervical scrapes | Multiplex-MSP | FAM19A4 and miR124-2 | QIAsure Methylation Test (Qiagen) | 67–100% Se 68% Sp | CE marked | [108] |
| Cervical cancer | Liquid-based cytology | q-MSP | POU4F3 | CONFIDENCE™ (Neumann Diagnostics) | 1.67–1.74 Relative sensitivity 0.98–1.01 Relative Specificity | CE marked | [109] |
| Cervical cancer | Cervical scrapes | MSP | PAX1 | Cervi-M® (ISTAT BIOMEDICAL Co.) | 73% Se 80% Sp | CE-marked | [110] |
| Oral cancer | Oral epithelial cells | MSP | ZNF582 and PAX1 | Oral-M® (ISTAT BIOMEDICAL Co.) | 72–85% Se 86% Sp | CE-marked | [111] |
| Esophageal cancer | Esophageal brushing | Bisulfite next-generation sequencing | CCNA1 and VIM | EsoGuard | 91% Sp 93% Se | CLIA | [112] |
| Type of Cancer | Sample | Technique | Epigenetic Alteration | Test Name | Sensitivity/Specificity 1 | Phase | Reference |
|---|---|---|---|---|---|---|---|
| Carcinoma of unknown primary | Tumor biopsy samples | Illumina 450/EPIC methylation arrays | Genome-wide methylation | EPICUP (Ferrer and IDIBELL) | 97.7% Se 99.6% Sp | CE-marked | [113] |
| Multiple types of tumors | Plasma | Algorithm established from Illumina 450 methylation arrays | Genome-wide methylation | CancerLOCATOR | NA | Laboratory-developed test | [114] |
| Multiple types of tumors | Plasma | Bisulfite next-generation sequencing | 477 genomic regions associated to 657 genes and covering 10,613 CpG sites | PanSeer (SINGLERA Genomics) | 75–96% Se 96% | Laboratory-developed test | [115] |
| Multiple types of tumors | Plasma | Whole genome bisulfite sequencing | Genome-wide methylation | Galleri (Grail) | 51.5–90.1% Se 99.00% Sp | Preclinical | [116] |
| Multiple types of tumors | Plasma | Bisulfite next-generation sequencing | CpG-rich cfDNA fragments | cfMethyl-Seq (EarlyDx) | 74.5–80.7% Se 97.7% Sp | Laboratory-developed test | [117] |
| Multiple types of tumors | Plasma | Bisulfite next-generation sequencing | Genome-wide methylation | OverC Multi-Cancer Detection Blood Test (MCDBT) | 52–81% Se 95–96% Sp | CE-marked | [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppedè, F.; Bhaduri, U.; Stoccoro, A.; Nicolì, V.; Di Venere, E.; Merla, G. DNA Methylation in the Fields of Prenatal Diagnosis and Early Detection of Cancers. Int. J. Mol. Sci. 2023, 24, 11715. https://doi.org/10.3390/ijms241411715
Coppedè F, Bhaduri U, Stoccoro A, Nicolì V, Di Venere E, Merla G. DNA Methylation in the Fields of Prenatal Diagnosis and Early Detection of Cancers. International Journal of Molecular Sciences. 2023; 24(14):11715. https://doi.org/10.3390/ijms241411715
Chicago/Turabian StyleCoppedè, Fabio, Utsa Bhaduri, Andrea Stoccoro, Vanessa Nicolì, Eleonora Di Venere, and Giuseppe Merla. 2023. "DNA Methylation in the Fields of Prenatal Diagnosis and Early Detection of Cancers" International Journal of Molecular Sciences 24, no. 14: 11715. https://doi.org/10.3390/ijms241411715
APA StyleCoppedè, F., Bhaduri, U., Stoccoro, A., Nicolì, V., Di Venere, E., & Merla, G. (2023). DNA Methylation in the Fields of Prenatal Diagnosis and Early Detection of Cancers. International Journal of Molecular Sciences, 24(14), 11715. https://doi.org/10.3390/ijms241411715