
Neurotrophin Analog ENT-A044 Activates the p75 Neurotrophin Receptor, Regulating Neuronal Survival in a Cell Context-Dependent Manner

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. ENT-A044 Induces Cell Death of PC12 Cells
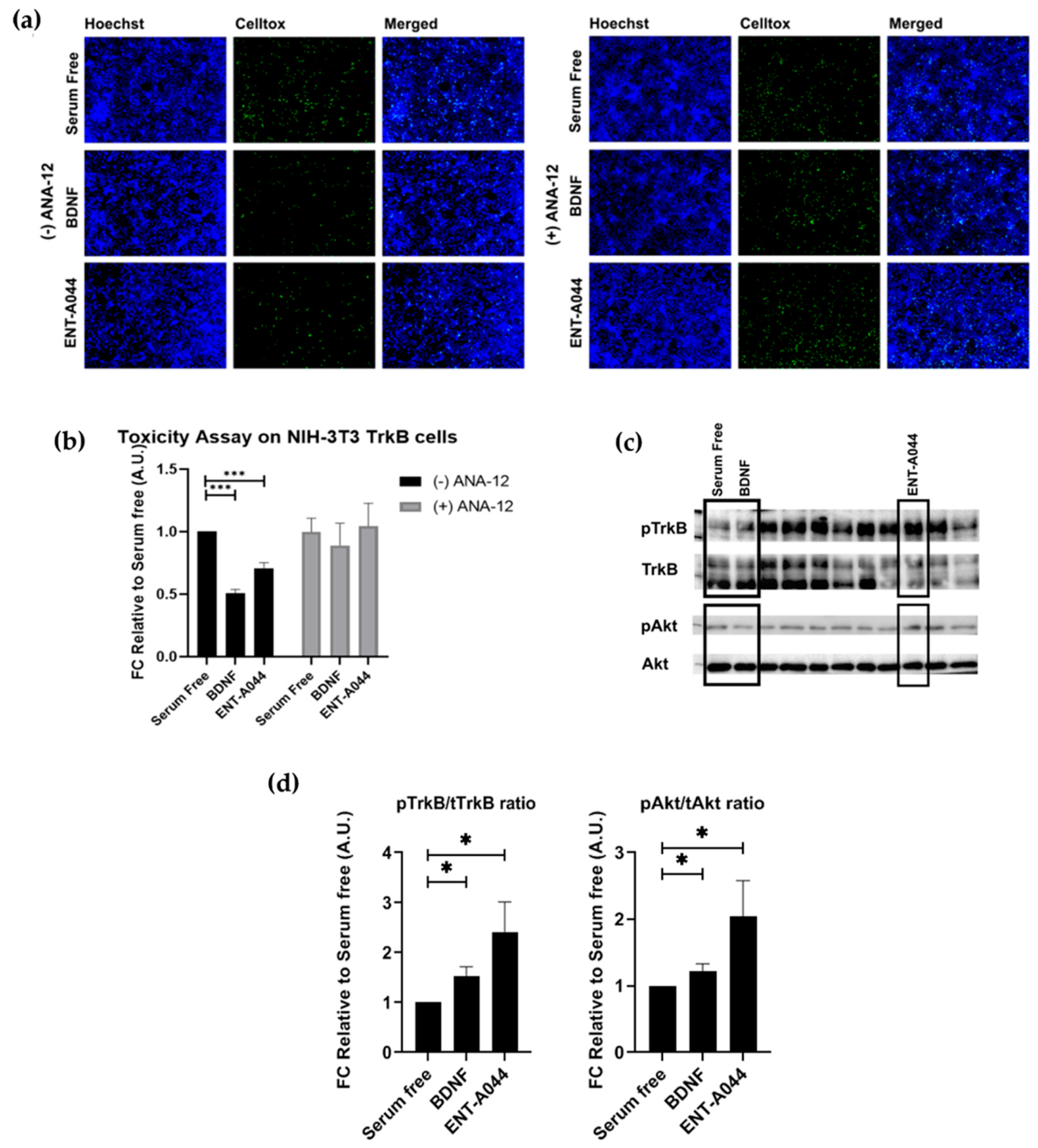
2.2. ENT-A044 Activates TrkB Receptor and Its Downstream Signaling Kinase Akt and Protects NIH-3T3-TrkB Cells from Serum Deprivation-Induced Cell Death
2.3. ENT-A044 Promotes Cell Death by Activating p75NTR in Transiently Transfected HEK293T Cells
2.4. ENT-A044 Promotes Cell Survival in P7 Mouse Hippocampal NSCs
2.5. ENT-A044 Shows a Strong Pro-Apoptotic Effect on Human NPCs
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Primary Cell Cultures
4.3. Generation and Culture of Human Neural Progenitor Cells (NPCs)
4.4. Treatment with Compound ENTA-044
4.5. Immunoprecipitation and Immunoblotting
4.6. Cell Tox Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Dawbarn, D. Clinical relevance of the neurotrophins and their receptors. Clin. Sci. 2006, 110, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.R.; Yoon, S.O.; Carter, B.D. The Biological Functions and Signaling Mechanisms of the p75 Neurotrophin Receptor. Handb. Exp. Pharmacol. 2014, 220, 121–164. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.; Williams, K. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen. Res. 2015, 10, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Gentry, J.J.; Barker, P.A.; Carter, B.D. The p75 neurotrophin receptor: Multiple interactors and numerous functions. Prog. Brain Res. 2004, 146, 25–39. [Google Scholar] [CrossRef]
- Defreitas, M.F.; Mcquillen, P.S.; Shatz, C.J. A Novel p75NTR Signaling Pathway Promotes Survival, Not Death, of Immunopurified Neocortical Subplate Neurons. J. Neurosci. 2001, 21, 5121–5129. [Google Scholar] [CrossRef]
- Vilar, M.; Charalampopoulos, I.; Kenchappa, R.S.; Simi, A.; Karaca, E.; Reversi, A.; Choi, S.; Bothwell, M.; Mingarro, I.; Friedman, W.J.; et al. Activation of the p75 Neurotrophin Receptor through Conformational Rearrangement of Disulphide-Linked Receptor Dimers. Neuron 2009, 62, 72–83. [Google Scholar] [CrossRef]
- Charalampopoulos, I.; Vicario, A.; Pediaditakis, I.; Gravanis, A.; Simi, A.; Ibáñez, C.F. Genetic Dissection of Neurotrophin Signaling through the p75 Neurotrophin Receptor. Cell Rep. 2012, 2, 1563–1570. [Google Scholar] [CrossRef]
- Zigova, T.; Pencea, V.; Wiegand, S.J.; Luskin, M.B. Intraventricular Administration of BDNF Increases the Number of Newly Generated Neurons in the Adult Olfactory Bulb. Mol. Cell. Neurosci. 1998, 11, 234–245. [Google Scholar] [CrossRef]
- Frielingsdorf, H.; Simpson, D.R.; Thal, L.J.; Pizzo, D.P. Nerve growth factor promotes survival of new neurons in the adult hippocampus. Neurobiol. Dis. 2007, 26, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Marinelli, S.; Ceci, M.; Vignone, D.; Amato, G.; Malerba, F.; Paoletti, F.; Meli, G.; Viegi, A.; Pavone, F.; et al. Intranasal “painless” Human Nerve Growth Factors Slows Amyloid Neurodegeneration and Prevents Memory Deficits in App X PS1 Mice. PLoS ONE 2012, 7, e37555. [Google Scholar] [CrossRef]
- Capsoni, S.; Cattaneo, A. Getting Into the Brain: The Intranasal Approach to Enhance the Delivery of Nerve Growth Factor and Its Painless Derivative in Alzheimer’s Disease and Down Syndrome. Front. Neurosci. 2022, 16, 773347. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, I.; Charalampopoulos, I.; Alexaki, V.-I.; Avlonitis, N.; Pediaditakis, I.; Efstathopoulos, P.; Calogeropoulou, T.; Castanas, E.; Gravanis, A. Neurosteroid Dehydroepiandrosterone Interacts with Nerve Growth Factor (NGF) Receptors, Preventing Neuronal Apoptosis. PLoS Biol. 2011, 9, e1001051. [Google Scholar] [CrossRef]
- Charalampopoulos, I.; Remboutsika, E.; Margioris, A.N.; Gravanis, A. Neurosteroids as modulators of neurogenesis and neuronal survival. Trends Endocrinol. Metab. 2008, 19, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Charalampopoulos, I.; Tsatsanis, C.; Dermitzaki, E.; Alexaki, V.-I.; Castanas, E.; Margioris, A.N.; Gravanis, A. Dehydroepiandrosterone and allopregnanolone protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 8209–8214. [Google Scholar] [CrossRef] [PubMed]
- Pediaditakis, I.; Kourgiantaki, A.; Prousis, K.C.; Potamitis, C.; Xanthopoulos, K.P.; Zervou, M.; Calogeropoulou, T.; Charalampopoulos, I.; Gravanis, A. BNN27, a 17-Spiroepoxy Steroid Derivative, Interacts With and Activates p75 Neurotrophin Receptor, Rescuing Cerebellar Granule Neurons from Apoptosis. Front. Pharmacol. 2016, 7, 512. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Efstathopoulos, P.; Prousis, K.C.; Zervou, M.; Arévalo, J.C.; Alexaki, V.I.; Nikoletopoulou, V.; Karagianni, E.; Potamitis, C.; Tavernarakis, N.; et al. Selective and differential interactions of BNN27, a novel C17-spiroepoxy steroid derivative, with TrkA receptors, regulating neuronal survival and differentiation. Neuropharmacology 2016, 111, 266–282. [Google Scholar] [CrossRef]
- Barker, P.A.; Shooter, E.M. Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA on PC12 cells. Neuron 1994, 13, 203–215. [Google Scholar] [CrossRef]
- Rogdakis, T.; Charou, D.; Latorrata, A.; Papadimitriou, E.; Tsengenes, A.; Athanasiou, C.; Papadopoulou, M.; Chalikiopoulou, C.; Katsila, T.; Ramos, I.; et al. Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity. Biomedicines 2022, 10, 614. [Google Scholar] [CrossRef]
- Estus, S.; Zaks, W.J.; Freeman, R.S.; Gruda, M.; Bravo, R.; Johnson, E.M. Altered gene expression in neurons during programmed cell death: Identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 1994, 127, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Palmada, M.; Kanwal, S.; Rutkoski, N.; Gustafson-Brown, C.; Johnson, R.; Wisdom, R.; Carter, B. c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J. Cell Biol. 2002, 158, 453–461. [Google Scholar] [CrossRef]
- Compagnone, N.A.; Mellon, S.H. Neurosteroids: Biosynthesis and Function of These Novel Neuromodulators. Front. Neuroendocr. 2000, 21, 1–56. [Google Scholar] [CrossRef]
- Calogeropoulou, T.; Avlonitis, N.; Minas, V.; Alexi, X.; Pantzou, A.; Charalampopoulos, I.; Zervou, M.; Vergou, V.; Katsanou, E.S.; Lazaridis, I.; et al. Novel Dehydroepiandrosterone Derivatives with Antiapoptotic, Neuroprotective Activity. J. Med. Chem. 2009, 52, 6569–6587. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Hamanoue, M.; Middleton, G.; Wyatt, S.; Jaffray, E.; Hay, R.T.; Davies, A.M. p75-Mediated NF-κB Activation Enhances the Survival Response of Developing Sensory Neurons to Nerve Growth Factor. Mol. Cell. Neurosci. 1999, 14, 28–40. [Google Scholar] [CrossRef]
- Carter, B.D.; Kaltschmidt, C.; Kaltschmidt, B.; Offenhäuser, N.; Böhm-Matthaei, R.; Baeuerle, P.A.; Barde, Y.-A. Selective Activation of NF-κB by Nerve Growth Factor Through the Neurotrophin Receptor p75. Science 1996, 272, 542–545. [Google Scholar] [CrossRef]
- Vicario, A.; Kisiswa, L.; Tann, J.Y.; Kelly, C.E.; Ibáñez, C.F. Neuron-type-specific signaling by the p75NTR death receptor regulated by differential proteolytic cleavage. J. Cell Sci. 2015, 128, 1507–1517. [Google Scholar] [CrossRef]
- Nykjaer, A.; Willnow, T.E.; Petersen, C.M. p75NTR—Live or let die. Curr. Opin. Neurobiol. 2005, 15, 49–57. [Google Scholar] [CrossRef]
- Okumura, T.; Shimada, Y.; Imamura, M.; Yasumoto, S. Neurotrophin receptor p75NTR characterizes human esophageal keratinocyte stem cells in vitro. Oncogene 2003, 22, 4017–4026. [Google Scholar] [CrossRef]
- Okumura, T.; Tsunoda, S.; Mori, Y.; Ito, T.; Kikuchi, K.; Wang, T.C.; Yasumoto, S.; Shimada, Y. The Biological Role of the Low-Affinity p75 Neurotrophin Receptor in Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. 2006, 12, 5096–5103. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Pan, Y.; Zhao, L.; Zhai, H.; Li, X.; Sun, L.; He, L.; Chen, Y.; Hong, L.; Du, Y.; et al. p75 Neurotrophin Receptor Suppresses the Proliferation of Human Gastric Cancer Cells. Neoplasia 2007, 9, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Yuanlong, H.; Haifeng, J.; Xiaoyin, Z.; Jialin, S.; Jie, L.; Li, Y.; Huahong, X.; Jiugang, S.; Yanglin, P.; Kaichun, W.; et al. The inhibitory effect of p75 neurotrophin receptor on growth of human hepatocellular carcinoma cells. Cancer Lett. 2008, 268, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-D.; Yuan, Y.; Liu, X.-H.; Gong, D.-J.; Bai, C.-G.; Wang, F.; Luo, J.-H.; Xu, Z.-Y. Self-renewal and chemotherapy resistance of p75NTR positive cells in esophageal squamous cell carcinomas. BMC Cancer 2009, 9, 9. [Google Scholar] [CrossRef]
- Blondy, S.; Christou, N.; David, V.; Verdier, M.; Jauberteau, M.-O.; Mathonnet, M.; Perraud, A. Neurotrophins and their involvement in digestive cancers. Cell Death Dis. 2019, 10, 123. [Google Scholar] [CrossRef]
- Goh, E.T.; Lin, Z.; Ahn, B.Y.; Lopes-Rodrigues, V.; Dang, N.H.; Salim, S.; Berger, B.; Dymock, B.; Senger, D.L.; Ibáñez, C.F. A Small Molecule Targeting the Transmembrane Domain of Death Receptor p75NTR Induces Melanoma Cell Death and Reduces Tumor Growth. Cell Chem. Biol. 2018, 25, 1485–1494.e5. [Google Scholar] [CrossRef]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.-L.; Cui, Q.-L.; Schambach, A.; Kim, K.-P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulou, M.A.; Rogdakis, T.; Charou, D.; Peteinareli, M.; Ntarntani, K.; Gravanis, A.; Chanoumidou, K.; Charalampopoulos, I. Neurotrophin Analog ENT-A044 Activates the p75 Neurotrophin Receptor, Regulating Neuronal Survival in a Cell Context-Dependent Manner. Int. J. Mol. Sci. 2023, 24, 11683. https://doi.org/10.3390/ijms241411683
Papadopoulou MA, Rogdakis T, Charou D, Peteinareli M, Ntarntani K, Gravanis A, Chanoumidou K, Charalampopoulos I. Neurotrophin Analog ENT-A044 Activates the p75 Neurotrophin Receptor, Regulating Neuronal Survival in a Cell Context-Dependent Manner. International Journal of Molecular Sciences. 2023; 24(14):11683. https://doi.org/10.3390/ijms241411683
Chicago/Turabian StylePapadopoulou, Maria Anna, Thanasis Rogdakis, Despoina Charou, Maria Peteinareli, Katerina Ntarntani, Achille Gravanis, Konstantina Chanoumidou, and Ioannis Charalampopoulos. 2023. "Neurotrophin Analog ENT-A044 Activates the p75 Neurotrophin Receptor, Regulating Neuronal Survival in a Cell Context-Dependent Manner" International Journal of Molecular Sciences 24, no. 14: 11683. https://doi.org/10.3390/ijms241411683
APA StylePapadopoulou, M. A., Rogdakis, T., Charou, D., Peteinareli, M., Ntarntani, K., Gravanis, A., Chanoumidou, K., & Charalampopoulos, I. (2023). Neurotrophin Analog ENT-A044 Activates the p75 Neurotrophin Receptor, Regulating Neuronal Survival in a Cell Context-Dependent Manner. International Journal of Molecular Sciences, 24(14), 11683. https://doi.org/10.3390/ijms241411683