
Transcriptomic Analysis Followed by the Isolation of Extracellular Bacteriolytic Proteases from Lysobacter capsici VKM B-2533T
 ,
,
Abstract
1. Introduction
2. Results
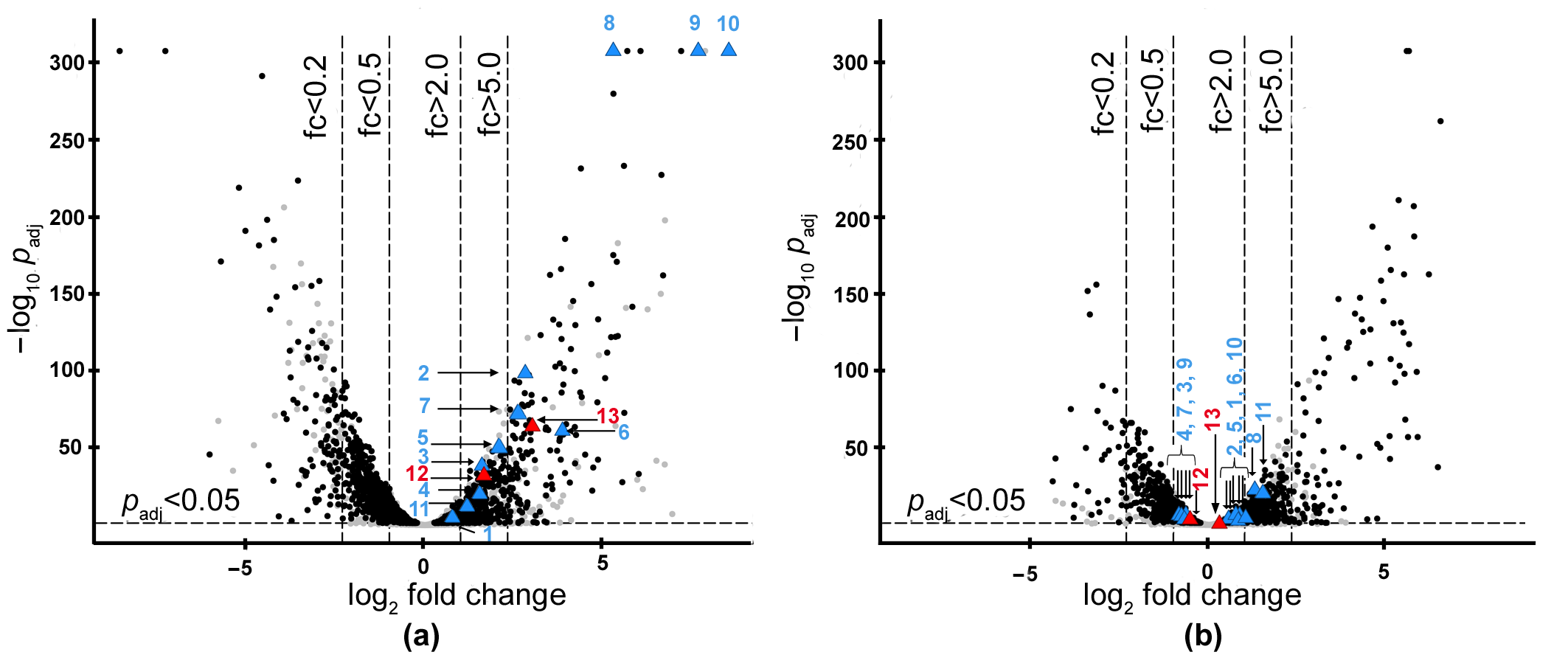
2.1. Search for the Genes of Bacteriolytic Enzymes Using the Transcriptomic Approach
2.2. Isolation and Characterization of L. capsici VKM B-2533T Bacteriolytic Enzymes
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Cultivation Conditions
4.2. Isolation of RNA
4.3. RNA-Seq Data Analysis
4.4. Molecular Genetic Kits and Equipment
4.5. Construction of Plasmids and Production of L. capsici Expression Strains
4.6. Purification of Bacteriolytic Enzymes
4.7. Electrophoresis of Proteins in PAG
4.8. Determination of Protein Concentration
4.9. Determination of Total Bacteriolytic Activity
4.10. Determination of Optimal Conditions for the Bacteriolytic Activities of Serp and Serp3 to Be Exhibited
4.11. Measurement of Proteolytic Activity
4.12. Determination of the Specificity of Action of Bacteriolytic Enzymes against Protein Substrates and Autoclaved Bacterial Target Cells
4.13. Determination of Antifungal Activity by the Spot Test
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitaker, D.R. Lytic Enzymes of Sorangium sp. Isolation and Enzymatic Properties of the Alpha- and Beta-Lytic Proteases. Can. J. Biochem. 1965, 43, 1935–1954. [Google Scholar] [CrossRef]
- Panthee, S.; Hamamoto, H.; Paudel, A.; Sekimizu, K. Lysobacter species: A Potential Source of Novel Antibiotics. Arch. Microbiol. 2016, 198, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Brescia, F.; Vlassi, A.; Bejarano, A.; Seidl, B.; Marchetti-Deschmann, M.; Schuhmacher, R.; Puopolo, G. Characterisation of the Antibiotic Profile of Lysobacter capsici AZ78, an Effective Biological Control Agent of Plant Pathogenic Microorganisms. Microorganisms 2021, 9, 1320. [Google Scholar] [CrossRef]
- Stepnaya, O.A.; Ledova, L.A.; Kulaev, I.S. Bacteriolytic Enzymes. Uspekhi Biol. Khimii 1999, 39, 327–354. [Google Scholar]
- Kulaev, I.S.; Stepnaya, O.A.; Tsfasman, I.M.; Tchermenskaja, T.S.; Ledova, L.A.; Zubrizkaja, L.G.; Akimenko, V.K. Bacteriolytic Complex, Method for Producing Said Complex and Strain for Carrying out Said Method. RF Patent No. 2,193,063, 20 November 2002. (In Russian). [Google Scholar]
- Afoshin, A.S.; Kudryakova, I.V.; Borovikova, A.O.; Suzina, N.E.; Toropygin, I.Y.; Shishkova, N.A.; Vasilyeva, N.V. Lytic Potential of Lysobacter capsici VKM B-2533T: Bacteriolytic Enzymes and Outer Membrane Vesicles. Sci. Rep. 2020, 10, 9944. [Google Scholar] [CrossRef] [PubMed]
- Afoshin, A.; Tishchenko, S.; Gabdulkhakov, A.; Kudryakova, I.; Galemina, I.; Zelenov, D.; Leontyevskaya, E.; Saharova, S.; Leontyevskaya, N. Structural and Functional Characterization of Β−lytic Protease from Lysobacter capsici VKM B−2533T. Int. J. Mol. Sci. 2022, 23, 16100. [Google Scholar] [CrossRef] [PubMed]
- Kudryakova, I.; Afoshin, A.; Leontyevskaya, E.; Leontyevskaya, N. The First Homologous Expression System for the β-Lytic Protease of Lysobacter capsici VKM B-2533T, a Promising Antimicrobial Agent. Int. J. Mol. Sci. 2022, 23, 5722. [Google Scholar] [CrossRef]
- Palumbo, J.D.; Sullivan, R.F.; Kobayashi, D.Y. Molecular Characterization and Expression in Escherichia coli of Three Beta-1,3-Glucanase Genes from Lysobacter enzymogenes Strain N4-7. J. Bacteriol. 2003, 185, 4362–4370. [Google Scholar] [CrossRef]
- Yano, S.; Kanno, H.; Tsuhako, H.; Ogasawara, S.; Suyotha, W.; Konno, H.; Makabe, K.; Uechi, K.; Taira, T. Cloning, Expression, and Characterization of a GH 19-Type Chitinase with Antifungal Activity from Lysobacter sp. MK9-1. J. Biosci. Bioeng. 2021, 131, 348–355. [Google Scholar] [CrossRef]
- Cimmino, A.; Bejarano, A.; Masi, M.; Puopolo, G.; Evidente, A. Isolation of 2,5-Diketopiperazines from Lysobacter capsici AZ78 with Activity against Rhodococcus fascians. Nat. Prod. Res. 2021, 35, 4969–4977. [Google Scholar] [CrossRef]
- Puopolo, G.; Cimmino, A.; Palmieri, M.C.; Giovannini, O.; Evidente, A.; Pertot, I. Lysobacter capsici AZ78 Produces Cyclo(L-Pro-L-Tyr), a 2,5-Diketopiperazine with Toxic Activity against Sporangia of Phytophthora infestans and Plasmopara viticola. J. Appl. Microbiol. 2014, 117, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Afoshin, A.S.; Konstantinov, M.A.; Toropygin, I.Y.; Kudryakova, I.V.; Vasilyeva, N.V. β-Lytic Protease of Lysobacter capsici VKM B-2533T. Antibiotics 2020, 9, 744. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, Y.S.; Zolova, O.E.; Shlyapnikov, M.G.; Tsfasman, I.M.; Muranova, T.A.; Stepnaya, O.A.; Kulaev, I.S.; Granovsky, I.E. Cloning and Expression Analysis of Genes Encoding Lytic Endopeptidases L1 and L5 from Lysobacter sp. Strain XL1. Appl. Environ. Microbiol. 2012, 78, 7082–7089. [Google Scholar] [CrossRef] [PubMed]
- Stepanaia, O.A.; Begunova, E.A.; Tsfasman, I.M.; Kulaev, I.S. Bacteriolytic enzyme preparation lysoamidase. Purification and some properties of bacteriolytic peptidase L1. Biokhimiia 1996, 61, 656–663. [Google Scholar]
- Begunova, E.A.; Stepnaya, O.A.; Lysanskaya, V.Y.; Kulaev, I.S. Specificity of the Action of Lysoamidase on Staphylococcus aureus 209P Cell Walls. Biochemistry 2003, 68, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Tishchenko, S.; Gabdulkhakov, A.; Melnik, B.; Kudryakova, I.; Latypov, O.; Vasilyeva, N.; Leontievsky, A. Structural Studies of Component of Lysoamidase Bacteriolytic Complex from Lysobacter sp. XL1. Protein J. 2016, 35, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Vasilyeva, N.V.; Tsfasman, I.M.; Suzina, N.E.; Stepnaya, O.A.; Kulaev, I.S. Secretion of Bacteriolytic Endopeptidase L5 of Lysobacter sp. XL1 into the Medium by Means of Outer Membrane Vesicles. FEBS J. 2008, 275, 3827–3835. [Google Scholar] [CrossRef]
- Vasilyeva, N.V.; Shishkova, N.A.; Marinin, L.I.; Ledova, L.A.; Tsfasman, I.M.; Muranova, T.A.; Stepnaya, O.A.; Kulaev, I.S. Lytic Peptidase L5 of Lysobacter sp. XL1 with Broad Antimicrobial Spectrum. J. Mol. Microbiol. Biotechnol. 2014, 24, 59–66. [Google Scholar] [CrossRef]
- Kudryakova, I.V.; Suzina, N.E.; Vasilyeva, N.V. Biogenesis of Lysobacter sp. XL1 Vesicles. FEMS Microbiol. Lett. 2015, 362, fnv137. [Google Scholar] [CrossRef]
- Kudryakova, I.V.; Gabdulkhakov, A.G.; Tishchenko, S.V.; Lysanskaya, V.Y.; Suzina, N.E.; Tsfasman, I.M.; Afoshin, A.S.; Vasilyeva, N.V. Structural and Functional Properties of Antimicrobial Protein L5 of Lysobacter sp. XL1. Appl. Microbiol. Biotechnol. 2018, 102, 10043–10053. [Google Scholar] [CrossRef]
- Kudryakova, I.V.; Afoshin, A.S.; Ivashina, T.V.; Suzina, N.E.; Leontyevskaya, E.A.; Leontyevskaya, N.V. Deletion of AlpB Gene Influences Outer Membrane Vesicles Biogenesis of Lysobacter sp. XL1. Front. Microbiol. 2021, 12, 715802. [Google Scholar] [CrossRef] [PubMed]
- Stepnaya, O.A.; Tsfasman, I.M.; Logvina, I.A.; Ryazanova, L.P.; Muranova, T.A.; Kulaev, I.S. Isolation and Characterization of a New Extracellular Bacteriolytic Endopeptidase of Lysobacter sp. XL1. Biochemistry 2005, 70, 1031–1037. [Google Scholar] [CrossRef]
- Ohara, T.; Makino, K.; Shinagawa, H.; Nakata, A.; Norioka, S.; Sakiyama, F. Cloning, Nucleotide Sequence, and Expression of Achromobacter Protease I Gene. J. Biol. Chem. 1989, 264, 20625–20631. [Google Scholar] [CrossRef] [PubMed]
- Barequet, I.S.; Ben Simon, G.J.; Safrin, M.; Ohman, D.E.; Kessler, E. Pseudomonas aeruginosa LasA Protease in Treatment of Experimental Staphylococcal Keratitis. Antimicrob. Agents Chemother. 2004, 48, 1681–1687. [Google Scholar] [CrossRef]
- Schindler, C.A.; Schuhardt, V.T. Lysostaphin: A New Bacteriolytic Agent for the Staphylococcus. Proc. Natl. Acad. Sci. USA 1964, 51, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Sugai, M.; Fujiwara, T.; Akiyama, T.; Ohara, M.; Komatsuzawa, H.; Inoue, S.; Suginaka, H. Purification and Molecular Characterization of Glycylglycine Endopeptidase Produced by Staphylococcus capitis EPK1. J. Bacteriol. 1997, 179, 1193–1202. [Google Scholar] [CrossRef]
- Lai, A.C.Y.; Tran, S.; Simmonds, R.S. Functional Characterization of Domains Found within a Lytic Enzyme Produced by Streptococcus equi subsp. zooepidemicus. FEMS Microbiol. Lett. 2002, 215, 133–138. [Google Scholar] [CrossRef]
- Nilsen, T.; Nes, I.F.; Holo, H. Enterolysin A, a Cell Wall-Degrading Bacteriocin from Enterococcus faecalis LMG 2333. Appl. Environ. Microbiol. 2003, 69, 2975–2984. [Google Scholar] [CrossRef]
- Beukes, M.; Bierbaum, G.; Sahl, H.G.; Hastings, J.W. Purification and Partial Characterization of a Murein Hydrolase, Millericin B, Produced by Streptococcus milleri NMSCC 061. Appl. Environ. Microbiol. 2000, 66, 23–28. [Google Scholar] [CrossRef]
- Tang, B.L.; Yang, J.; Chen, X.L.; Wang, P.; Zhao, H.L.; Su, H.N.; Li, C.Y.; Yu, Y.; Zhong, S.; Wang, L.; et al. A Predator-Prey Interaction between a Marine Pseudoalteromonas sp. and Gram-Positive Bacteria. Nat. Commun. 2020, 11, 285. [Google Scholar] [CrossRef]
- Wang, P.; Chen, H.; Qian, G.; Liu, F. LetR Is a TetR Family Transcription Factor from Lysobacter Controlling Antifungal Antibiotic Biosynthesis. Appl. Microbiol. Biotechnol. 2017, 101, 3273–3282. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Jiang, J.; Taylor, A.J.; Leite, A.D.L.; Dodds, E.D.; Du, L. Outer Membrane Vesicle-Mediated Codelivery of the Antifungal HSAF Metabolites and Lytic Polysaccharide Monooxygenase in the Predatory Lysobacter enzymogenes. ACS Chem. Biol. 2021, 16, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- van Staden, A.D.P.; van Zyl, W.F.; Trindade, M.; Dicks, L.M.T.; Smith, C. Therapeutic Application of Lantibiotics and Other Lanthipeptides: Old and New Findings. Appl. Environ. Microbiol. 2021, 87, e00186-21. [Google Scholar] [CrossRef] [PubMed]
- DebRoy, S.; Dao, J.; Söderberg, M.; Rossier, O.; Cianciotto, N.P. Legionella Pneumophila Type II Secretome Reveals Unique Exoproteins and a Chitinase That Promotes Bacterial Persistence in the Lung. Proc. Natl. Acad. Sci. USA 2006, 103, 19146–19151. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, K.V.; Sandkvist, M. Architecture, Function, and Substrates of the Type II Secretion System. EcoSal Plus 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Sgro, G.G.; Oka, G.U.; Souza, D.P.; Cenens, W.; Bayer-Santos, E.; Matsuyama, B.Y.; Bueno, N.F.; dos Santos, T.R.; Alvarez-Martinez, C.E.; Salinas, R.K.; et al. Bacteria-Killing Type IV Secretion Systems. Front. Microbiol. 2019, 10, 1078. [Google Scholar] [CrossRef]
- Trunk, K.; Coulthurst, S.J.; Quinn, J. A New Front in Microbial Warfare—Delivery of Antifungal Effectors by the Type VI Secretion System. J. Fungi 2019, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Xu, K.; Shen, D.; Chou, S.H.; Gomelsky, M.; Qian, G. Antifungal Weapons of Lysobacter, a Mighty Biocontrol Agent. Environ. Microbiol. 2021, 23, 5704–5715. [Google Scholar] [CrossRef]
- Souza, D.P.; Oka, G.U.; Alvarez-Martinez, C.E.; Bisson-Filho, A.W.; Dunger, G.; Hobeika, L.; Cavalcante, N.S.; Alegria, M.C.; Barbosa, L.R.S.; Salinas, R.K.; et al. Bacterial Killing via a Type IV Secretion System. Nat. Commun. 2015, 6, 6453. [Google Scholar] [CrossRef]
- Shen, X.; Wang, B.; Yang, N.; Zhang, L.; Shen, D.; Wu, H.; Dong, Y.; Niu, B.; Chou, S.H.; Puopolo, G.; et al. Lysobacter enzymogenes Antagonizes Soilborne Bacteria Using the Type IV Secretion System. Environ. Microbiol. 2021, 23, 4673–4688. [Google Scholar] [CrossRef]
- Tomada, S.; Sonego, P.; Moretto, M.; Engelen, K.; Pertot, I.; Perazzolli, M.; Puopolo, G. Dual RNA-Seq of Lysobacter capsici AZ78—Phytophthora Infestans Interaction Shows the Implementation of Attack Strategies by the Bacterium and Unsuccessful Oomycete Defense Responses. Environ. Microbiol. 2017, 19, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Bayer-Santos, E.; Ceseti, L.d.M.; Farah, C.S.; Alvarez-Martinez, C.E. Distribution, Function and Regulation of Type 6 Secretion Systems of Xanthomonadales. Front. Microbiol. 2019, 10, 1635. [Google Scholar] [CrossRef] [PubMed]
- Trunk, K.; Peltier, J.; Liu, Y.C.; Dill, B.D.; Walker, L.; Gow, N.A.R.; Stark, M.J.R.; Quinn, J.; Strahl, H.; Trost, M.; et al. The Type VI Secretion System Deploys Antifungal Effectors against Microbial Competitors. Nat. Microbiol. 2018, 3, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, R.; Aslam, Z.; Jeon, C.O.; Chung, Y.R. Lysobacter capsici sp. nov., with Antimicrobial Activity, Isolated from the Rhizosphere of Pepper, and Emended Description of the Genus Lysobacter. Int. J. Syst. Evol. Microbiol. 2008, 58, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Bullock, W.O.; Fernandez, J.M.; Short, J.M. XL1-Blue: A High Efficiency Plasmid Transforming RecA Escherichia coli Strain with Beta-Galactosidase Selection. BioTecchniques 1987, 5, 376–379. [Google Scholar]
- Kovach, M.E.; Elzer, P.H.; Hill, D.S.; Robertson, G.T.; Farris, M.A.; Roop, R.M.; Peterson, K.M. Four New Derivatives of the Broad-Host-Range Cloning Vector PBBR1MCS, Carrying Different Antibiotic-Resistance Cassettes. Gene 1995, 166, 175–176. [Google Scholar] [CrossRef]
- Segarra, G.; Puopolo, G.; Giovannini, O.; Pertot, I. Stepwise Flow Diagram for the Development of Formulations of Non Spore-Forming Bacteria against Foliar Pathogens: The Case of Lysobacter capsici AZ78. J. Biotechnol. 2015, 216, 56–64. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 2 February 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.F.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; ISBN 0-87969-576-5. [Google Scholar]
- Hanahan, D. Studies on Transformation of Escherichia coli with Plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Fernandez-Patron, C.; Castellanos-Serra, L.; Rodriguez, P. Reverse Staining of Sodium Dodecyl Sulfate Polyacrylamide Gels by Imidazole-Zinc Salts: Sensitive Detection of Unmodified Proteins. Biotechniques 1992, 12, 564–573. [Google Scholar]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Hull, M.E. Studies on Milk Proteins. II. Colorimetric Determination of the Partial Hydrolysis of the Proteins in Milk. J. Dairy Sci. 1947, 30, 881–884. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes (Protein_Id)/(Locus_Tag) | Change of Expression * | ||
|---|---|---|---|
| RM | SYM | ||
| Bacteriolytic enzymes | |||
| Protease Blp (UOF15870.1)/(IEQ11_04180) | 7.0 | 1.5 | |
| Protease L1 (UOF13170.1)/(IEQ11_15580) | 39.1 | 2.4 | |
| Protease L4 (UOF16892.1)/(IEQ11_09745) | 2.9 | 0.6 | |
| Protease L5 (UOF13168.1)/(IEQ11_15570) | 6.0 | 0.6 | |
| Protease Serp (UOF16681.1)/(IEQ11_08595) | 3.2 | 0.7 | |
| Protease Serp6 (UOF14691.1)/(IEQ11_23755) | 2.2 | 2.8 | |
| Protease Serp7 (UOF17397.1)/(IEQ11_12530) | 4.2 | 1.7 | |
| N-acetylglucosaminidase (UOF15745.1)/(IEQ11_03495) | 1.7 | 1.8 | |
| Antifungal enzymes | |||
| Glucanase GluA (UOF14439.1)/(IEQ11_22400) | 370.9 | 2.0 | |
| Glucanase GluB (UOF13513.1)/(IEQ11_17420) | 204.6 | 0.6 | |
| Glucanase GluC (UOF16369.1)/(IEQ11_06885) | 3.0 | 0.6 | |
| Chitinase (UOF12917.1)/(IEQ11_14225) | 14.5 | 1.8 | |
| Enzymes (Protein_Id)/(Locus_Tag) | Change of Expression of Genes | |
|---|---|---|
| RM | SYM | |
| Secondary metabolites (antibiotics and peptides) | ||
| Class III lanthipeptide (UOF16985.1)/(IEQ11_10290) | 7.7 | 4.0 |
| Class III lanthionine synthetase LanKC (UOF16986.1)/(IEQ11_10295) | 8.5 | 3.6 |
| Class 2 lanthipeptide synthetase LanM family protein (UOF15613.1)/(IEQ11_02800) | 1.7 | 3.5 |
| Class 2 lanthipeptide synthetase LanM family protein (UOF15622.1)/(IEQ11_02845) | 5.2 | 2.0 |
| Non-ribosomal peptide synthetase (UOF17373.1)/(IEQ11_12390) | – | 1.4 |
| Non-ribosomal peptide synthetase (UOF17381.1)/(IEQ11_12435) | – | 2.4 |
| Non-ribosomal peptide synthetase (UOF17380.1)/(IEQ11_12430) | – | 1.7 |
| Class 2 lanthipeptide synthetase LanM family protein (UOF14422.1)/(IEQ11_22315) | 3.1 | 2.2 |
| Non-ribosomal peptide synthetase (UOF14553.1)/(IEQ11_23005) | 1.5 | – |
| Non-ribosomal peptide synthetase (UOF14554.1)/(IEQ11_23010) | 1.8 | – |
| HSAF biosynthetic non-ribosomal peptide synthetase/polyketide synthase (UOF16764.1)/(IEQ11_09055) | 3.8 | 1.4 |
| Bacterial Substrates | Micrococcus luteus Ac-2230T | Kocuria rosea Ac-2200T | Bacillus cereus 217 | Proteus vulgaris H-19 | Pseudomonas aeruginosa | S. aureus 209P | |
|---|---|---|---|---|---|---|---|
| Enzymes | Bacteriolytic Activity, LU/mg | ||||||
| Serp | 7700 ± 346 | 0 | 1492 ± 142 | 2533 ± 126 | 806 ± 63 | 1422 ± 258 | |
| Serp3 | 24,700 ± 816 | 0 | 0 | 7114 ± 251 | 5536 ± 286 | 3562 ± 112 | |
| Bacteriolytic Enzymes | Casein | Azofibrin | Hemoglobin | Gelatin | Elastin | Collagen |
|---|---|---|---|---|---|---|
| Serp | + | + | – | – | – | – |
| Serp3 | + | + | – | – | – | – |
| Bacteriolytic Enzyme (Protein_Id)/(Locus_Tag) | Identity with Earlier Isolated Lysobacter Proteins | References |
|---|---|---|
| Blp (UOF15870.1)/(IEQ11_04180) | Isolated from L. capsici VKM B-2533T and characterized | [6,13] |
| L1 (UOF13170.1)/(IEQ11_15580) | 99.25% identical with L1 (ACZ72924.1) of L. capsici XL1 [14]; has been investigated | [15,16,17] |
| L5 (UOF13168.1)/(IEQ11_15570) | 99.00% identical with L5 (ACZ72925.1) of L. capsici XL1; has been investigated but was identified in strain VKM B-2533T for the first time | [18,19,20,21,22] |
| L4 (UOF16892.1)/(IEQ11_09745) | 99.7% identical with L4 of L. capsici XL1; has been investigated | [23] |
| Serp (UOF16681.1) */(IEQ11_08595) | New enzyme, isolated from L. capsici VKM B-2533T, investigated in the present work | [6] |
| Serp6 (UOF14691.1)/(IEQ11_23755) | New enzyme, isolated from L. capsici VKM B-2533T, but has not been investigated | [6] |
| Serp7 (UOF17397.1)/(IEQ11_12530) | 76.70% identical with lysin-specific serine protease (P15636.1) of L. enzymogenes, isolated from L. capsici VKM B-2533T, but has not been investigated as a bacteriolytic enzyme | [6,24] |
| Serp3 (UOF12968.1) */(IEQ11_14490) | New enzyme, identified and investigated in the present work | |
| N-acetylglucosaminidase (UOF15745.1)/(IEQ11_03495) | New enzyme, isolated from L. capsici VKM B-2533T, but has not been investigated | [6] |
| Strains and Plasmids | Characteristic | Refs |
|---|---|---|
| Lysobacter capsici VKM B-2533T | Wild type | [45] |
| L. capsici PT5–serp(6his) | Strain L. capsici VKM B-2533T with plasmid pBBR1-MCS5 PT5–serp(6his) | This work |
| L. capsici PGroEL(A)–serp3(6his) | Strain L. capsici VKM B-2533T with plasmid pBBR1-MCS5 PGroEL(A)–serp3(6his) | This work |
| Escherichia coli XL1–Blue | recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 lac/[F′::Tn10 proAB+ lacIq lacZΔM15 traD36] | [46] |
| pBBR1-MCS5 * | Broad-host vector, GmR | [47] |
| pBBR1-MCS5 PT5–gfp | Plasmid pBBR1-MCS5 with gene gfp under the regulation of bacteriophage T5 promoter | [8] |
| pBBR1-MCS5 PGroEL(A)–gfp | Plasmid pBBR1-MCS5 with gene gfp under the regulation of GroEL promoter of L. enzymogenes with modification | [8] |
| pBBR1-MCS5 PT5–serp(6his) | Plasmid pBBR1-MCS5 with gene serp and a tag of 18 nucleotides encoding His6-tag, under the regulation of bacteriophage T5 promoter | This work |
| pBBR1-MCS5 PGroEL(A)–serp3(6his) | Plasmid pBBR1-MCS5 with gene serp3 and a tag of 18 nucleotides encoding His6-tag, under the regulation of GroE promoter of L. enzymogenes with modification | This work |
| Oligonucleotides | Sequence | Goal |
|---|---|---|
| Oligonucleotides used for cloning | ||
| Serp_BamHI (for) | GGATCCATGATCCGCAAGAACGCACTTTG | To amplify gene serp(6his) 1395 bp long with DNA of L. capsici VKM B-2533T |
| Serp_HindIII (rev) | AAGCTTTCAATGATGATGATGATGATGGGGATTGAAATAGCTCGACACG | |
| Serp3_HindIII (for) | AAGCTTATGCGTAAGTTCAGTTTGTCGATCCTC | To amplify gene serp3(6his) 1233 bp long with DNA of L. capsici VKM B-2533T |
| Serp3_BamHI (rev) | GGATCCTTAATGATGATGATGATGATGCGGACTGGTGCG | |
| Oligonucleotides used for sequencing | ||
| T5_KpnI (for) | GGTACCGTGCCACCTGACGTCTAAG | To confirm absence of mutation in the cloned sequence in the assembled pBBR1-MCS5 PT5–serp(6his) constructs |
| T5_XbaI (rev) | TCTAGACTGAAAATCTCGCCAAGCTAGC | |
| Gro_KpnI (for) | GGTACCCGGACCGACGCCTGTCA | To confirm absence of mutation in the cloned sequence in the assembled pBBR1-MCS5 pBBR1-MCS5 PGroEL(A)–serp3(6his) constructs |
| Term_XbaI (rev) | TCTAGAAGAGTTTGTAGAAACGCAAAAAGGC | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afoshin, A.; Kudryakova, I.; Tarlachkov, S.; Leontyevskaya, E.; Zelenov, D.; Rudenko, P.; Leontyevskaya, N. Transcriptomic Analysis Followed by the Isolation of Extracellular Bacteriolytic Proteases from Lysobacter capsici VKM B-2533T. Int. J. Mol. Sci. 2023, 24, 11652. https://doi.org/10.3390/ijms241411652
Afoshin A, Kudryakova I, Tarlachkov S, Leontyevskaya E, Zelenov D, Rudenko P, Leontyevskaya N. Transcriptomic Analysis Followed by the Isolation of Extracellular Bacteriolytic Proteases from Lysobacter capsici VKM B-2533T. International Journal of Molecular Sciences. 2023; 24(14):11652. https://doi.org/10.3390/ijms241411652
Chicago/Turabian StyleAfoshin, Alexey, Irina Kudryakova, Sergey Tarlachkov, Elena Leontyevskaya, Dmitry Zelenov, Pavel Rudenko, and Natalya Leontyevskaya (Vasilyeva). 2023. "Transcriptomic Analysis Followed by the Isolation of Extracellular Bacteriolytic Proteases from Lysobacter capsici VKM B-2533T" International Journal of Molecular Sciences 24, no. 14: 11652. https://doi.org/10.3390/ijms241411652
APA StyleAfoshin, A., Kudryakova, I., Tarlachkov, S., Leontyevskaya, E., Zelenov, D., Rudenko, P., & Leontyevskaya, N. (2023). Transcriptomic Analysis Followed by the Isolation of Extracellular Bacteriolytic Proteases from Lysobacter capsici VKM B-2533T. International Journal of Molecular Sciences, 24(14), 11652. https://doi.org/10.3390/ijms241411652