
T1AM/TAAR1 System Reduces Inflammatory Response and β-Amyloid Toxicity in Human Microglial HMC3 Cell Line

 ,
,  , ,
, , 
Abstract
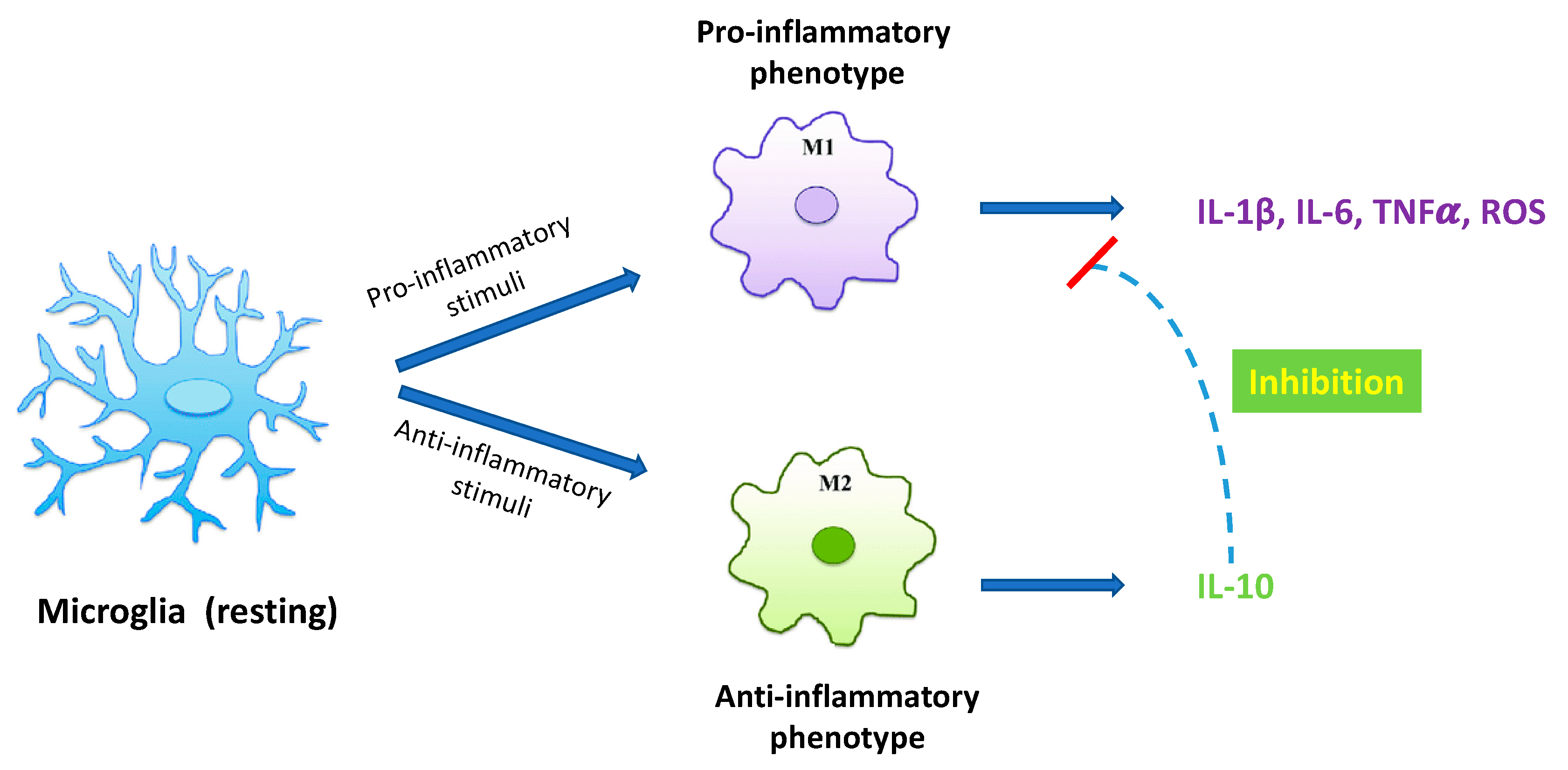
1. Introduction
2. Results
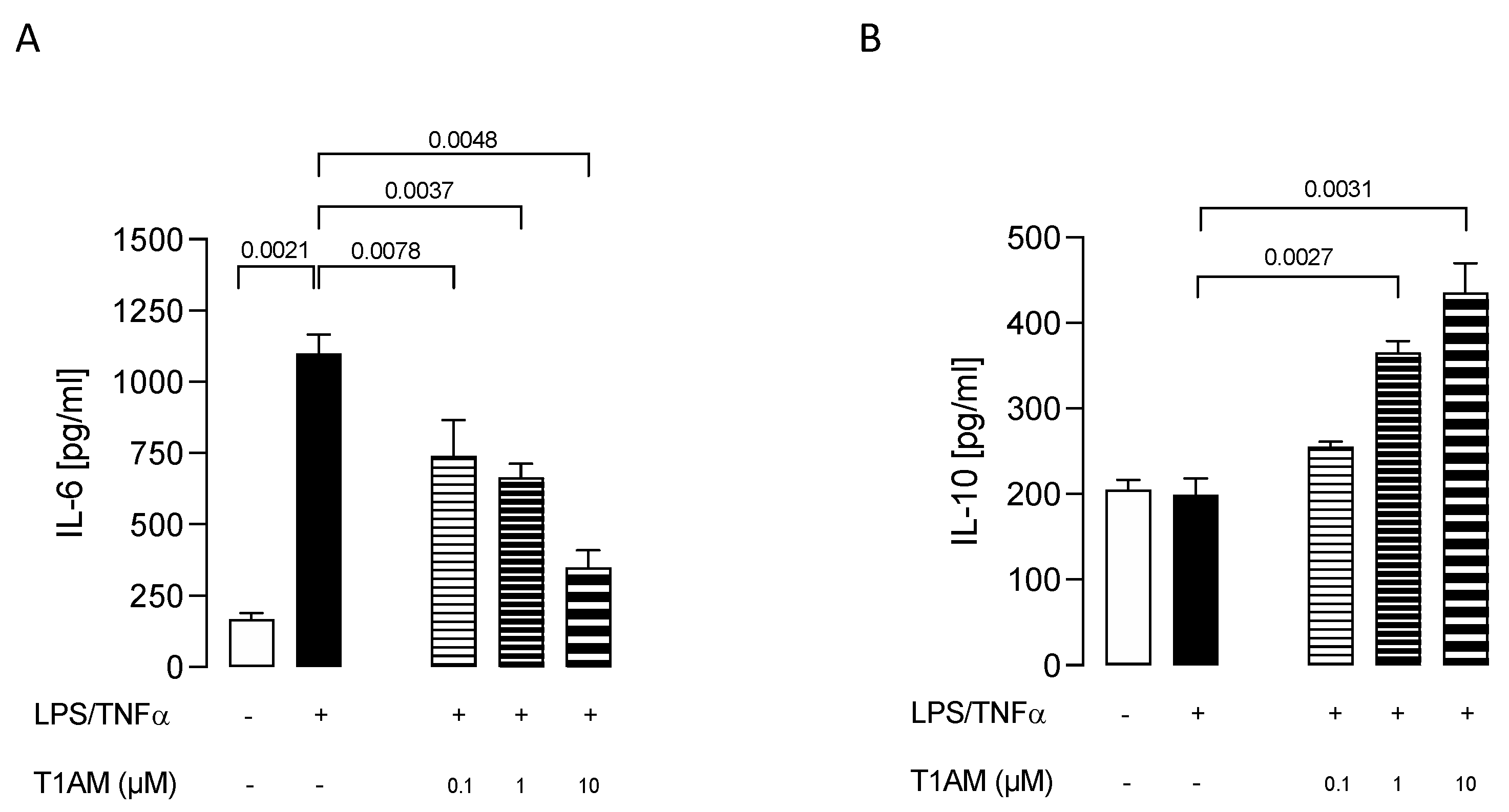
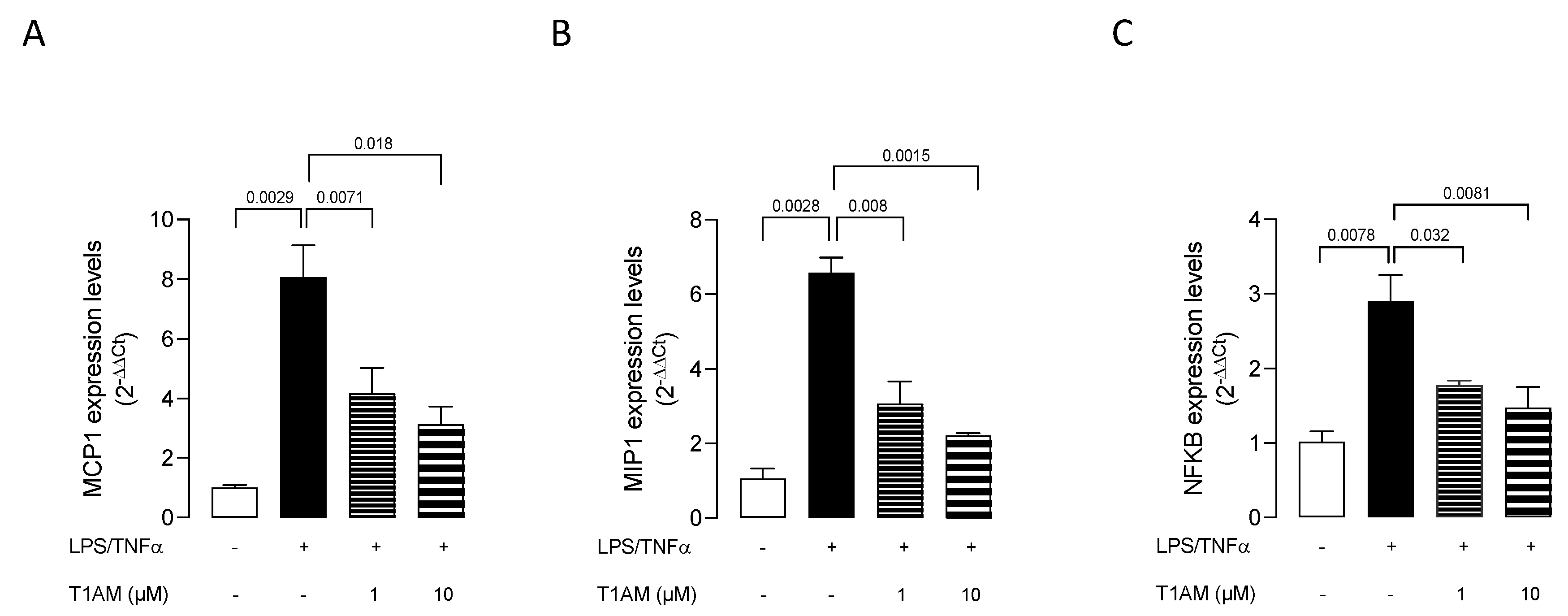
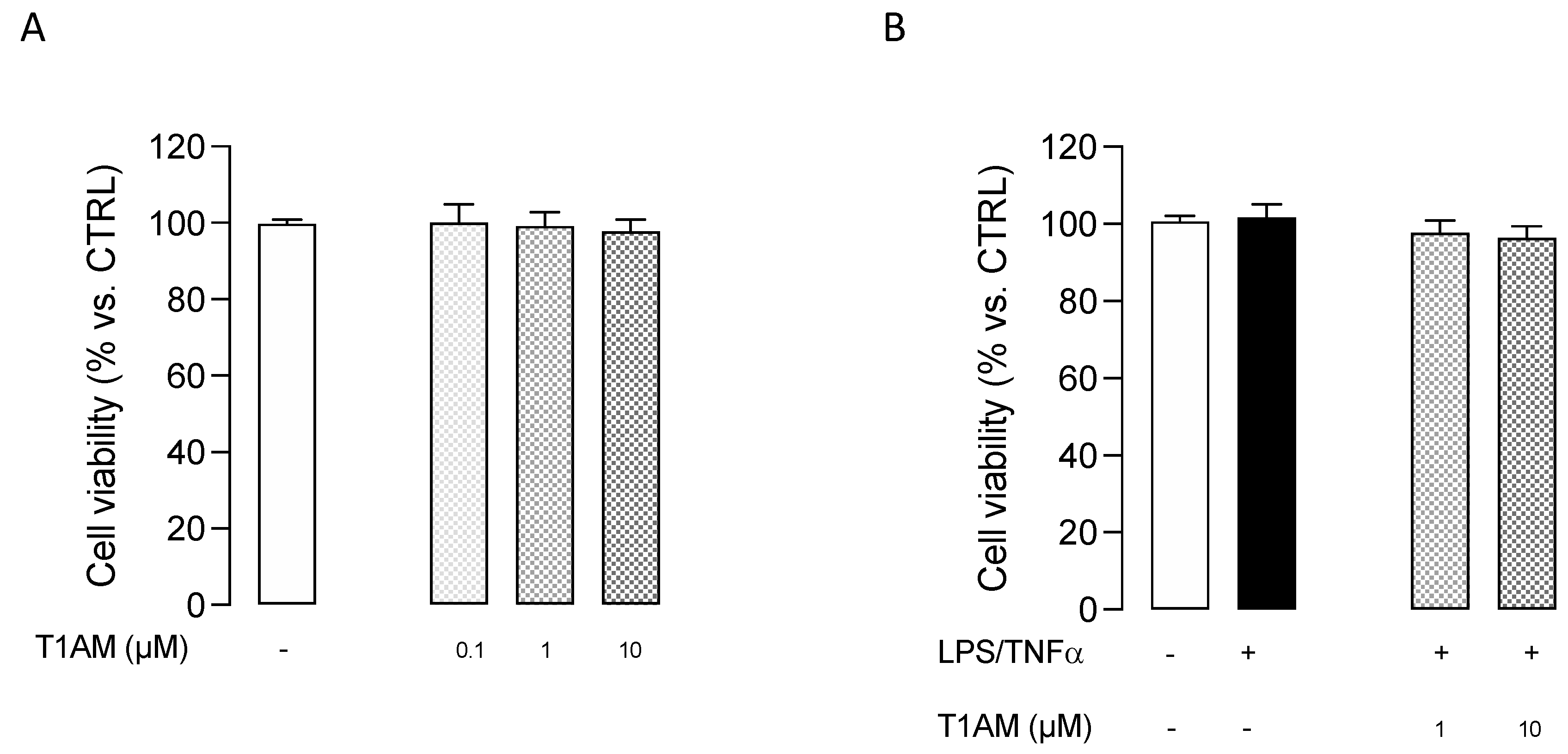
2.1. T1AM Decreased the Inflammatory Phenotype of LPS/TNFα-Stimulated HMC3 Human Microglial Cells
2.2. T1AM Uptake and Metabolism in HMC3 Human Microglial Cells
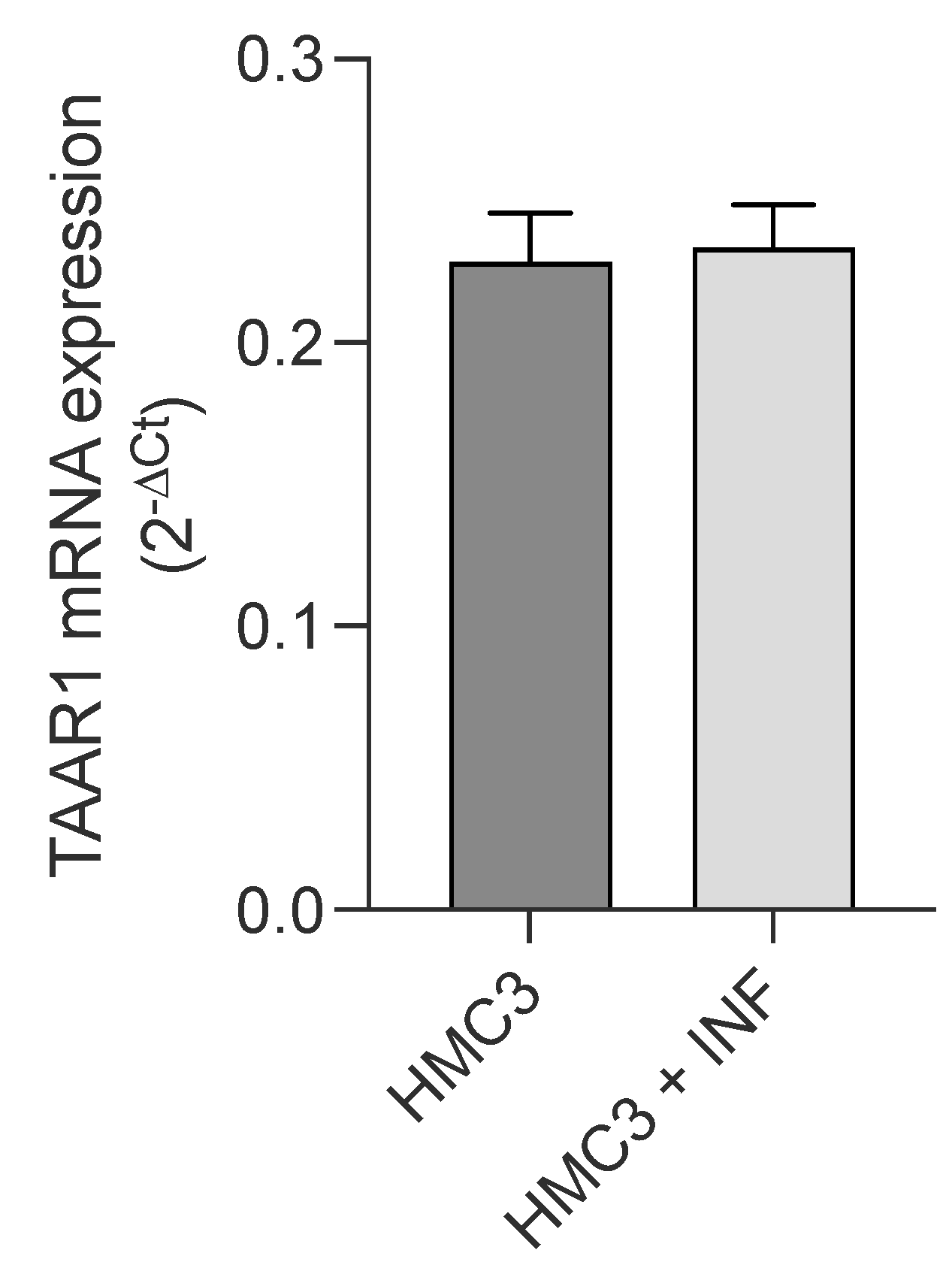
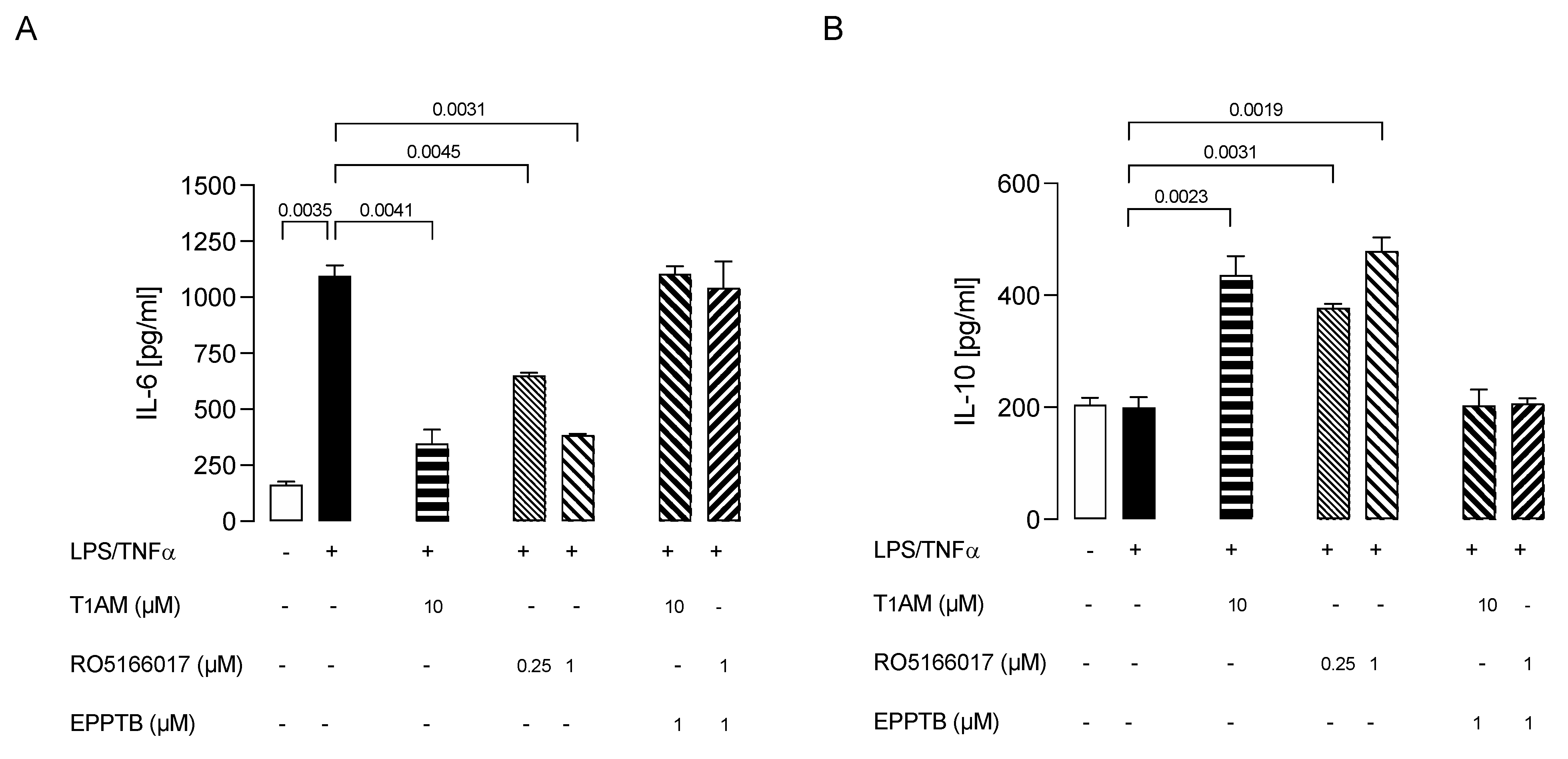
2.3. Trace Amine-Associated Receptor TAAR1 Is Involved in T1AM-Mediated Anti-Inflammatory Response of Microglial Cells
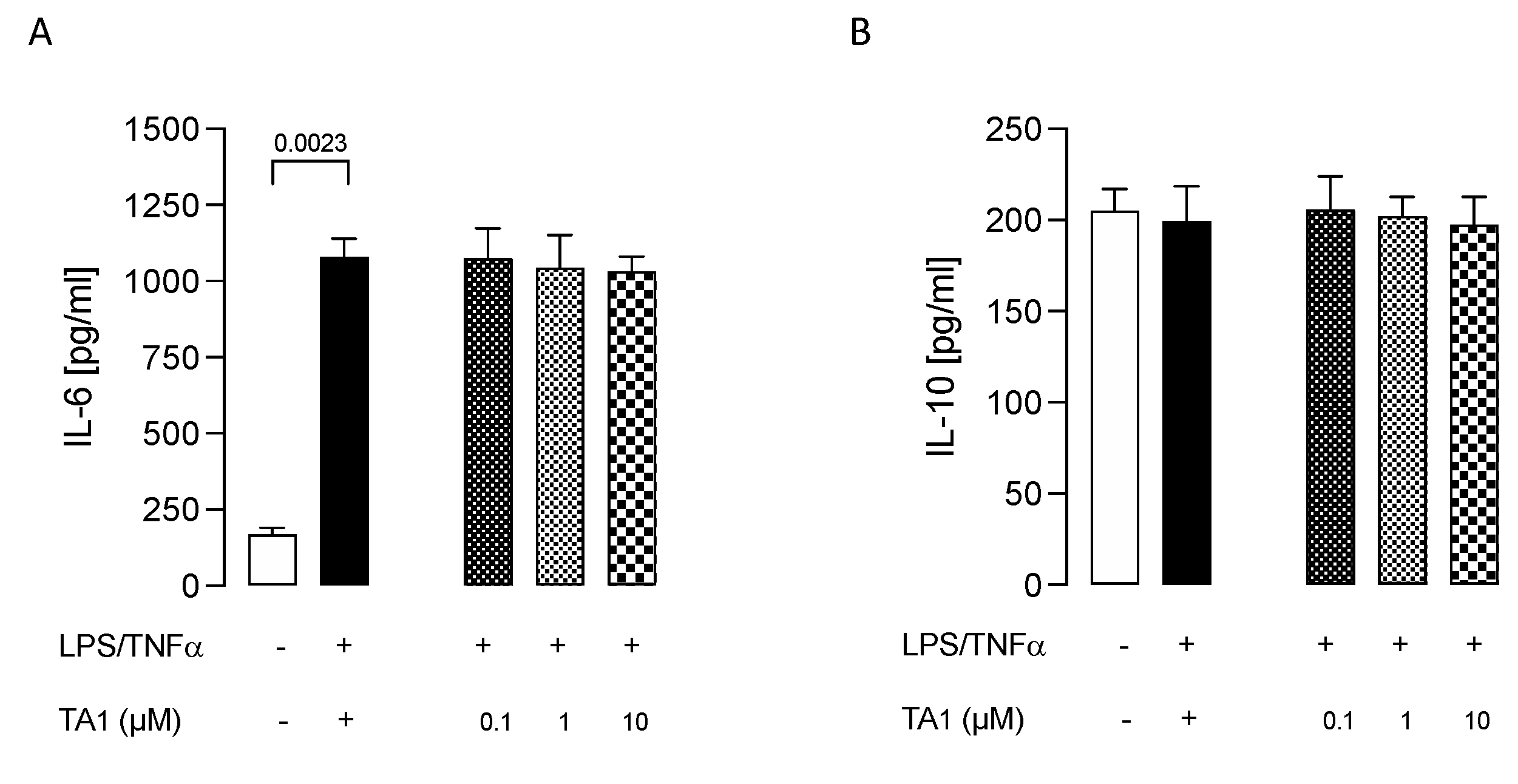
2.4. 3-Iodothyroacetic Acid (TA1) Was Not Able to Decrease the Inflammatory Phenotype of LPS/TNFα-Stimulated HMC3 Human Microglial Cells
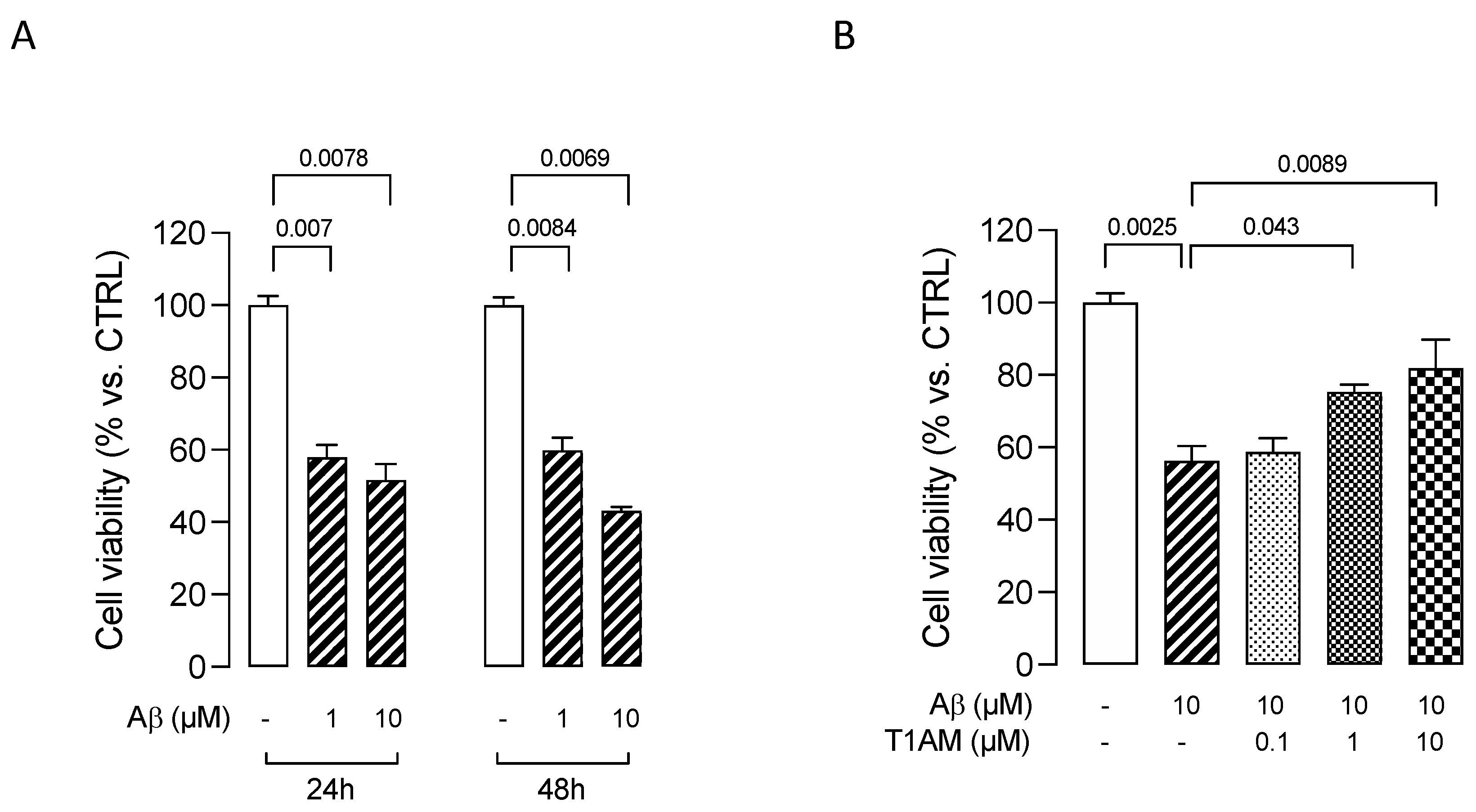
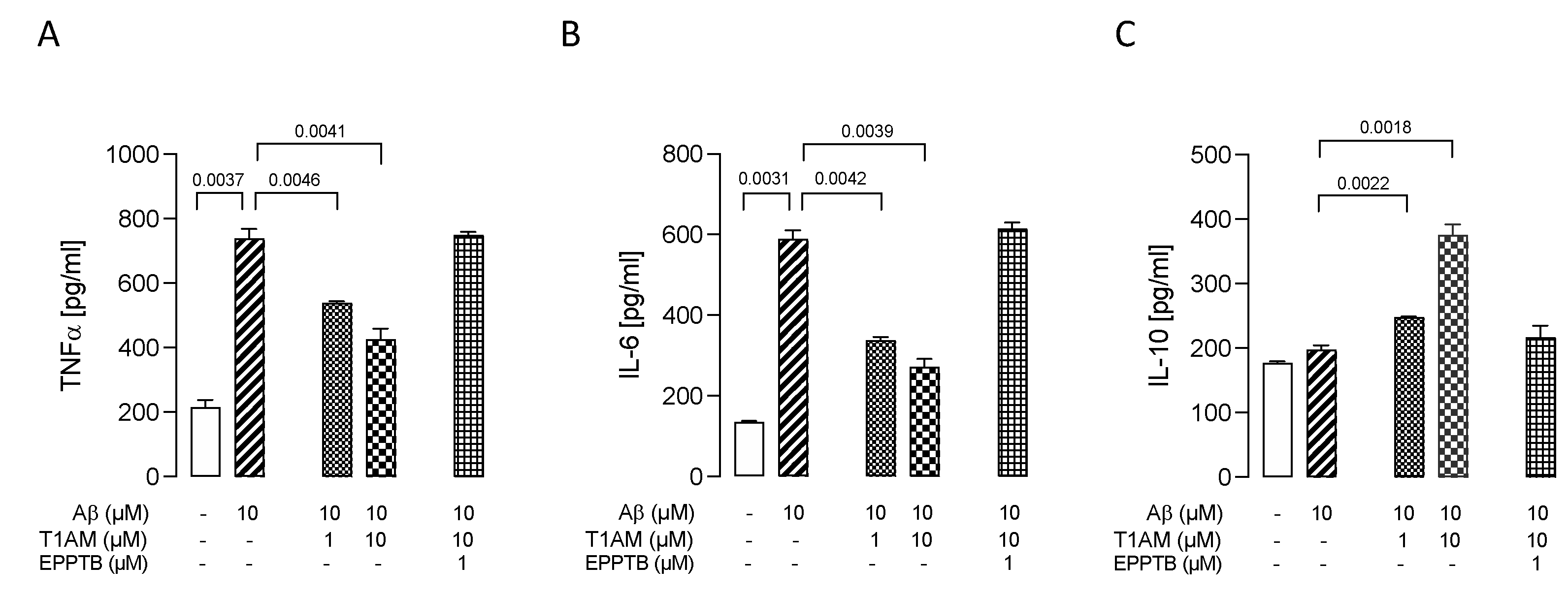
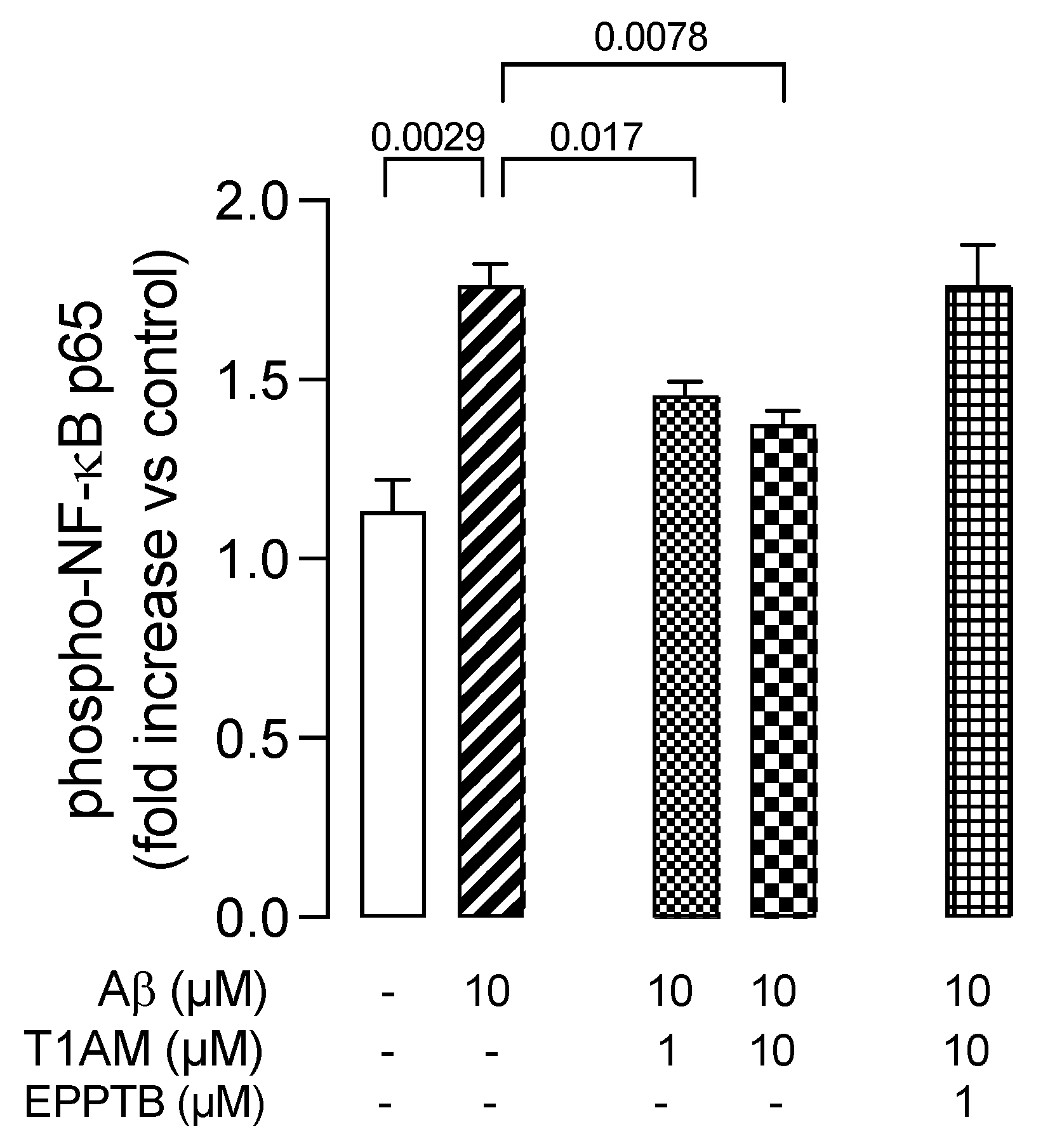
2.5. T1AM-TAAR1 System Protects against Aβ25–35-Mediated Release of Proinflammatory Factors in HMC3 Cells
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Analysis of T1AM and TA1
4.3. Cell Cultures and Reagents
4.4. MTT (Cell Viability Assay)
4.5. Release of Inflammatory Molecules HMC3 Cells Treated with LPS/TNFα
4.6. Release of Inflammatory Molecules from Aβ25–35-Induced HMC3 Cells
4.7. Gene Expression Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Mecha, M.; Feliú, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutiérrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids Drive the Acquisition of an Alternative Phenotype in Microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef]
- Reith, W. Neurodegenerative Erkrankungen. Radiologe 2018, 58, 241–258. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-Mediated Neuroinflammation in Neurodegenerative Diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of Neuroinflammation in Neurodegenerative Diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-Propagation of Pathogenic Protein Aggregates in Neurodegenerative Diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic Mechanisms of Neurodegeneration: A Critical Update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Thameem Dheen, S.; Kaur, C.; Ling, E.-A. Microglial Activation and Its Implications in the Brain Diseases. CMC 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial Activation and Chronic Neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Scanlan, T.S.; Suchland, K.L.; Hart, M.E.; Chiellini, G.; Huang, Y.; Kruzich, P.J.; Frascarelli, S.; Crossley, D.A.; Bunzow, J.R.; Ronca-Testoni, S.; et al. 3-Iodothyronamine Is an Endogenous and Rapid-Acting Derivative of Thyroid Hormone. Nat. Med. 2004, 10, 638–642. [Google Scholar] [CrossRef]
- Barak, L.S.; Salahpour, A.; Zhang, X.; Masri, B.; Sotnikova, T.D.; Ramsey, A.J.; Violin, J.D.; Lefkowitz, R.J.; Caron, M.G.; Gainetdinov, R.R. Pharmacological Characterization of Membrane-Expressed Human Trace Amine-Associated Receptor 1 (TAAR1) by a Bioluminescence Resonance Energy Transfer CAMP Biosensor. Mol. Pharmacol. 2008, 74, 585–594. [Google Scholar] [CrossRef]
- Bellusci, L.; Laurino, A.; Sabatini, M.; Sestito, S.; Lenzi, P.; Raimondi, L.; Rapposelli, S.; Biagioni, F.; Fornai, F.; Salvetti, A.; et al. New Insights into the Potential Roles of 3-Iodothyronamine (T1AM) and Newly Developed Thyronamine-Like TAAR1 Agonists in Neuroprotection. Front. Pharmacol. 2017, 8, 905. [Google Scholar] [CrossRef]
- Bellusci, L.; Runfola, M.; Carnicelli, V.; Sestito, S.; Fulceri, F.; Santucci, F.; Lenzi, P.; Fornai, F.; Rapposelli, S.; Origlia, N.; et al. Endogenous 3-Iodothyronamine (T1AM) and Synthetic Thyronamine-Like Analog SG-2 Act as Novel Pleiotropic Neuroprotective Agents through the Modulation of SIRT6. Molecules 2020, 25, 1054. [Google Scholar] [CrossRef]
- Tozzi, F.; Rutigliano, G.; Borsò, M.; Falcicchia, C.; Zucchi, R.; Origlia, N. T1AM-TAAR1 Signalling Protects against OGD-Induced Synaptic Dysfunction in the Entorhinal Cortex. Neurobiol. Dis. 2021, 151, 105271. [Google Scholar] [CrossRef]
- Landucci, E.; Gencarelli, M.; Mazzantini, C.; Laurino, A.; Pellegrini-Giampietro, D.E.; Raimondi, L. N-(3-Ethoxy-Phenyl)-4-Pyrrolidin-1-Yl-3-Trifluoromethyl-Benzamide (EPPTB) Prevents 3-Iodothyronamine (T1AM)-Induced Neuroprotection against Kainic Acid Toxicity. Neurochem. Int. 2019, 129, 104460. [Google Scholar] [CrossRef]
- Di Leo, N.; Moscato, S.; Borso’, M.; Sestito, S.; Polini, B.; Bandini, L.; Grillone, A.; Battaglini, M.; Saba, A.; Mattii, L.; et al. Delivery of Thyronamines (TAMs) to the Brain: A Preliminary Study. Molecules 2021, 26, 1616. [Google Scholar] [CrossRef]
- Xie, Z.; Miller, G.M. Trace Amine-Associated Receptor 1 as a Monoaminergic Modulator in Brain. Biochem. Pharmacol. 2009, 78, 1095–1104. [Google Scholar] [CrossRef]
- Lindemann, L.; Meyer, C.A.; Jeanneau, K.; Bradaia, A.; Ozmen, L.; Bluethmann, H.; Bettler, B.; Wettstein, J.G.; Borroni, E.; Moreau, J.-L.; et al. Trace Amine-Associated Receptor 1 Modulates Dopaminergic Activity. J. Pharmacol. Exp. Ther. 2008, 324, 948–956. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.-L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 Activation Modulates Monoaminergic Neurotransmission, Preventing Hyperdopaminergic and Hypoglutamatergic Activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef]
- Al-Sabri, M.H.; Nikpour, M.; Clemensson, L.E.; Attwood, M.M.; Williams, M.J.; Rask-Anderson, M.; Mwinyi, J.; Schiöth, H.B. The Regulatory Role of AP-2β in Monoaminergic Neurotransmitter Systems: Insights on Its Signalling Pathway, Linked Disorders and Theragnostic Potential. Cell Biosci. 2022, 12, 151. [Google Scholar] [CrossRef]
- Schwartz, M.D.; Canales, J.J.; Zucchi, R.; Espinoza, S.; Sukhanov, I.; Gainetdinov, R.R. Trace Amine-Associated Receptor 1: A Multimodal Therapeutic Target for Neuropsychiatric Diseases. Expert Opin. Ther. Targets 2018, 22, 513–526. [Google Scholar] [CrossRef]
- Koblan, K.S.; Kent, J.; Hopkins, S.C.; Krystal, J.H.; Cheng, H.; Goldman, R.; Loebel, A. A Non–D2-Receptor-Binding Drug for the Treatment of Schizophrenia. N. Engl. J. Med. 2020, 382, 1497–1506. [Google Scholar] [CrossRef]
- Nair, P.C.; Chalker, J.M.; McKinnon, R.A.; Langmead, C.J.; Gregory, K.J.; Bastiampillai, T. Trace Amine-Associated Receptor 1 (TAAR1): Molecular and Clinical Insights for the Treatment of Schizophrenia and Related Comorbidities. ACS Pharmacol. Transl. Sci. 2022, 5, 183–188. [Google Scholar] [CrossRef]
- Murtazina, R.Z.; Kuvarzin, S.R.; Gainetdinov, R.R. TAARs and Neurodegenerative and Psychiatric Disorders. In Handbook of Neurotoxicity; Kostrzewa, R.M., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 1–18. ISBN 978-3-030-71519-9. [Google Scholar]
- Fleischer, L.M.; Somaiya, R.D.; Miller, G.M. Review and Meta-Analyses of TAAR1 Expression in the Immune System and Cancers. Front. Pharmacol. 2018, 9, 683. [Google Scholar] [CrossRef]
- Cisneros, I.E.; Ghorpade, A. Methamphetamine and HIV-1-Induced Neurotoxicity: Role of Trace Amine Associated Receptor 1 CAMP Signaling in Astrocytes. Neuropharmacology 2014, 85, 499–507. [Google Scholar] [CrossRef]
- Dave, S.; Chen, L.; Yu, C.; Seaton, M.; Khodr, C.E.; Al-Harthi, L.; Hu, X.-T. Methamphetamine Decreases K+ Channel Function in Human Fetal Astrocytes by Activating the Trace Amine-Associated Receptor Type-1. J. Neurochem. 2019, 148, 29–45. [Google Scholar] [CrossRef]
- Barnes, D.A.; Galloway, D.A.; Hoener, M.C.; Berry, M.D.; Moore, C.S. TAAR1 Expression in Human Macrophages and Brain Tissue: A Potential Novel Facet of MS Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 11576. [Google Scholar] [CrossRef]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The Human Microglial HMC3 Cell Line: Where Do We Stand? A Systematic Literature Review. J. Neuroinflamm. 2018, 15, 259. [Google Scholar] [CrossRef]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12990. [Google Scholar] [CrossRef]
- Gado, F.; Ferrisi, R.; Polini, B.; Mohamed, K.A.; Ricardi, C.; Lucarini, E.; Carpi, S.; Domenichini, F.; Stevenson, L.A.; Rapposelli, S.; et al. Design, Synthesis, and Biological Activity of New CB2 Receptor Ligands: From Orthosteric and Allosteric Modulators to Dualsteric/Bitopic Ligands. J. Med. Chem. 2022, 65, 9918–9938. [Google Scholar] [CrossRef]
- Ferrisi, R.; Gado, F.; Polini, B.; Ricardi, C.; Mohamed, K.A.; Stevenson, L.A.; Ortore, G.; Rapposelli, S.; Saccomanni, G.; Pertwee, R.G.; et al. Design, Synthesis and Biological Evaluation of Novel Orthosteric-Allosteric Ligands of the Cannabinoid Receptor Type 2 (CB2R). Front. Chem. 2022, 10, 984069. [Google Scholar] [CrossRef]
- Ferrisi, R.; Polini, B.; Ricardi, C.; Gado, F.; Mohamed, K.A.; Baron, G.; Faiella, S.; Poli, G.; Rapposelli, S.; Saccomanni, G.; et al. New Insights into Bitopic Orthosteric/Allosteric Ligands of Cannabinoid Receptor Type 2. Int. J. Mol. Sci. 2023, 24, 2135. [Google Scholar] [CrossRef]
- Ghelardoni, S.; Chiellini, G.; Frascarelli, S.; Saba, A.; Zucchi, R. Uptake and Metabolic Effects of 3-Iodothyronamine in Hepatocytes. J. Endocrinol. 2014, 221, 101–110. [Google Scholar] [CrossRef]
- Assadi-Porter, F.; Reiland, H.; Sabatini, M.; Lorenzini, L.; Carnicelli, V.; Rogowski, M.; Selen Alpergin, E.; Tonelli, M.; Ghelardoni, S.; Saba, A.; et al. Metabolic Reprogramming by 3-Iodothyronamine (T1AM): A New Perspective to Reverse Obesity through Co-Regulation of Sirtuin 4 and 6 Expression. Int. J. Mol. Sci. 2018, 19, 1535. [Google Scholar] [CrossRef]
- Saba, A.; Chiellini, G.; Frascarelli, S.; Marchini, M.; Ghelardoni, S.; Raffaelli, A.; Tonacchera, M.; Vitti, P.; Scanlan, T.S.; Zucchi, R. Tissue Distribution and Cardiac Metabolism of 3-Iodothyronamine. Endocrinology 2010, 151, 5063–5073. [Google Scholar] [CrossRef]
- Accorroni, A.; Rutigliano, G.; Sabatini, M.; Frascarelli, S.; Borsò, M.; Novelli, E.; Bandini, L.; Ghelardoni, S.; Saba, A.; Zucchi, R.; et al. Exogenous 3-Iodothyronamine Rescues the Entorhinal Cortex from β-Amyloid Toxicity. Thyroid 2020, 30, 147–160. [Google Scholar] [CrossRef]
- Siemian, J.N.; Zhang, Y.; Li, J.-X. Trace Amine-Associated Receptor 1 Agonists RO5263397 and RO5166017 Attenuate Quinpirole-Induced Yawning but Not Hypothermia in Rats. Behav. Pharmacol. 2017, 28, 590–593. [Google Scholar] [CrossRef]
- Laurino, A.; Landucci, E.; Raimondi, L. Central Effects of 3-Iodothyronamine Reveal a Novel Role for Mitochondrial Monoamine Oxidases. Front. Endocrinol. 2018, 9, 290. [Google Scholar] [CrossRef]
- Laurino, A.; De Siena, G.; Saba, A.; Chiellini, G.; Landucci, E.; Zucchi, R.; Raimondi, L. In the Brain of Mice, 3-Iodothyronamine (T1AM) Is Converted into 3-Iodothyroacetic Acid (TA1) and It Is Included within the Signaling Network Connecting Thyroid Hormone Metabolites with Histamine. Eur. J. Pharmacol. 2015, 761, 130–134. [Google Scholar] [CrossRef]
- Musilli, C.; De Siena, G.; Manni, M.E.; Logli, A.; Landucci, E.; Zucchi, R.; Saba, A.; Donzelli, R.; Passani, M.B.; Provensi, G.; et al. Histamine Mediates Behavioural and Metabolic Effects of 3-Iodothyroacetic Acid, an Endogenous End Product of Thyroid Hormone Metabolism: A Novel Link between Thyroid and Histamine. Br. J. Pharmacol. 2014, 171, 3476–3484. [Google Scholar] [CrossRef]
- Shi, S.; Liang, D.; Chen, Y.; Xie, Y.; Wang, Y.; Wang, L.; Wang, Z.; Qiao, Z. Gx-50 Reduces β-Amyloid-Induced TNF-α, IL-1β, NO, and PGE2 Expression and Inhibits NF-ΚB Signaling in a Mouse Model of Alzheimer’s Disease. Eur. J. Immunol. 2016, 46, 665–676. [Google Scholar] [CrossRef]
- Cisbani, G.; Rivest, S. Targeting Innate Immunity to Protect and Cure Alzheimer’s Disease: Opportunities and Pitfalls. Mol. Psychiatry 2021, 26, 5504–5515. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Bian, J.-S. Hydrogen Sulfide Protects Amyloid-β Induced Cell Toxicity in Microglia. JAD 2011, 22, 1189–1200. [Google Scholar] [CrossRef]
- Ranaivo, H.R.; Craft, J.M.; Hu, W.; Guo, L.; Wing, L.K.; Van Eldik, L.J.; Watterson, D.M. Glia as a Therapeutic Target: Selective Suppression of Human Amyloid-β-Induced Upregulation of Brain Proinflammatory Cytokine Production Attenuates Neurodegeneration. J. Neurosci. 2006, 26, 662–670. [Google Scholar] [CrossRef]
- Naldi, M.; Fiori, J.; Pistolozzi, M.; Drake, A.F.; Bertucci, C.; Wu, R.; Mlynarczyk, K.; Filipek, S.; De Simone, A.; Andrisano, V. Amyloid β-Peptide 25–35 Self-Assembly and Its Inhibition: A Model Undecapeptide System to Gain Atomistic and Secondary Structure Details of the Alzheimer’s Disease Process and Treatment. ACS Chem. Neurosci. 2012, 3, 952–962. [Google Scholar] [CrossRef]
- Mattson, M.P.; Camandola, S. NF-ΚB in Neuronal Plasticity and Neurodegenerative Disorders. J. Clin. Investig. 2001, 107, 247–254. [Google Scholar] [CrossRef]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-ΚB Drives Tau Spreading and Toxicity in a Mouse Model of Tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef]
- Christian, S.L.; Berry, M.D. Trace Amine-Associated Receptors as Novel Therapeutic Targets for Immunomodulatory Disorders. Front. Pharmacol. 2018, 9, 680. [Google Scholar] [CrossRef]
- Rutigliano, G.; Accorroni, A.; Zucchi, R. The Case for TAAR1 as a Modulator of Central Nervous System Function. Front. Pharmacol. 2018, 8, 987. [Google Scholar] [CrossRef]
- Atienza, M.; Ziontz, J.; Cantero, J.L. Low-Grade Inflammation in the Relationship between Sleep Disruption, Dysfunctional Adiposity, and Cognitive Decline in Aging. Sleep Med. Rev. 2018, 42, 171–183. [Google Scholar] [CrossRef]
- Sama, D.M.; Norris, C.M. Calcium Dysregulation and Neuroinflammation: Discrete and Integrated Mechanisms for Age-Related Synaptic Dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef]
- Akiyama, H. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Liang, J.; Takeuchi, H.; Doi, Y.; Kawanokuchi, J.; Sonobe, Y.; Jin, S.; Yawata, I.; Li, H.; Yasuoka, S.; Mizuno, T.; et al. Excitatory Amino Acid Transporter Expression by Astrocytes Is Neuroprotective against Microglial Excitotoxicity. Brain Res. 2008, 1210, 11–19. [Google Scholar] [CrossRef]
- Pubill, D.; Canudas, A.M.; Pallàs, M.; Camins, A.; Camarasa, J.; Escubedo, E. Different Glial Response to Methamphetamine- and Methylenedioxymethamphetamine-Induced Neurotoxicity. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 367, 490–499. [Google Scholar] [CrossRef]
- Tremblay, M.-È. Microglial Functional Alteration and Increased Diversity in the Challenged Brain: Insights into Novel Targets for Intervention. Brain Behav. Immun. Health 2021, 16, 100301. [Google Scholar] [CrossRef]
- Chiellini, G.; Nesi, G.; Digiacomo, M.; Malvasi, R.; Espinoza, S.; Sabatini, M.; Frascarelli, S.; Laurino, A.; Cichero, E.; Macchia, M.; et al. Design, Synthesis, and Evaluation of Thyronamine Analogues as Novel Potent Mouse Trace Amine Associated Receptor 1 (mTAAR1) Agonists. J. Med. Chem. 2015, 58, 5096–5107. [Google Scholar] [CrossRef]
- Laurino, A.; Gencarelli, M.; Raimondi, L. The 3-Iodothyronamine (T1AM) and the 3-Iodothyroacetic Acid (TA1) Indicate a Novel Connection with the Histamine System for Neuroprotection. Eur. J. Pharmacol. 2021, 912, 174606. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Arai, T.; Kondo, H.; Tanno, E.; Haga, C.; Ikeda, K. Cell Mediators of Inflammation in the Alzheimer Disease Brain. Alzheimer Dis. Assoc. Disord. 2000, 14, S47–S53. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, M.; Prada, I.; Joshi, P.; Falcicchia, C.; D’Arrigo, G.; Rutigliano, G.; Battocchio, E.; Zenatelli, R.; Tozzi, F.; Radeghieri, A.; et al. Microglial Large Extracellular Vesicles Propagate Early Synaptic Dysfunction in Alzheimer’s Disease. Brain 2022, 145, 2849–2868. [Google Scholar] [CrossRef]
- Hauss-Wegrzyniak, B.; Lynch, M.A.; Vraniak, P.D.; Wenk, G.L. Chronic Brain Inflammation Results in Cell Loss in the Entorhinal Cortex and Impaired LTP in Perforant Path-Granule Cell Synapses. Exp. Neurol. 2002, 176, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Kelly, Á.; Vereker, E.; Nolan, Y.; Brady, M.; Barry, C.; Loscher, C.E.; Mills, K.H.G.; Lynch, M.A. Activation of P38 Plays a Pivotal Role in the Inhibitory Effect of Lipopolysaccharide and Interleukin-1β on Long Term Potentiation in Rat Dentate Gyrus. J. Biol. Chem. 2003, 278, 19453–19462. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Medium | Cell Lysates | ||
|---|---|---|---|---|
| T1AM (nM) | TA1 (nM) | T1AM (nM) | TA1 (nM) | |
| 0 | N.D. | N.D. | N.D. | N.D. |
| 5 | 64.1 ± 2.70 | 0.31 ± 0.020 | 26.0 ± 0.28 | 0.06 ± 0.01 |
| 15 | 57.2 ± 5.40 | 0.66 ± 0.20 | 24.9 ± 0.74 | 0.12 ± 0.02 |
| 30 | 56.7 ± 8.10 | 1.33 ± 0.15 | 27.0 ± 0.30 | 0.14 ± 0.02 |
| 60 | 53.4 ± 3.60 | 3.81 ± 0.80 | 24.5 ± 0.81 | 0.23 ± 0.04 |
| Reference Sequence (RefSeq) RNA | Gene Symbol | Primer Sequences |
|---|---|---|
| NM_002046 | GAPDH | (F) 5′-CCCTTCATTGACCTCAACTACATG |
| (R) 5′-TGGGATTTCCATTGATGACAAGC | ||
| NM_002982.4 | MCP1 | (F) 5′-GAGAGGCTGAGACTAACC |
| (R) 5′-TGATTGCATCTGGCTGAG | ||
| NM_002983 | MIP1 | (F) 5′-ACTTTGAGACGAGCAGCCAGTG |
| (R) 5′-TTTCTGGACCCACTCCTCACTG | ||
| NM_001404662 | NFKB | (F) 5′-CCTTTCTCATCCCATCTTT |
| (R) 5′-CCTCAATGTCCTCTTTCTG | ||
| NM_138327 | TAAR1 | (F) 5′-GAGATCTGCTGAGCACTGTTGG |
| (R) 5′-CAGCATAGTAGCGGTCAATGGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polini, B.; Ricardi, C.; Bertolini, A.; Carnicelli, V.; Rutigliano, G.; Saponaro, F.; Zucchi, R.; Chiellini, G. T1AM/TAAR1 System Reduces Inflammatory Response and β-Amyloid Toxicity in Human Microglial HMC3 Cell Line. Int. J. Mol. Sci. 2023, 24, 11569. https://doi.org/10.3390/ijms241411569
Polini B, Ricardi C, Bertolini A, Carnicelli V, Rutigliano G, Saponaro F, Zucchi R, Chiellini G. T1AM/TAAR1 System Reduces Inflammatory Response and β-Amyloid Toxicity in Human Microglial HMC3 Cell Line. International Journal of Molecular Sciences. 2023; 24(14):11569. https://doi.org/10.3390/ijms241411569
Chicago/Turabian StylePolini, Beatrice, Caterina Ricardi, Andrea Bertolini, Vittoria Carnicelli, Grazia Rutigliano, Federica Saponaro, Riccardo Zucchi, and Grazia Chiellini. 2023. "T1AM/TAAR1 System Reduces Inflammatory Response and β-Amyloid Toxicity in Human Microglial HMC3 Cell Line" International Journal of Molecular Sciences 24, no. 14: 11569. https://doi.org/10.3390/ijms241411569
APA StylePolini, B., Ricardi, C., Bertolini, A., Carnicelli, V., Rutigliano, G., Saponaro, F., Zucchi, R., & Chiellini, G. (2023). T1AM/TAAR1 System Reduces Inflammatory Response and β-Amyloid Toxicity in Human Microglial HMC3 Cell Line. International Journal of Molecular Sciences, 24(14), 11569. https://doi.org/10.3390/ijms241411569