

Transglutaminase 2 Facilitates Murine Wound Healing in a Strain-Dependent Manner


Abstract
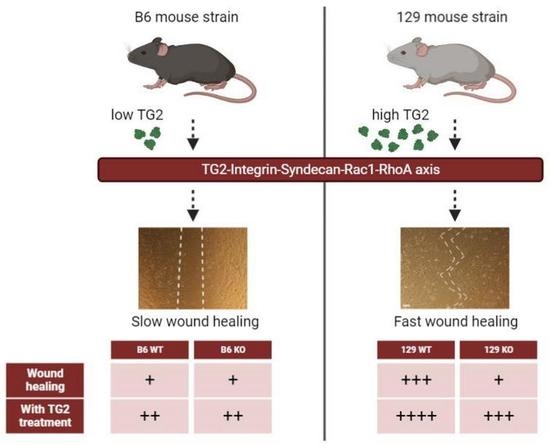
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Early Healing of Cutaneous Wounds Was Slower in 129 TG2−/− Mice Than in 129 TG2+/+, B6 TG2+/+ or B6 TG2−/− Mice
2.2. Bupivacaine Injection Prior to Skin Punch Biopsy Abrogated the Difference in Wound Healing between 129 TG2−/− and 129 TG2+/+ Mice and Delayed Wound Healing
2.3. Neutrophil and Monocyte Recruitment to Cutaneous Wounds Was No Different between 129 TG2−/− Mice and TG2+/+ Mice up to 5 Days Post-Wounding
2.4. mRNA Abundance of Wound-Related Genes Was Increased to the Same Levels in 129 TG2−/− and TG2+/+ Mice on Day 2 Post-Wounding
2.5. A Single Dose of TG2 Protein to Cutaneous Wounds in 129 TG2−/− Mice Restored Rate of Wound Healing to That in 129 TG2+/+ Mice
2.6. The Number of 129 TG2−/− MEFs Adherent to an Fn Matrix Was Less Compared to 129 TG2+/+ MEFs, but Was No Different from B6 TG2+/+ or B6 TG2−/− MEFs
2.7. The Number of MEFs Adhered to Fn Was Increased by the Exogenous Addition of Recombinant TG2 and Required the β-Sandwich and Core Domains but Not the Transamidase or GTP-Binding Activity of TG2
2.8. Adhesion of 129 TG2+/+ or 129 TG2−/− MEFs Was Dependent on Syndecan Binding in the Presence, but Not Absence, of RGD Peptides, and the Exogenous Addition of Recombinant Wild-Type TG2 to an Fn Matrix Rescued RGD-Impaired MEF Adhesion
2.9. Spreading of 129 TG2−/−, B6 TG2+/+ and B6 TG2−/− MEFs Was Equivalent but Delayed Relative to 129 TG2+/+ MEFs, and Was Accelerated in All Genotypes by Either the Exogenous Addition of Recombinant TG2 or Transfection of cDNAs Encoding TG2, Independent of Transamidase or GTP-Binding Activity
2.10. Activation Dynamics of the GTPases RhoA and Rac1 That Control Cell Adhesion and Spreading on Fn Were Altered in 129 TG2−/− Relative to TG2+/+ MEFs
2.11. Closure of an In Vitro Scratch Wound in Confluent Cell Monolayers Was Delayed in 129 but Not B6 TG2−/− MEFs, Relative to Their TG2+/+ Counterparts, and the Exogenous Addition of Recombinant Wild-Type TG2 Immediately Post-Wounding Accelerated Wound Closure in All Genotypes
2.12. TG2 Expression Levels Are Higher in 129 Than in B6 Mice
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animal and Cell Culture
4.3. Recombinant Rat TG2 Production
4.4. In-Vivo Wound Healing Assay
4.5. Histological Analysis of Cutaneous Wounds
4.6. Wound RNA Extraction
4.7. Microarray Analysis of Total RNA Isolated from Cutaneous Wounds
4.8. Quantitative PCR (qPCR) Analysis of Wound RNA
4.9. Isolation and Maintenance of Mouse Embryonic Fibroblasts
4.10. Cell Proliferation Assay
4.11. In Vitro Cell Adhesion Assay
4.12. In Vitro Cell Spreading Assay
4.13. Transfection of MEFs with TG2-IRES-GFP Expression Vectors
4.14. RhoA and Rac1 Profiling in MEFs
4.15. In Vitro Scratch Wound Assay
4.16. Western Blotting
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.R.; Humphries, M.J.; Bass, M.D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Tong, X.; You, S.; Mao, R.; Cai, E.; Pan, W.; Zhang, C.; Hu, R.; Shen, J. Mild Hyperthermia-Assisted ROS Scavenging Hydrogels Achieve Diabetic Wound Healing. ACS Macro Lett. 2022, 11, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.J.; Qi, X.L.; Cai, E.; Zhang, C.F.; Wang, J.J.; Lan, Y.L.; Deng, H.; Shen, J.L.; Hu, R.D. Highly efficient bacteria-infected diabetic wound healing employing a melanin-reinforced biopolymer hydrogel. Chem. Eng. J. 2023, 460, 141852. [Google Scholar] [CrossRef]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Sane, D.C.; Moser, T.L.; Pippen, A.M.; Parker, C.J.; Achyuthan, K.E.; Greenberg, C.S. Vitronectin is a substrate for transglutaminases. Biochem. Biophys. Res. Commun. 1988, 157, 115–120. [Google Scholar] [CrossRef]
- Achyuthan, K.E.; Rowland, T.C.; Birckbichler, P.J.; Lee, K.N.; Bishop, P.D.; Achyuthan, A.M. Hierarchies in the binding of human factor XIII, factor XIIIa, and endothelial cell transglutaminase to human plasma fibrinogen, fibrin, and fibronectin. Mol. Cell Biochem. 1996, 162, 43–49. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Paulsson, M. Cross-linking of laminin-nidogen complexes by tissue transglutaminase. A novel mechanism for basement membrane stabilisation. J. Biol. Chem. 1991, 266, 15308–15317. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Wetterwald, A.; Fleisch, H.; Paulsson, M. Expression of tissue transglutaminase in skeletal tissues correlates with events of terminal differentiation of chondrocytes. J. Cell Biol. 1993, 120, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.S.; Achyuthan, K.E.; Borowitz, M.J.; Shuman, M.A. The transglutaminase in vascular cells and tissues could provide an alternate pathway for fibrin stabilization. Blood 1987, 70, 702–709. [Google Scholar] [CrossRef]
- Kaartinen, M.T.; Pirhonen, A.; LinnalaKankkunen, A.; Maenpaa, P.H. Transglutaminase-catalyzed cross-linking of osteopontin is inhibited by osteocalcin. J. Biol. Chem. 1997, 272, 22736–22741. [Google Scholar] [CrossRef]
- Kleman, J.P.; Aeschlimann, D.; Paulsson, M.; van der Rest, M. Transglutaminase-catalyzed cross-linking of fibrils of collagen V/XI in A204 rhabdomyosarcoma cells. Biochemistry 1995, 34, 13768–13775. [Google Scholar] [CrossRef]
- Im, M.-J.; Riek, R.P.; Graham, R.M. A novel guanine nucleotide-binding protein coupled to the α1-adrenergic receptor. II. Purification, characterization, and reconstitution. J. Biol. Chem. 1990, 265, 18952–18960. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, H.; Perez, D.M.; Baek, K.J.; Das, T.; Husain, A.; Misono, K.; Im, M.J.; Graham, R.M. Gh: A GTP-binding protein with transglutaminase activity and receptor signaling function. Science 1994, 264, 1593–1596. [Google Scholar] [CrossRef]
- Begg, G.E.; Carrington, L.; Stokes, P.H.; Matthews, J.M.; Wouters, M.A.; Husain, A.; Lorand, L.; Iismaa, S.E.; Graham, R.M. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc. Natl. Acad. Sci. USA 2006, 103, 19683–19688. [Google Scholar] [CrossRef] [PubMed]
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Gaudry, C.A.; Verderio, E.; Aeschlimann, D.; Cox, A.; Smith, C.; Griffin, M. Cell surface localization of tissue transglutaminase is dependent on a fibronectin-binding site in its N-terminal b-sandwich domain. J. Biol. Chem. 1999, 274, 30707–30714. [Google Scholar] [CrossRef] [PubMed]
- Telci, D.; Wang, Z.; Li, X.; Verderio, E.A.; Humphries, M.J.; Baccarini, M.; Basaga, H.; Griffin, M. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and beta1 integrin co-signaling. J. Biol. Chem. 2008, 283, 20937–20947. [Google Scholar] [CrossRef]
- Wang, Z.; Collighan, R.J.; Gross, S.R.; Danen, E.H.; Orend, G.; Telci, D.; Griffin, M. RGD-independent cell adhesion via a tissue transglutaminase-fibronectin matrix promotes fibronectin fibril deposition and requires syndecan-4/2 alpha5beta1 integrin co-signaling. J. Biol. Chem. 2010, 285, 40212–40229. [Google Scholar] [CrossRef]
- Greenberg, C.S.; Birckbichler, P.J.; Rice, R.H. Transglutaminases—Multifunctional Cross-Linking Enzymes That Stabilize Tissues. FASEB J. 1991, 5, 3071–3077. [Google Scholar] [CrossRef] [PubMed]
- Gaudry, C.A.; Verderio, E.; Jones, R.A.; Smith, C.; Griffin, M. Tissue transglutaminase is an important player at the surface of human endothelial cells: Evidence for its externalization and its colocalization with the beta(1) integrin. Exp. Cell Res. 1999, 252, 104–113. [Google Scholar] [CrossRef]
- Verderio, E.A.; Johnson, T.; Griffin, M. Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids 2004, 26, 387–404. [Google Scholar] [CrossRef]
- Verderio, E.A.; Johnson, T.S.; Griffin, M. Transglutaminases in wound healing and inflammation. Prog. Exp. Tumor Res. 2005, 38, 89–114. [Google Scholar] [CrossRef] [PubMed]
- Haroon, Z.A.; Hettasch, J.M.; Lai, T.S.; Dewhirst, M.W.; Greenberg, C.S. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999, 13, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Bowness, J.M.; Henteleff, H.; Dolynchuk, K.N. Components of increased labelling with putrescine and fucose during healing of skin wounds. Connect. Tissue Res. 1987, 16, 57–70. [Google Scholar] [CrossRef]
- Bowness, J.M.; Tarr, A.H.; Wong, T. Increased transglutaminase activity during skin wound healing in rats. Biochim. Biophys. Acta 1988, 967, 234–240. [Google Scholar] [CrossRef]
- Gentile, V.; Thomazy, V.; Piacentini, M.; Fesus, L.; Davies, P.J. Expression of tissue transglutaminase in Balb-C 3T3 fibroblasts: Effects on cellular morphology and adhesion. J. Cell Biol. 1992, 119, 463–474. [Google Scholar] [CrossRef]
- Jones, R.A.; Nicholas, B.; Mian, S.; Davies, P.J.; Griffin, M. Reduced expression of tissue transglutaminase in a human endothelial cell line leads to changes in cell spreading, cell adhesion and reduced polymerisation of fibronectin. J. Cell Sci. 1997, 110, 2461–2472. [Google Scholar] [CrossRef]
- Stephens, P.; Grenard, P.; Aeschlimann, P.; Langley, M.; Blain, E.; Errington, R.; Kipling, D.; Thomas, D.; Aeschlimann, D. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 2004, 117, 3389–3403. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Celep, I.; Nihan Kilinc, A.; Griffin, M.; Telci, D. Nitrosylation of tissue transglutaminase enhances fibroblast migration and regulates MMP activation. Matrix Biol. 2022, 105, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Aplin, M.; Holman, S.; Yiu, T.W.; Jackson, K.; Burchfield, J.G.; Mitchell, C.J.; O’Reilly, L.; Davenport, A.; Cantley, J.; et al. Glucose homeostasis in mice is transglutaminase 2 independent. PLoS ONE 2013, 8, e63346. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 2004, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Tsai, P.S.; Huang, C.J. Bupivacaine inhibits COX-2 expression, PGE2, and cytokine production in endotoxin-activated macrophages. Acta Anaesthesiol. Scand. 2008, 52, 530–535. [Google Scholar] [CrossRef]
- Chen, L.; DiPietro, L.A. Toll-Like Receptor Function in Acute Wounds. Adv. Wound Care 2017, 6, 344–355. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takeda, K.; Akira, S. TIR domain-containing adaptors define the specificity of TLR signaling. Mol. Immunol. 2004, 40, 861–868. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.S.; Choi, D.H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.Y. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735. [Google Scholar] [CrossRef]
- Iismaa, S.E.; Holman, S.; Wouters, M.A.; Lorand, L.; Graham, R.M.; Husain, A. Evolutionary specialization of a tryptophan indole group for transition-state stabilization by eukaryotic transglutaminases. Proc. Natl. Acad. Sci. USA 2003, 100, 12636–12641. [Google Scholar] [CrossRef]
- Begg, G.E.; Holman, S.R.; Stokes, P.H.; Matthews, J.M.; Graham, R.M.; Iismaa, S.E. Mutation of a critical arginine in the GTP-binding site of transglutaminase 2 disinhibits intracellular cross-linking activity. J. Biol. Chem. 2006, 281, 12603–12609. [Google Scholar] [CrossRef]
- Iismaa, S.E.; Chung, L.; Wu, M.J.; Teller, D.C.; Yee, V.C.; Graham, R.M. The core domain of the tissue transglutaminase Gh hydrolyzes GTP and ATP. Biochemistry 1997, 36, 11655–11664. [Google Scholar] [CrossRef] [PubMed]
- Radek, J.T.; Jeong, J.M.; Murthy, S.N.; Ingham, K.C.; Lorand, L. Affinity of human erythrocyte transglutaminase for a 42-kDa gelatin-binding fragment of human plasma fibronectin. Proc. Natl. Acad. Sci. USA 1993, 90, 3152–3156. [Google Scholar] [CrossRef] [PubMed]
- Verderio, E.A.; Telci, D.; Okoye, A.; Melino, G.; Griffin, M. A novel RGD-independent cel adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J. Biol. Chem. 2003, 278, 42604–42614. [Google Scholar] [CrossRef]
- Ren, X.D.; Kiosses, W.B.; Schwartz, M.A. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999, 18, 578–585. [Google Scholar] [CrossRef]
- Price, L.S.; Leng, J.; Schwartz, M.A.; Bokoch, G.M. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell 1998, 9, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Gutierrez-Fernandez, A.; Inada, M.; Balbin, M.; Fueyo, A.; Pitiot, A.S.; Astudillo, A.; Hirose, K.; Hirata, M.; Shapiro, S.D.; Noel, A.; et al. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8). FASEB J. 2007, 21, 2580–2591. [Google Scholar] [CrossRef]
- Michalik, L.; Desvergne, B.; Tan, N.S.; Basu-Modak, S.; Escher, P.; Rieusset, J.; Peters, J.M.; Kaya, G.; Gonzalez, F.J.; Zakany, J.; et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J. Cell Biol. 2001, 154, 799–814. [Google Scholar] [CrossRef]
- Auld, G.C.; Ritchie, H.; Robbie, L.A.; Booth, N.A. Thrombin upregulates tissue transglutaminase in endothelial cells: A potential role for tissue transglutaminase in stability of atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1689–1694. [Google Scholar] [CrossRef]
- Kuncio, G.S.; Tsyganskaya, M.; Zhu, J.; Liu, S.L.; Nagy, L.; Thomazy, V.; Davies, P.J.; Zern, M.A. TNF-alpha modulates expression of the tissue transglutaminase gene in liver cells. Am. J. Physiol. 1998, 274, G240–G245. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeong, E.J.; Steinert, P.M. IFN-gamma induces transglutaminase 2 expression in rat small intestinal cells. J. Interferon Cytokine Res. 2002, 22, 677–682. [Google Scholar] [CrossRef]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef]
- Ffrench-Constant, C.; Van de Water, L.; Dvorak, H.F.; Hynes, R.O. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J. Cell Biol. 1989, 109, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.F.; Dubin, D.; Lavigne, L.; Logan, B.; Dvorak, H.F.; Van de Water, L. Macrophages and fibroblasts express embryonic fibronectins during cutaneous wound healing. Am. J. Pathol. 1993, 142, 793–801. [Google Scholar] [PubMed]
- George, J.; Wang, S.S.; Sevcsik, A.M.; Sanicola, M.; Cate, R.L.; Koteliansky, V.E.; Bissell, D.M. Transforming growth factor-beta initiates wound repair in rat liver through induction of the EIIIA-fibronectin splice isoform. Am. J. Pathol. 2000, 156, 115–124. [Google Scholar] [CrossRef]
- Xia, P.; Culp, L.A. Adhesion activity in fibronectin’s alternatively spliced domain EDa (EIIIA): Complementarity to plasma fibronectin functions. Exp. Cell Res. 1995, 217, 517–527. [Google Scholar] [CrossRef]
- Liao, Y.F.; Gotwals, P.J.; Koteliansky, V.E.; Sheppard, D.; Van De Water, L. The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J. Biol. Chem. 2002, 277, 14467–14474. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Isobe, T.; Horibe, S.; Takagi, J.; Yokosaki, Y.; Sheppard, D.; Saito, Y. Tissue transglutaminase, coagulation factor XIII, and the pro-polypeptide of von Willebrand factor are all ligands for the integrins alpha 9beta 1 and alpha 4beta 1. J. Biol. Chem. 2000, 275, 23589–23595. [Google Scholar] [CrossRef]
- Bass, M.D.; Roach, K.A.; Morgan, M.R.; Mostafavi-Pour, Z.; Schoen, T.; Muramatsu, T.; Mayer, U.; Ballestrem, C.; Spatz, J.P.; Humphries, M.J. Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. J. Cell Biol. 2007, 177, 527–538. [Google Scholar] [CrossRef]
- Echtermeyer, F.; Streit, M.; Wilcox-Adelman, S.; Saoncella, S.; Denhez, F.; Detmar, M.; Goetinck, P. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J. Clin. Investig. 2001, 107, R9–R14. [Google Scholar] [CrossRef]
- Nanda, N.; Iismaa, S.E.; Owens, W.A.; Husain, A.; Mackay, F.; Graham, R.M. Targeted inactivation of Gh/tissue transglutaminase II. J. Biol. Chem. 2001, 276, 20673–20678. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.N.; Iismaa, S.; Begg, G.; Freymann, D.M.; Graham, R.M.; Lorand, L. Conserved tryptophan in the core domain of transglutaminase is essential for catalytic activity. Proc. Natl. Acad. Sci. USA 2002, 99, 2738–2742. [Google Scholar] [CrossRef] [PubMed]
- Benard, V.; Bohl, B.P.; Bokoch, G.M. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 1999, 274, 13198–13204. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yiu, T.W.; Holman, S.R.; Kaidonis, X.; Graham, R.M.; Iismaa, S.E. Transglutaminase 2 Facilitates Murine Wound Healing in a Strain-Dependent Manner. Int. J. Mol. Sci. 2023, 24, 11475. https://doi.org/10.3390/ijms241411475
Yiu TW, Holman SR, Kaidonis X, Graham RM, Iismaa SE. Transglutaminase 2 Facilitates Murine Wound Healing in a Strain-Dependent Manner. International Journal of Molecular Sciences. 2023; 24(14):11475. https://doi.org/10.3390/ijms241411475
Chicago/Turabian StyleYiu, Ting W., Sara R. Holman, Xenia Kaidonis, Robert M. Graham, and Siiri E. Iismaa. 2023. "Transglutaminase 2 Facilitates Murine Wound Healing in a Strain-Dependent Manner" International Journal of Molecular Sciences 24, no. 14: 11475. https://doi.org/10.3390/ijms241411475
APA StyleYiu, T. W., Holman, S. R., Kaidonis, X., Graham, R. M., & Iismaa, S. E. (2023). Transglutaminase 2 Facilitates Murine Wound Healing in a Strain-Dependent Manner. International Journal of Molecular Sciences, 24(14), 11475. https://doi.org/10.3390/ijms241411475